
Abstract
There are numerous treatment options currently available for patients with type 2 diabetes mellitus; however, a multitude of patients continue to have inadequately controlled glycemic levels with their current antihyperglycemic regimen. Furthermore, the American Diabetes Association guidelines increasingly highlight the importance of multifactorial management and optimizing medication regimens that include cardiovascular, renal, and/or weight benefits in patients with type 2 diabetes mellitus. Glucagon-like peptide-1 receptor agonists belong to a novel class of type 2 diabetes mellitus agents that are becoming increasingly prevalent owing to their ability to improve glycemic status without the risk of hypoglycemia. Currently, there are three US Food and Drug Administration-approved glucagon-like peptide-1 receptor agonists, subcutaneous semaglutide, dulaglutide, and liraglutide, that also have an indication for reducing major adverse cardiovascular events in patients with type 2 diabetes mellitus and established cardiovascular disease. However, these agents are not often the first options because of their subcutaneous administration. Nevertheless, co-formulation of oral semaglutide with an absorption enhancer has shown to increase its bioavailability and has made its oral absorption possible. In the PIONEER trials, oral semaglutide effectively lowered blood glucose levels, and showed benefits on weight and cardiovascular outcomes; however, there is no Food and Drug Administration indication approved yet as the SOUL trial is still ongoing. Such characteristics of oral semaglutide may improve and increase its use compared to subcutaneous agents and possibly lead to earlier cardiovascular protection in addition to achieving glycemic control.
Similar content being viewed by others

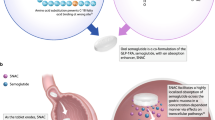
Oral semaglutide is a novel glucagon-like peptide-1 receptor agonist tablet co-formulated with the absorption enhancer sodium N-(8-[2-hydroxybenzoyl] amino) caprylate. |
Oral semaglutide was found to be safe and well tolerated across all phase III trials with the main adverse reactions being nausea, vomiting, and diarrhea, similar to the subcutaneous glucagon-like peptide-1 receptor agonist formulations. |
Oral semaglutide demonstrated good efficacy regarding glycosylated hemoglobin reduction, similar to the subcutaneous glucagon-like peptide-1 receptor agonists and superior to sitagliptin and empagliflozin in phase III trials. |
1 Background
In the USA, approximately 34 million individuals are diagnosed with diabetes mellitus, of whom 95% have type 2 diabetes mellitus (T2DM) [1]. In healthy individuals, insulin is produced by the pancreatic beta cells and is a key component in maintaining glucose homeostasis. However, in patients with T2DM, the development of insulin resistance as well as impaired insulin secretion due to beta-cell dysfunction inhibits this homeostasis [2]. In patients with uncontrolled T2DM, prolonged hyperglycemia can cause macrovascular complications, such as coronary artery disease, peripheral arterial disease, cerebrovascular accidents, cardiovascular (CV) death, and microvascular complications, such as diabetic nephropathy, diabetic peripheral neuropathy, and diabetic retinopathy. Furthermore, hyperglycemia leads to decreased production of nitric oxide from the endothelial lining of blood vessels. Nitric oxide is a vasodilator that allows blood vessels to widen, a reduction in nitric oxide can inhibit blood vessels from widening causing the pressure to increase in blood vessels and may result in stiffening and, therefore, increases the risk of hypertension [3]. To reduce the occurrence and severity of these comorbidities, proper management of elevated plasma glucose levels in patients with T2DM is mandated.
Management of T2DM begins with healthy lifestyle changes such as diet modifications and exercise [4, 5]. While this may be enough for some individuals to achieve adequate blood glucose control, many individuals with T2DM will require antihyperglycemic medications to achieve and sustain appropriate glycemic control. Metformin has been and continues to remain the preferred first-line treatment option for most patients with T2DM [4]. In the case where metformin monotherapy does not achieve a desired glycemic target of glycosylated hemoglobin (HbA1c) ≤ 7%, the 2021 American Diabetes Association Guideline for Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes recommends an add-on treatment with a glucagon-like peptide-1 receptor agonist (GLP-1RA) and/or a sodium-glucose cotransporter-2 inhibitor when CV disease (CVD), congestive heart failure, chronic kidney disease (CKD), and/or a need for weight management are present, owing to their documented weight loss benefits and potential CVD and CKD benefits, in addition to their blood glucose-lowering capabilities [4].
Currently, there are five GLP-1RAs, exenatide, liraglutide, dulaglutide, lixisenatide, and subcutaneous semaglutide, that are US Food and Drug Administration (FDA) approved and commercially available for the long-term management of T2DM. Overall, GLP-1RAs substantially lower HbA1c by up to 1.9% and reduce weight by 5–10 lbs with some having favorable effects on CVD, myocardial infarction prevention, and cerebrovascular accident prevention [4, 6,7,8,9]. However, within the GLP-RA drug class, substantial differences in the molecular structure, dosing interval, glycemic control, weight loss, immunogenicity, and tolerability profiles of each agent can cause efficacy in glycemic reduction to vary [10, 11]. In addition, some GLP‐1RAs, such as dulaglutide, liraglutide, and subcutaneous semaglutide, have shown positive results and clinical benefits in patients with renal impairment who have an estimated glomerular filtration rate (eGFR) ≥ 15 mL/min per 1.73 m2 [12, 13]. Nevertheless, despite the abundant complementary benefits of GLP-RAs, their subcutaneous administration limits their usage amongst patients [14, 15]. With the introduction of an oral option to this drug class, such as oral semaglutide, there is potential to eliminate the limitation of subcutaneous administration providing more therapeutic options for patients with T2DM.
In numerous clinical studies, subcutaneous semaglutide was demonstrated to be efficacious and safe in patients with T2DM. In the SUSTAIN 3 and SUSTAIN 7 studies, subcutaneous semaglutide once weekly exhibited superior glycemic control and weight loss compared with exenatide and dulaglutide (p < 0.0001) [13, 16]. Furthermore, in the SUSTAIN‐6 study, in patients with T2DM with CVD and/or CKD, subcutaneous semaglutide compared with placebo was associated with a significant reduction in the first occurrence of CV-related death, nonfatal myocardial infarction, or nonfatal cerebrovascular accidents (p < 0.001) [17].
The efficacy of subcutaneous semaglutide on HbA1c reduction, bodyweight (BW) reduction, and CV outcomes demonstrated in the SUSTAIN trials was anticipated to correspond with an oral formulation of semaglutide and was substantiated by the PIONEER studies described in detail further in this paper. In the PIONEER trials, the oral formulation of semaglutide has also shown efficacy and safety in patients with an eGFR as low as 30 mL/min per 1.73 m2 and has also shown improvement in the urine albumin creatinine ratio [18, 19]. Furthermore, the positive results from the PIONEER studies granted oral semaglutide (Rybelsus®) its FDA approval on 20 September, 2019, and it became the first oral GLP-1RA. There are a few good reviews on oral semaglutide including Bucheit et al. and Anderson et al. and this review hopes to add to the gaining literature and, more specifically, the safety and efficacy [20, 21].
2 Mechanism of Action
Endogenous GLP-1 is secreted by the enteroendocrine L cells in the gastrointestinal (GI) tract and is responsible for potentiating glucose-induced insulin secretion and most postprandial insulin secretion [22,23,24,25]. Glucagon-like peptide-1 receptor agonists are synthetically modified peptides similar to endogenous GLP-1 and affect the brain, pancreas, liver, heart, gut, kidneys, muscles, and vasculature causing numerous pleiotropic effects [24, 26]. These effects include signals sent to the hypothalamus reducing appetite, stimulating gluconeogenesis, lowering hepatic glucose output, amplifying glucose-dependent insulin release, inhibiting glucagon release, increasing cardiac output and cardioprotection, and decreasing high blood pressure [23, 24].
Glucagon-like peptide-1 receptor agonists have also been shown to slow gastric emptying and reduce glucagon levels by the same mechanism as endogenous GLP-1 [27]. The exposure of the small intestine to nutrients triggers a powerful inhibitory feedback mechanism to control the transit of a meal through the GI tract to optimize nutrient digestion and absorption [7, 27]. This results in highly regulated gastric emptying of nutrients, including carbohydrates from the stomach to the small intestine, which is a major determinant of postprandial glycemic excursions. Because of the effects of the GLP-1RAs on the GI tract involving satiety and slowed gastric emptying, GI side effects such as nausea are common in patients taking these agents [7, 27].
Oral semaglutide is different from other GLP-1RAs currently on the market because of its oral route of administration. The inherent physicochemical properties of peptides such as high molecular weight, enzymatically labile, increased hydrophilicity, and low permeability have hampered attempts to deliver peptides such as GLP-1 through the oral route previously [28]. Oral semaglutide undergoes gastric absorption, which is unique to this medication as most oral medications are absorbed in the intestines [21]. The enzymatic degradation of peptides in the GI tract, which often play a major role in hindering absorption leading to low bioavailability, hampers the ability to deliver peptides through the oral route. Uniquely, fatty acid acylation can achieve a prolongation of the half-life (t½) independently while having no appreciable impact on the function of the peptide. These characteristics make the semaglutide molecule able to be orally formulated when co-formulated with an absorption enhancer, which can sufficiently augment its absorption [28]. Oral semaglutide was co-formulated into a tablet with an absorption enhancer, sodium N-(8-[2-hydroxybenzoyl] amino) caprylate (SNAC) [29]. These enhancers prevent enzyme degradation and facilitate absorption because they can transiently open the inter-epithelial tight junctions and allow paracellular transport, which protects the peptide from proteolytic degradation by exerting a buffering action in the stomach, decreasing the efficacy of digestive enzymes and promoting absorption across the gastric mucosa. Once SNAC is incorporated into the gastric epithelium, it fluidizes the lipid membrane facilitating semaglutide transport and entry into the systemic circulation. When semaglutide and SNAC reach the bloodstream, the two molecules readily dissociate, allowing oral semaglutide to interact with the body in the same manner as subcutaneous semaglutide [20]. However, as the absorption enhancers can facilitate the penetration of all GI tract contents, including toxins and pathogens into the systemic circulation, when used long term, absorption enhancers may damage the bio-membrane, which can lead to local GI inflammation. [20, 30].
3 Preclinical Studies
A preclinical study by Buckley et al. explored the absorption of oral semaglutide when co-formulated with an absorption enhancer, SNAC, in humans and beagle dog models [28]. In the study, a total of 26 healthy male subjects in the fasted state received a single-dose tablet containing oral semaglutide 10 mg with SNAC 300 mg as well as 240 mL of water. A scintigraphic scan image was used to visualize the complete tablet erosion and revealed a mean complete tablet erosion time of 57 min within the stomachs. In one subject, the scintigraphic images at 2 min after dosing exhibited no tablet erosion and at minute 140, no intact tablet core remained. Similarly, in dog models, they tracked tablet disintegration through magnetic monitoring, which confirmed the average duration of absorption and time to complete tablet erosion. Plasma concentrations of oral semaglutide indicated that its absorption occurred early on, and once in the systemic circulation, it had a slow elimination rate. Sodium N-(8-[2-hydroxybenzoyl] amino) caprylate was also absorbed into the systemic circulation soon after its administration, but had a significantly faster rate of elimination, about 4–6 h. The pharmacokinetic parameters of oral semaglutide exhibited a significantly long t½ of about 1 week. However, the study demonstrated that the exposure of oral semaglutide in the body after once-daily dosing did not result in an increased variable effect on blood glucose levels at steady state after 26 weeks [28].
To study the potential impact of food in the stomach, an analysis was carried out in 78 healthy subjects aged between 18 and 75 years. Subjects received SNAC/oral semaglutide once daily in the fed or fasted state for 10 days with a dose escalation from 5 mg/300 mg the first 5 days to 10 mg/300 mg in the last 5 days to reduce the risk of GI adverse events (AEs) [28]. When subjects were dosed in the fed state, a measurable concentration of oral semaglutide was noted in 14 out of 25 subjects whereas in the remaining 11 subjects, the measurable concentration of oral semaglutide was limited at day 10. For all subjects dosed in the fasted state (n = 26), a measurable concentration of oral semaglutide was noted at day 10. The measurable concentrations of oral semaglutide of those in the fasted state compared to those in the fed state indicate that the absorption of oral semaglutide is hindered by the presence of food in the stomach and to improve its absorption and overall exposure, oral semaglutide should be taken in the fasted state [28].
To determine the specific concentration of SNAC that would be needed to allow measurable plasma concentrations of oral semaglutide in the body, multiple doses of SNAC were given to a total of 155 healthy male subjects. A single dose of oral semaglutide 5 mg was given with either 150 mg, 300 mg, or 600 mg of SNAC. The results of the single doses showed that oral semaglutide plasma concentrations were higher when oral semaglutide was co-formulated with 300 mg of SNAC compared with 600 mg of SNAC. This determined that 300 mg of SNAC proved the most appropriate amount of SNAC to enhance absorption of the oral semaglutide formulation [28].
In addition to the previously discussed benefits of the addition of the SNAC to oral semaglutide, it was also introduced to protect oral semaglutide against the effects of pepsin, which is one of the primary digestive enzymes. Optimal pepsin activity in the body is at a low pH (2–4), such as that found in gastric fluid of the stomach. To determine the effects of pepsin on oral semaglutide, oral semaglutide was incubated with pepsin at pH values of 2.6, 5.0, and 7.4, and the t½ was calculated. The effect of pepsin on oral semaglutide stability was most profound at low pH, with oral semaglutide being most labile toward pepsin at pH 2.6 (t½ = 16 min). In contrast, increasing the pH to 5.0 extended the t½ of oral semaglutide to 34 minutes, and at neutral pH, oral semaglutide was almost entirely stabilized (t½ >100 min) [28].
3.1 Phase I Clinical Studies
A phase I study completed by Granhall et al. evaluated the safety, tolerability, pharmacokinetics, and pharmacodynamics of multiple doses of oral semaglutide and SNAC in healthy subjects and subjects with T2DM in both a single- and multi-dose study [31]. The primary outcome studied was the number of treatment-emergent adverse effects in each group, and secondary outcomes included area under the concentration–time curve (AUC), plasma concentration, and change from baseline in fasting plasma glucose, C-peptide, insulin, glucagon, and HbA1c.
The single-dose study utilized varying combinations of SNAC and oral semaglutide (2–20 mg) to determine the amount of SNAC that provided the largest systemic absorption of oral semaglutide. In this study, healthy male subjects (Table 1) were randomized to one of three parts; part 1a consisted of four ascending dose groups of SNAC, part 1b included three additional dose groups, and part 2 included three of the doses utilized in part 1 that were selected to be repeated in part 2 in a parallel design. Within each dose group, subjects were randomized to receive either oral semaglutide or placebo containing matching amounts of SNAC. In the single-dose study, most AEs were mild, with the most common AEs being GI disorders and headache. More subjects in the oral semaglutide group experienced GI disorders and headache compared with those in the placebo group with 15% in the oral semaglutide group vs 4% in the placebo group reporting headaches and 14% in the oral semaglutide vs 13% in the placebo group reporting GI disorders. Results from Part 1a and 1b determined 300 mg of SNAC as the most optimal fixed dose for a range of oral semaglutide concentrations and demonstrated oral semaglutide exposure increased in a dose-dependent manner. Part 2 also included two additional dose groups (intravenous and subcutaneous semaglutide) to investigate the absolute and relative bioavailability of oral semaglutide. However, bioavailability could not be reliably estimated because the chosen dose concentration for intravenous administration was too low for the terminal part of the concentration–time curve to be apparent. The plasma semaglutide exposure concentrations in this single-dose study were generally insufficient for a robust estimation of bioavailability, therefore, the data were not shown.
The multiple-dose study included two sets of patients. The study consisted of 84 healthy male individuals and 23 male individuals with T2DM treated with diet/exercise and/or metformin [31]. The healthy subjects were randomized to oral semaglutide 20 mg and 40 mg once daily with SNAC 300 mg and subjects with T2DM received oral semaglutide 40 mg once daily with SNAC 300 mg. Oral semaglutide doses were initiated at 5 mg during week 1, increasing to 10 mg during week 2, to 20 mg at week 3, and to 40 mg at week 5. Overall, 92 subjects completed the study and all 107 subjects were included in the safety analyses and full analysis [31]. In the study, oral semaglutide exposure was approximately two-fold higher in the 40-mg group vs the healthy male individual oral semaglutide 20-mg group with similar results in the group of male individuals with T2DM. Furthermore, oral semaglutide plasma exposure did not differ between healthy subjects receiving 40 mg and subjects with T2DM receiving 40 mg. The t½ was approximately 1 week and was comparable between all treatment groups. In subjects with T2DM, a statistically significant decrease in HbA1c was seen after 10 weeks of treatment with 40 mg of oral semaglutide vs placebo with a mean reduction of 1.5% (p < 0.001). A reduction in BW was also observed after 10 weeks of oral semaglutide once daily in both healthy subjects and subjects with T2DM. The reduction in BW was statistically more significant for oral semaglutide vs placebo with a mean decrease of 4.3 kg in the 20-mg healthy subject group (p < 0.001), 7.2 kg for the 40-mg healthy subject group (p < 0.001), and 5.4 kg in the T2DM group (p < 0.001) [31].
The multiple-dose study consisted of 662 AEs that were reported in 99 subjects. A similar proportion of subjects reported AEs across treatment groups. The most reported AEs were GI disorders. Among the healthy subjects, 50%, 84%, 28%, and 72% of subjects reported GI disorders for oral semaglutide 20 mg, oral semaglutide 40 mg, placebo alone, and placebo with SNAC respectively. Among subjects with T2DM, 73%, 50%, and 33% of subjects reported GI disorders for oral semaglutide 40 mg, placebo alone, and placebo with SNAC, respectively. Most AEs were mild; however, the severity of AEs increased with an increasing dose of oral semaglutide. Overall, no systematic differences in the proportion of subjects reporting AEs were observed between treatment groups [31]. Two different pharmacokinetic studies in subjects with renal or hepatic impairment were conducted for oral semaglutide. A study conducted by Granhall et al. [12] evaluated the pharmacokinetics, safety, and tolerability of oral semaglutide in patients with T2DM and renal impairment. A total of 71 subjects were randomized (1:1:1:1:1) based on their renal function (Table 2). All subjects initially received oral semaglutide 5 mg daily for 5 days followed by oral semaglutide 10 mg for 5 additional days with 120 mL of water to reduce the risk of GI adverse events. Subjects took their dose of oral semaglutide after an overnight fast that included no liquid or food intake in addition to no liquid or food for 30 minutes after dosing. The results showed that there was no consistent pattern of increase or decrease in oral semaglutide exposure (area under the plasma concentration–time curve from time zero to 24 h after the tenth dose [AUC24,Day10] and maximum plasma concentration 0–24 h after the tenth dose [Cmax,Day10]) by renal function group on day 10 (Table 3). Oral semaglutide exposure was similar for the groups with moderate renal impairment and normal renal function (AUC24,Day10, nmol·h/L) (Table 3). No consistent or clinically relevant pattern of increase or decrease in oral semaglutide exposure was observed when subjects were categorized into renal function groups by eGFR based on the Modification of Diet in Renal Disease formula, with similar exposure in all groups except for higher apparent mean exposure in the moderately renally impaired group. Compared with the group with normal renal function, the mean exposure of oral semaglutide appeared to be higher in the group with mild renal impairment (estimated ratio: AUC24,Day10 1.37 [90% confidence interval (CI) 0.91–2.06] and Cmax,Day10 1.39 [0.93–2.06]), whereas lower exposure was observed in the group with severe renal impairment (estimated ratio: AUC24,Day10 0.61 [90% CI 0.42–0.88] and Cmax,Day10 0.61 [0.42–0.87]). Additionally, the median time for oral semaglutide to reach its Cmax was noted to be similar between all groups and ranged from 1.0 to 1.5 h, whereas the mean t1/2 was similar among the groups when compared to normal renal function (152 h) except for subjects with severally impaired renal function, which appeared to be higher (165 h). Furthermore, hemodialysis did not show any significant effect on the exposure of oral semaglutide exposure when compared to normal renal function and the estimated ratio of AUC was AUC24,day 10 1.02 (90% CI 0.59–1.79), Cmax,Day10 1.06 (90% CI 0.61–1.84) [12].
3.1.1 Safety
Throughout the study, oral semaglutide was well tolerated with a total of 53 AEs documented from 25 subjects across all renal function groups. Of those events, 48 were classified as mild and five were moderate in severity. The proportion of subjects with AEs was higher in the groups with renal impairment (25.0–58.3%) than in the group with normal renal function (20.8%); however, the overall occurrence of AEs did not increase with increasing renal impairment. Gastrointestinal AEs such as abdominal distension (n = 11), vomiting (n = 6), and nausea (n = 4) were the most frequently reported AEs with no severe hypoglycemia episodes. Overall, the results from this study appeared to match previous subcutaneous semaglutide studies and concluded that the oral administration of semaglutide does not affect the pharmacokinetics in subjects with renal impairment [12].
A study conducted by Baekdal et al. assessed the pharmacokinetics, safety, and tolerability of oral semaglutide in subjects with hepatic impairment. A total of 56 subjects with a BMI of 18.5–40.0 kg/m2 were stratified based on their hepatic function according to the Child-Pugh criteria and comprised four treatment groups; normal hepatic function < 5 points with no Child-Pugh classification (n = 24), mild (Grade A; 5–6 points, n = 12), moderate (Grade B; 7–9 points, n = 12), and severe (Grade C; 10–15 points, n = 12). These subjects received oral semaglutide 5 mg for 5 days followed by oral semaglutide 10 mg for 5 days. The primary endpoint of AUC0–24 at day 10 was similar across all four groups where the estimated ratio of mean AUC in relation to normal hepatic function appeared to be 0.91 (90% CI 0.60–1.40) in the mild group, 0.87 (90% CI 0.57–1.31), in the moderate group, and 0.90 (90% CI 0.61–1.32) in the severe group. A similar trend was observed in the Cmax of oral semaglutide at day 10 where the mean estimated ratio was 0.92 (90% CI 0.60–1.40) for the mild group, 0.85 (90% CI 0.55–1.30) for the moderate group, and 0.88 (90% CI 0.61–0.28) for the severe group when compared to the normal hepatic group. Furthermore, no difference was noted in the time to Cmax and the t1/2 of oral semaglutide across the groups. Safety and tolerability of the oral semaglutide appeared to be similar across all four groups with headache being the most frequently reported AE (14.3%) as well as GI AEs such as dyspepsia (8.9%), vomiting (7.1%), decreased appetite (7.1%), and diarrhea (5.4%) [32].
3.2 Phase II Clinical Studies
A phase II study by Davies et al. evaluated the efficacy of oral semaglutide compared to subcutaneous semaglutide on glycemic control in 632 subjects with T2DM [29]. Subjects with an HbA1c of 7.0–9.5% who were treated with diet and exercise or metformin were randomized in an equal ratio to one of the nine treatment groups (oral semaglutide 2.5 mg, 5 mg, 10 mg, 20 mg, 40 mg, and 40 mg once daily with a slow 8-week dose escalation, and 40 mg fast 2-week dose-escalation groups, oral placebo group, and open-label subcutaneous semaglutide 1-mg once-weekly group). This study met its primary endpoint of achieving a mean reduction in HbA1c of 1.8% from baseline to week 26 with oral semaglutide 40 mg compared with 0.3% with placebo (p < 0.001). All oral semaglutide groups had a significant reduction in HBA1c levels in a dose-dependent manner with reductions of 0.4%, 0.9%, 1.2%, 1.4%, and 1.6% for the 2.5-mg, 5-mg, 10-mg, 20-mg, and 40-mg standard groups, respectively. A greater number of subjects achieved an HbA1c < 7.0% with oral semaglutide compared with placebo with a total of 44% in the 2.5-mg group, 81% in the 5-mg group, 84% in the 10-mg group, 86% in the 20-mg group, 90% in the 40-mg standard group, and 93% in the subcutaneous semaglutide compared with 28% in the placebo group at week 26 (p < 0.001 for all). Subjects in the oral and the subcutaneous semaglutide groups showed a significant mean reduction in BW compared with placebo (p < 0.001) and at week 26, the decrease from baseline in mean BW in the oral semaglutide groups was dose dependent and significantly greater than placebo with weight reductions of 2.1 kg (p = 0.25), 2.7 kg (p = 0.06), 4.8 kg (p < 0.001), 6.1 kg (p < 0.001), and 6.9 kg (p < 0.001) for oral semaglutide 2.5-mg, 5-mg, 10-mg, 20-mg, and 40-mg standard escalation compared with 1.2 kg with placebo. While the proportion of patients achieving a 5% weight loss was significantly greater for oral semaglutide dosage groups of 10 mg and higher (p < 0.001), no significant difference in weight loss was observed between the 20- and 40-mg standard escalation groups of oral semaglutide and the subcutaneous semaglutide.
3.2.1 Safety
The most frequently reported AEs were GI related and were mild to moderate in severity and occurred at a higher frequency with oral semaglutide (31–77%) and subcutaneous semaglutide (54%) compared with placebo (28%). Two episodes of severe hypoglycemia were reported (one in the subcutaneous semaglutide group; one in the oral semaglutide 40-mg fast-escalation group) and a total of three cases of pancreatitis that were mild to moderate in severity (one in the subcutaneous semaglutide group; one in the oral semaglutide 40-mg standard escalation group; 1 in the oral semaglutide 20-mg group). Heart rate was significantly higher in subjects in the oral semaglutide group (ranged from + 0.6 to + 3 beats per minute [bpm]) [p < 0.001 for oral semaglutide 5 mg, 10 mg, 20 mg, and 40 mg) and subcutaneous semaglutide group (+ 2.6 bpm) compared with placebo (− 4 bpm) except for the oral semaglutide 2.5-mg group (− 1.7 bpm). Overall, this study demonstrated that oral semaglutide significantly lowered HbA1c when compared with placebo and the degree of change in HbA1c with oral semaglutide 20 mg and 40 mg was not significantly different compared to subcutaneous semaglutide [29].
3.3 Phase III Clinical Studies
The global oral semaglutide phase III program, PIONEER, consists of 12 phase III clinical studies and has enrolled a total of 9542 subjects with T2DM as of 2020. Enrollment for the PIONEER studies 1 through 10 are complete and the currently enrolling PIONEER 11 and PIONEER 12 clinical studies are expected to conclude in late 2021. The PIONEER trial program used two estimands, a treatment policy estimand and a trial product estimand, to evaluate efficacy. The treatment policy estimand describes the treatment effect in patients regardless of trial product (oral semaglutide) discontinuation or use of rescue medication, while the trial product estimand describes the treatment effect in patients if all patients had continued use of the trial product (oral semaglutide) and did not use rescue medication. For the purposes of this review, only the treatment policy estimand data are presented here, which reflects the intention-to-treat concept.
3.3.1 PIONEER 1
PIONEER 1, which completed in February 2020, evaluated the safety and efficacy of oral semaglutide vs placebo in subjects with T2DM currently treated with diet and exercise alone. Subjects with a mean age of 55 years and an HbA1c of 7.0–9.5%, were randomized (1:1:1:1) to receive oral semaglutide 3 mg, 7 mg, or 14 mg once daily or placebo for 26 weeks (Table 1) [6]. All subjects randomized to oral semaglutide were initiated with 3 mg once daily and those patients randomized to 7 mg and 14 mg had dose escalations every 4 weeks until their designated randomized maintenance dose was achieved. All subjects randomized to oral semaglutide demonstrated greater reductions in HbA1c compared with placebo after 26 weeks (p < 0.001 for all) (Table 4). In addition, a greater percentage of subjects administered oral semaglutide 3 mg, 7 mg, or 14 mg achieved an HbA1c of < 7% compared with placebo (55.1%, 68.8%, or 76.9%, respectively vs 31.0%; p < 0.001 for all) with similar results for an HbA1c ≤ 6.5% (35.9%, 47.5%, or 63.8%, respectively vs 17.9%; p < 0.001 for all). When evaluating fasting plasma glucose (FPG), reductions were greater for oral semaglutide 3 mg, 7 mg, and 14 mg when compared with placebo with a mean change in FPG from baseline of − 16.2%, − 27.9%, − 32.9% , and − 3.2% (p = 0.003 for 3 mg, p < 0.001 for 7 mg and 14 mg), respectively. However, superior efficacy of oral semaglutide on BW was only observed with oral semaglutide 14 mg compared with placebo with a placebo-adjusted treatment difference of − 2.3 kg (p < 0.001) (Table 4). Bodyweight loss of ≥ 5% was seen more commonly with oral semaglutide with a dose-dependent effect. At week 26, oral semaglutide 3 mg, 7 mg, and 14 mg showed 19.6%, 26.9%, and 41.3% of subjects achieved a BW loss of ≥ 5% compared with those in the placebo group (14.9%; p < 0.001). A significant mean increase in lipase levels was noted with oral semaglutide (13–34%) compared with placebo with a mean lipase at week 26 of 32 U/L, 36 U/L, 38 U/L, and 28 U/L for oral semaglutide 3 mg, 7 mg, 14 mg, and placebo (p = 0.007 for 3 mg, p < 0.001 for 7 mg and 14 mg), respectively. In addition, mean heart rate increased significantly (3 bpm; p = 0.003) for oral semaglutide 14 mg compared with placebo, but was not statistically significant in oral semaglutide 3 mg or 7 mg.
3.3.1.1 Safety
Overall, oral semaglutide and placebo exhibited a similar safety profile with 57.7%, 53.1%, 56.6%, and 55.6% of subjects taking oral semaglutide 3 mg, 7 mg, 14 mg, and placebo, respectively experiencing any AE. Severe AEs (SAEs) were most reported in the oral semaglutide 3-mg group at 4.6% compared with 0.6%, 1.7%, and 2.8% for oral semaglutide 7 mg, 14 mg, and placebo, respectively. The most frequent AEs reported, with a 5.1–16.0% occurrence across all groups, were GI related, including nausea and diarrhea, which were mild to moderate in severity. Adverse events leading to premature trial product discontinuation were highest with oral semaglutide 14 mg (7.4%) compared with oral semaglutide 3 mg (2.3%), 7 mg (4.0%), and placebo (2.2%), respectively, and were due to GI disorders. The occurrence of hypoglycemic events with subjects experiencing at least one severe or symptomatic hypoglycemic event was similar between oral semaglutide and placebo with 2.9%, 1.1%, and 0.6% in the oral semaglutide 3-mg, 7-mg, and 14-mg groups, respectively, compared with 0.6% on placebo. Overall, the PIONEER 1 study demonstrated superiority of oral semaglutide 3 mg, 7 mg, and 14 mg compared with placebo with regard to HbA1c and BW reduction and was considered safe and well tolerated [6].
3.3.2 PIONEER 2
A randomized open-label study comparing oral semaglutide once daily to empagliflozin once daily, also known as the PIONEER 2 study, included subjects with T2DM and an HbA1c between 7.0 and 10.5% who were treated with metformin ≥1500 mg daily. Subjects (Table 4) received either oral semaglutide 14 mg once daily or empagliflozin 25 mg once daily for 52 weeks. Both treatment arms had dose-escalation periods. Oral semaglutide started at 3 mg once daily for 4 weeks, then increased to 7 mg once daily for 4 weeks, and then increased to 14 mg starting after week 8, while empagliflozin was initiated at 10 mg once daily and increased to 25 mg at week 8. Oral semaglutide demonstrated a superior reduction in HbA1c at week 26 with a decrease of − 1.3% compared with a decrease of − 0.9% for empagliflozin (p < 0.0001). Additionally, 47.4% of subjects taking oral semaglutide reached an HbA1c target of ≤ 6.5% compared with 17.2% of subjects taking empagliflozin at week 26, which was sustained through week 52 (47.4% and 21.7%, respectively; p < 0.0001). Results were similar for the estimated mean change from baseline in FPG with − 1.99 mg/dL, − 2.01 mg/dL, − 2.01 mg/dL, and − 2.09 mg/dL for oral semaglutide at 26 weeks, empagliflozin at 26 weeks (p = 0.8812 at 26 weeks), oral semaglutide at 52 weeks, and empagliflozin at 52 weeks (p = 0.5759 at 52 weeks), respectively. Oral semaglutide showed significantly greater reductions in mean 7-point self-monitoring blood glucose profiles compared with empagliflozin at both week 26 (− 0.5 mg/dL vs − 0.3 mg/dL [p = 0.0267], respectively) and week 52 (− 0.7 mg/dL vs − 0.3 mg/dL [p = 0.0328], respectively). Bodyweight reductions of > 10% were also higher in the oral semaglutide group (12.5%) at week 26 vs the empagliflozin group (6.8%; p = 0.0066), which was continued through week 52 (15.0% and 7.8%; p = 0.0028, respectively). Overall, superiority in HbA1c and BW reduction in the oral semaglutide group compared with the empagliflozin group was confirmed.
3.3.2.1 Safety
Rescue medication was more commonly initiated at week 26 with oral semaglutide (1.9%) compared with empagliflozin (1.2% p < 0.23), but more subjects taking empagliflozin required rescue treatment (10.7%) compared with oral semaglutide (7.5%) at week 52. At 26 weeks, a total of 237 subjects or 60.5% of those taking oral semaglutide achieved an HbA1c of < 7% without hypoglycemia and no weight gain compared with 35.7% of subjects taking empagliflozin. The most reported AEs were mild to moderate in severity with the most common being nausea (19.8% for oral semaglutide and 2.4% for empagliflozin), which occurred in < 10% of patients. Genital mycotic infection, which was mild to moderate in severity, occurred more frequently with empagliflozin compared with oral semaglutide (8.5% in female subjects and 6.7% in male subjects in the empagliflozin group vs 2.0% in female subjects and 0% in male subjects in the oral semaglutide group). Furthermore, AEs resulting in premature discontinuation of the study drug occurred more frequently with oral semaglutide (8.0%) than with empagliflozin (0.7%) and were mainly due to symptoms related to GI disorders. The safety profile for oral semaglutide in the PIONEER 2 study is consistent with previous studies with oral semaglutide and fewer patients in the oral semaglutide group reported SAEs compared with empagliflozin (6.6% vs 9.0%, respectively) [33].
3.3.3 PIONEER 3
A 78-week study included subjects with T2DM uncontrolled with metformin with or without a sulfonylurea and evaluated the efficacy of oral semaglutide compared to sitagliptin. This randomized, double-blind, double-dummy, parallel-group study, PIONEER 3, included 1864 subjects with a mean age of 58 years and an HbA1c of 7.0–10.5% and randomized them (1:1:1:1) to oral semaglutide 3 mg, 7 mg, and 14 mg once daily or sitagliptin 100 mg once daily. Sitagliptin was initiated and maintained at the 100-mg daily dose while oral semaglutide was initiated at 3 mg and increased at 4 weeks to 7 mg and then to 14 mg after another 4 weeks until the randomized dose was achieved. Subjects receiving oral semaglutide achieved a change in HbA1c from baseline to week 26 of − 0.6%, − 1.0%, and − 1.3% for 3 mg, 7 mg, and 14 mg, respectively, and − 0.8% for sitagliptin. Further, the percentage of patients who achieved an HbA1c < 7.0% was highest for those randomized to oral semaglutide 14 mg with 55% of subjects achieving this goal (p < 0.001) by week 26 compared with 27% (p = 0.07) with 3 mg, 42% (p < 0.001) with 7 mg, and 32% of subjects in the sitagliptin group. The estimated mean change in FPG from baseline to week 26 was also highest with oral semaglutide 14 mg, which showed a reduction of 30.5 mg/dL (p < 0.001), followed by oral semaglutide 7 mg with 21.3 mg/dL (p = 0.04), sitagliptin with 15.4 mg/dL, and oral semaglutide 3 mg with 13.6 mg/dL (p = 0.50). By week 78, subjects receiving any dose of oral semaglutide had greater reductions in FPG than sitagliptin with − 17.1 mg/dL, − 18.1 mg/dL, − 30.8 mg/dL, and − 15.0 mg/dL for oral semaglutide 3 mg, 7 mg, and 14 mg (p = 0.50, p = 0.33, p < 0.001) and sitagliptin 100 mg respectively. Additionally, oral semaglutide showed greater mean changes from baseline in BW at week 26 compared with sitagliptin (Table 1). Correspondingly, a significantly greater proportion of subjects in the oral semaglutide 7-mg and 14-mg groups achieved a BW loss of ≥ 5% compared with the sitagliptin group at week 26 and week 78 (p < 0.001) with 19% and 30%, respectively, at week 26 compared with 10% for sitagliptin and 27% and 33%, respectively, at week 78 compared with only 12% for sitagliptin.
3.3.3.1 Safety
At week 26, 34% and 46% of subjects administered oral semaglutide 7 mg and 14 mg achieved an HbA1c < 7.0% without hypoglycemic episodes and without BW gain respectively compared with 20% of subjects in the sitagliptin group (p < 0.001). Rescue medication was initiated by 5.4%, 2.4%, and 1.1% of subjects with oral semaglutide 3 mg, 7 mg, and 14 mg and by 2.8% of subjects for the sitagliptin at week 26 and increased through the remainder of the trial to 26.0%, 15.7%, and 6.7% in subjects administered oral semaglutide 3 mg, 7 mg, and 14 mg, respectively, and 20.1% for sitagliptin at week 52, and 34.3%, 22.2%, 10.1%, and 27.6%, respectively, at week 78. For both the oral semaglutide 7-mg and 14-mg groups, the time to rescue medication was longer compared with sitagliptin, p = 0.002 and p < 0.001 respectively. The most frequently reported AEs were GI in nature with nausea and diarrhea being the most common with oral semaglutide 7 mg and 14 mg. Gastrointestinal AEs were also the primary cause of premature trial product discontinuation with 5.6%, 5.8%, and 11.6% of subjects receiving oral semaglutide 3 mg, 7 mg, and 14 mg, respectively, compared with 5.2% receiving sitagliptin therapy. Subjects randomized to sitagliptin showed a higher number experiencing any AE and a higher number experiencing severe or whole-blood glucose-confirmed symptomatic hypoglycemia at 8.4% compared with 4.9%, 5.2%, and 7.7% for oral semaglutide 3 mg, 7 mg, and 14 mg, respectively. Overall, oral semaglutide 7 mg and 14 mg proved to be superior to sitagliptin for a reductions in HbA1c and BW in this study [8].
3.3.4 PIONEER 4
Pioneer 4 was the first study to evaluate the efficacy and safety of oral semaglutide compared to another GLP-1RA, subcutaneous liraglutide. Subjects had a mean age of 55 years, an HbA1c between 7.0 and 9.5%, and received the maximally tolerated dose of metformin with or without a sodium-glucose cotransporter-2 inhibitor (Table 4). Subjects randomized to oral semaglutide or liraglutide groups underwent a dose-escalation process. Oral semaglutide was initiated at 3 mg and escalated to 7 mg at 4 weeks and to the maintenance dose of 14 mg at 8 weeks, whereas the liraglutide group initiated treatment at 0.6 mg once daily with a dose escalation to 1.2 mg after 1 week and to the maintenance dose of 1.8 mg after 2 weeks. Oral semaglutide demonstrated non-inferiority to subcutaneous liraglutide (p < 0.0001) and superiority to placebo (p < 0.0001) in decreasing HbA1c (Table 1). Additionally, at week 52, oral semaglutide demonstrated a significantly greater decrease in HbA1c (− 1.6%) compared with both liraglutide (− 1.1%, p = 0.0002) and placebo (− 0.2%, p < 0.0001). The percentage of subjects achieving HbA1c targets of < 7.0% or < 6.5% did not differentiate between oral semaglutide and liraglutide at week 26; however, 43% of subjects receiving oral semaglutide therapy achieved an HbA1c of 6.5% or less at week 52 while only 33% receiving liraglutide and 4% receiving placebo achieved the same goal (p = 0.0084, p < 0.0001). Oral semaglutide also resulted in superior weight loss compared with subcutaneous liraglutide and placebo (Table 1) and continued to show significantly greater decreases in BW at 52 weeks. Subjects were more likely to achieve a BW loss of ≥ 5% or ≥ 10% with oral semaglutide than with liraglutide or placebo at both 26 and 52 weeks, with 14%, 6%, and 0%, respectively achieving a ≥ 10% weight loss at week 26 (p = 0.0032, p = 0.0083) and 16%, 7%, and 3%, respectively, at week 52 (p = 0.0028, p = 0.0005). Oral semaglutide showed a statistically significant decrease in mean FPG when compared with placebo (2.0 mmol/L vs 0.36 mmol/L (p < 0.0001) but was not statistically significant compared to the mean FPG decrease with liraglutide (1.87 mmol/L, p = 0.3422).
3.3.4.1 Safety
Overall, 80% of subjects taking oral semaglutide reported an occurrence of AE compared with 74% receiving liraglutide and 67% receiving placebo. The reported AEs were largely attributable to GI events, with the most frequent being transient nausea, which is a known side effect with the GLP-1RA class. Discontinuation from study treatment was similar between oral semaglutide and liraglutide with 11% and 9% of subjects discontinuing study treatment while only 4% receiving placebo discontinued study treatment early because of AEs. Conversely, SAEs were similar for oral semaglutide and placebo and lower with the liraglutide group with 11%, 11%, and 8%, respectively. Study treatment was able to be completed without rescue medication for 78% of subjects taking oral semaglutide and 81% taking liraglutide compared with only 58% in the placebo group. Results from this study demonstrated that oral semaglutide was non-inferior to subcutaneous liraglutide once daily and superior to placebo in decreasing HbA1c at week 26. In addition, oral semaglutide showed significant weight loss compared with both liraglutide and placebo. Plasma glucose levels in subjects taking oral semaglutide were significantly lower than in those taking liraglutide at week 52, potentially suggesting a long-term benefit with continued oral semaglutide therapy [10].
3.3.5 PIONEER 5
The PIONEER 5 study assessed the safety and efficacy of oral semaglutide on renal dysfunction. Subjects were ≥ 18 years old, had a diagnosis of T2DM with an HbA1c between 7.0 and 9.5%, and an eGFR between 30 and 59 mL/min. Subjects were randomly assigned (1:1) to receive oral semaglutide 14 mg or matching placebo in addition to their background medication of metformin and/or sulfonylurea, or basal insulin for 26 weeks. After 26 weeks of treatment, a mean change from baseline in HbA1c of −1.0% was observed for oral semaglutide compared with − 0.2% for placebo (p < 0.0001). Similarly, 93 of the 154 subjects taking oral semaglutide (60%) achieved at least a 1% decrease in their HbA1c compared with only 31 of the 155 subjects (20%) taking placebo. Furthermore, more subjects were able to achieve an HbA1c of < 7.0% or < 6.5% with oral semaglutide compared with placebo, 58% compared with 23% respectively, achieving an HbA1c of < 7.0% and 39% compared with 8% respectively reaching an HbA1c of < 6.5% (p < 0.0001 for both). Thus, in patients with renal impairment, oral semaglutide maintained superiority vs placebo in HbA1c lowering. Similarly, oral semaglutide demonstrated superiority on a reduction in BW compared with placebo (p < 0.001). Likewise, a higher percentage of subjects achieved ≥ 5% and ≥ 10% weight loss taking oral semaglutide with 36% compared with 10% of patients taking placebo (p < 0.0001) and 8% of subjects taking oral semaglutide vs 0% of patients taking placebo (p = 0.0086), respectively. Fewer subjects required the use of rescue medication while taking oral semaglutide with only 4% of subjects needing rescue medication compared with 10% of subjects taking placebo throughout the duration of treatment. A health-related quality-of-life questionnaire was utilized in this study and included a physical component summary, bodily pain, and social functioning. All of those sections of the questionnaire favored oral semaglutide compared with placebo (p = 0.0058, p = 0.0326, p = 0.035, respectively). Additionally, the questionnaire addressed subject-perceived hyperglycemia, and showed a significantly lower frequency in those subjects taking oral semaglutide compared with those taking placebo (p < 0.0001).
3.3.5.1 Safety
A greater number of AEs were reported for oral semaglutide compared with placebo with 74% and 65% of subjects, respectively, although a similar number of subjects in each group reported SAEs with 10% taking oral semaglutide compared with 11% taking placebo. As seen in the previous PIONEER studies, the most frequent AEs reported were mild-to-moderate GI events with nausea being the most common (19% vs 7% for oral semaglutide vs placebo). Most AEs that lead to discontinuation were GI events; however, this was low with only 15% in subjects taking oral semaglutide and 5% of subjects taking placebo. Similarly, a small number of symptomatic hypoglycemic episodes occurred during the study with nine subjects (6%) taking oral semaglutide and three subjects (2%) taking placebo that were confirmed by a blood glucose test that concluded that none was severe. Renal function was followed throughout the study and remained unchanged throughout the 26 weeks in both treatment groups. Median eGFR ratios from baseline to follow-up were similar between both groups with 1.02 (range 0.27–1.96) for oral semaglutide and 1.00 (0.68–2.17) for placebo. Urine albumin creatinine ratios was also measured throughout the study. From baseline to week 26, there was a decrease in the mean urine albumin creatinine ratio with oral semaglutide − 0.86 (range of 0.04–56.71) while placebo experienced an increase of + 1.19 (range of 0.01–79.59). At 26 weeks, oral semaglutide demonstrated a mean systolic blood pressure decrease by 7 mmHg from baseline and a diastolic blood pressure decrease of 2 mmHg from baseline compared with no change in the placebo group (p < 0.0001 and p = 0.0018, respectively). PIONEER 5 demonstrated superiority for oral semaglutide 14 mg once daily compared with placebo in decreasing HbA1c and BW in patients with T2DM and moderate renal impairment. The overall safety profile, including renal safety, demonstrated no effect on kidney function and was consistent with the other subcutaneous GLP-1RAs on the market. Oral semaglutide appears to provide an important addition to the currently suboptimal treatment options for patients with T2DM and moderate renal impairment [18].
3.3.6 PIONEER 6
The oral semaglutide CV outcome trial, PIONEER 6, studied the safety of oral semaglutide in subjects with either established CV risk or at high risk of CV events. Subjects with T2DM were included if they were aged 50 years or older with established CV disease and/or CKD or if they were aged greater than 60 years with CV risk factors only. The study had 2695 subjects (84.7%) who met the criteria of 50 years of age or older and had established CVD or CKD. A total of 3183 subjects were randomized 1:1 to receive once-daily treatment with either oral semaglutide 14 mg or placebo in addition to standard of care. Subjects randomized to oral semaglutide followed a dose-escalation process receiving oral semaglutide 3 mg once daily for 4 weeks, increasing to 7 mg for 4 weeks, and then increasing to 14 mg for the remainder of the study. This study was event-driven and lasted about 20 months, until over 122 first major adverse cardiovascular events (MACEs) had occurred. The first occurrence of MACEs included CV death, non-fatal MI, or non-fatal stroke, which occurred in 3.8% of subjects in the oral semaglutide group and 4.8% of subjects in the placebo group (p < 0.001), indicating that subjects receiving oral semaglutide had a lower risk of developing a MACE than those receiving placebo. In addition, death from any cause occurred at a slightly higher rate in the placebo group with 2.8% of subjects vs 1.4% of those taking oral semaglutide. Death specifically from CV causes occurred in 0.9% in the oral semaglutide group and 1.9% in the placebo group. The composite of death from any cause including nonfatal myocardial infarction or nonfatal stroke occurred in 4.3% of the oral semaglutide group and 5.6% in the placebo group. A first event of unstable angina resulting in hospitalization occurred in 0.7% of subjects taking oral semaglutide and 0.4% taking placebo, while events of heart failure resulting in hospitalization occurred in 1.3% and 1.5%, respectively. Other outcomes that were evaluated were a significant decrease in BW in the oral semaglutide group than in the placebo group (Table 2). Additionally, a greater decrease in systolic blood pressure in the oral semaglutide group was seen with a decrease of 5 mmHg compared with 2 mmHg in the placebo group.
3.3.7 Safety
Overall, a small number of subjects in both groups permanently discontinued the study with more subjects taking oral semaglutide discontinuing (11.6%) than those taking placebo (5%). The cause of discontinuation was similar as the studies above and were mostly GI in nature, which occurred in 6.8% of subjects taking oral semaglutide vs 1.6% taking placebo. Nausea, vomiting, and diarrhea were more common for those taking oral semaglutide with nausea being the most common GI AE with 2.9% receiving oral semaglutide vs 0.5% receiving placebo and vomiting 1.5% vs 0.3%, and diarrhea 1.4% vs 0.4%, respectively. A small number of SAEs occurred throughout the study that led to permanent discontinuation and were similar between both groups with 2.6% of those receiving oral semaglutide and 3.0% of those receiving placebo. Overall, PIONEER 6 demonstrated safety and non-inferiority to placebo in the time to the first MACE [34].
3.3.8 PIONEER 7
PIONEER 7 evaluated the safety and efficacy of flexible dosing of oral semaglutide compared to sitagliptin 100 mg enrolled subjects aged ≥ 18 years with T2DM with an HbA1c of 7.5–9.5%. Five hundred and four subjects were randomized 1:1 to receive either oral semaglutide or sitagliptin. The oral semaglutide group went through a dose-escalation process starting at 3 mg and utilized a flexible dose-adjustment escalation up to the 14-mg maintenance dose. Subject dosing of oral semaglutide was adjusted based on their HbA1c and tolerability of medication throughout the study to imitate patient-centered care that would be seen in a clinical practice setting. At 52 weeks, 19% of subjects were receiving oral semaglutide 3 mg, 30% were receiving oral semaglutide 7 mg, and 59% were receiving oral semaglutide 14 mg. A greater percentage of subjects achieved an HbA1c of < 7% at 52 weeks with oral semaglutide (58%) compared with sitagliptin (25%) [p < 0.0001] and overall, the mean HbA1c was lower with oral semaglutide (7.0%) compared with sitagliptin (7.5%) at 52 weeks. Both endpoints of BW reductions ≥ 5% (p < 0.0001) as well as ≥ 10% (p = 0.0065) were statistically significantly greater with oral semaglutide compared with sitagliptin. The mean change in BW from baseline to 26 weeks was − 2.6 kg for oral semaglutide vs − 0.7 kg for sitagliptin (p < 0.0001) and was maintained through week 52 with mean changes in BW of − 2.9 kg and − 0.8 kg respectively (p = 0.0065). Time to the first dose of rescue medication was significantly longer with oral semaglutide than with the sitagliptin and with fewer subjects requiring rescue medication with only 3.2% of subjects taking oral semaglutide and 15.9% taking sitagliptin requiring rescue medication (p < 0.0001). Similarly, twice as many subjects were given additional glucose-lowering drugs and more than four times as many subjects were given rescue medication in the sitagliptin group compared with the oral semaglutide group. No other clinically relevant changes in blood pressure, pulse rate, or eGFR were reported throughout the study. Oral semaglutide with a flexible dose adjustment (3, 7, or 14 mg) was found to be superior to sitagliptin 100 mg for achievement of an HbA1c target of less than 7%. The proportion of participants who achieved this target with oral semaglutide was over twice the number of those receiving sitagliptin despite the flexible dose-adjustment approach and despite twice as many participants taking sitagliptin also receiving additional glucose-lowering medications. Additionally, oral semaglutide was superior to sitagliptin in decreasing BW [35].
3.3.8.1 Safety
The percentage of AEs in those receiving oral semaglutide was higher (78%) than in those receiving sitagliptin (69%) with the most common AEs reported being nausea and diarrhea. Additionally, no severe hypoglycemic episodes were seen during the study and no cases of pancreatitis were reported.
3.3.9 PIONEER 8
PIONEER 8 evaluated the efficacy, safety, and tolerability of multiple doses of oral semaglutide in addition to insulin. Subjects ≥ 18 years of age with T2DM and an HbA1c of 7.0–9.5% were randomized 1:1:1:1 to oral semaglutide 3 mg, 7 mg, 14 mg once daily or placebo given in addition to their stable regimen of basal, basal-bolus (in any combination), or premixed insulin that was at least 10 units/day. At randomization, a 20% reduction in total daily insulin dosage was recommended to be initiated and maintained to week 8 to reduce the chance of subjects experiencing hypoglycemic events. At week 26, the mean change in HbA1c from baseline was − 0.6%, − 0.9%, − 1.3%, and − 0.1% for oral semaglutide 3 mg, 7 mg, 14 mg, and placebo, respectively (p < 0.0001 for all), showing that HbA1c reductions were superior for all doses of oral semaglutide compared with placebo. In addition, subjects in all oral semaglutide groups observed a higher rate of achieving an HbA1c of ≤ 7.0% or ≤ 6.5% than the placebo group with 28.4%, 42.5%, 58.4%, and 6.8% reaching an HbA1c of < 7% for oral semaglutide 3 mg, 7 mg, 14 mg, and placebo (p < 0.0001 or all), respectively. Changes in mean BW occurred in a dose-dependent manner with − 1.4 kg (p = 0.032), − 2.4 kg (p = 0.0001), − 3.7 kg (p < 0.0001), and − 0.4 kg for oral semaglutide 3 mg, 7 mg, 14 mg, and placebo, respectively. A number of subjects in each group initiated rescue medication during the study, with the highest occurrence in subjects receiving placebo, with 4.9% of subjects at week 26 and 36.4% of subjects at week 52 compared with 2.7%, 1.1%, 2.2%, and 4.9% of subjects at week 26 taking oral semaglutide 3 mg, 7 mg, and 14mg respectively, and, by week 52, 29.3%, 18.1%, and 17.1%of subjects had initiated rescue medication for oral semaglutide 3 mg, 7 mg, and 14 mg, respectively. By week 26, 75.3% of all subjects had their insulin dosage reduced by 15–25%, 3.4% of subjects had a dose reduction in insulin of >25%. For the remaining subjects, 8.4% had their total daily insulin dosage reduced by < 15% and 12.4% remained unchanged. Bodyweight reductions were greater for subjects taking all doses of oral semaglutide, with a difference of − 0.9 kg (p = 0.0392), −2.0 kg (p = 0.0001), and −3.3 kg (p < 0.0001) for the oral semaglutide 3-mg, 7-mg, and 14-mg doses, respectively. Similarly, all subjects taking oral semaglutide had a greater reduction in BMI than those receiving placebo at weeks 26 and 52. Oral semaglutide treatment improved the fasting lipid profile from baseline and reductions in total cholesterol were statistically significantly greater with the oral semaglutide 7-mg and 14-mg doses compared with placebo. For all doses of oral semaglutide, there was an increase in heart rate of 2–4 bpm at week 26 (p < 0.05) and 1–2 bpm at week 52 (p < 0.05 for oral semaglutide 14 mg only) compared with placebo.
3.3.10 Safety
Gastrointestinal events were the most frequently reported AEs overall and occurred in a dose-dependent manner, with 39.1%, 44.8%, and 50.3% for the 3-mg, 7-mg, and 14-mg groups, respectively. Infections and infestations were the most reported AEs in those receiving oral semaglutide 3 mg (39.7%) and placebo (43.5%). The proportions of subjects who experienced a severe or blood glucose-confirmed symptomatic hypoglycemic episode were similar between subjects receiving oral semaglutide and placebo with 28.3% in the oral semaglutide 3-mg group; 26.0% in the oral semaglutide 7-mg group, 26.5% in the oral semaglutide 14-mg group, and 29.3% in the placebo group; hypoglycemic events occurred irrespective of oral semaglutide dosage and across all treatment arms, with the greatest number of hypoglycemic episodes occurring in patients receiving basal-bolus insulin. These findings support the addition of GLP-1RAs as an effective treatment intensification strategy for patients who are unable to reach, or maintain, HbA1c targets with insulin alone [36].
3.3.11 PIONEER 9
PIONEER 9 evaluated the safety and efficacy of multiple doses of oral semaglutide compared to subcutaneous liraglutide as monotherapy in Japanese subjects. Subjects were enrolled if they were ≥ 20 years of age, of Japanese descent, had T2DM with an HbA1c of 7.0–10% managed with diet and exercise alone or an HbA1c of 6.5–9.5% being treated with oral antihyperglycemic therapy. Two hundred and forty-three subjects were randomized 1:1:1:1:1 to receive oral semaglutide 3 mg, 7 mg, and 14 mg, subcutaneous liraglutide 0.9 mg, or placebo. Subjects taking an oral antihyperglycemic medication underwent an 8-week washout period before randomization. All subjects randomized to a GLP-1RA had a dose-escalation period. Oral semaglutide was initiated at 3 mg for 4 weeks, increased to 7 mg for 4 weeks, then increased to 14 mg for those randomized to that dose. Liraglutide was initiated at 0.3 mg and increased after 1 and 2 weeks to 0.9 mg for all. From a mean baseline HbA1c of 8.2%, subjects treated with oral semaglutide 3 mg, 7 mg, and 14 mg experienced statistically significant reductions in HbA1c of − 1.1%, − 1.6%, and − 1.8%, respectively, compared with a reduction of − 1.4 % with liraglutide and − 0.4% with placebo after 26 weeks. Furthermore, the 14-mg dose of oral semaglutide achieved a statistically significantly greater reduction in HbA1c with a difference of − 0.3 (p = 0.0272) compared with liraglutide. This trend continued at 52 weeks with subjects treated with oral semaglutide 3 mg, 7 mg, and 14 mg experiencing statistically significantly greater reductions in HbA1c compared with subjects treated with placebo (p < 0.0001 for all) (Table 2). The target of an HbA1c < 7.0% was achieved by 50%, 67%, and 80% of subjects treated with oral semaglutide 3 mg, 7 mg, and 14 mg, respectively, compared witho 49% of subjects treated with liraglutide and 12% of subjects treated with placebo at week 52 (p < 0.05 for all). Subjects treated with oral semaglutide 14 mg experienced a statistically significantly greater weight reduction of −2.8 kg after 52 weeks compared with − 1.0 kg with placebo (p = 0.0019) and a weight increase of +0.4 kg with liraglutide (p < 0.0001). Furthermore, significantly more subjects in the oral semaglutide 14-mg group achieved a weight loss of ≥ 5% compared with the liraglutide group or the placebo group at both 26 and 52 weeks (p < 0.01 for all). An increase in heart rate was observed for all subjects randomized to GLP-1RA treatment with an increase in 1 bpm with the oral semaglutide 3-mg dose, 3 bpm with the oral semaglutide 7-mg dose, and 4 bpm with the oral semaglutide 14-mg dose and 3 bpm with liraglutide.
3.3.11.1 Safety
Adverse effects were similar across all oral semaglutide groups with 71–76% reporting AEs. There was a slightly greater percentage of AEs reported in the placebo group of 80% when compared with the oral semaglutide groups, and a slightly lower number of AEs in the liraglutide group with 67% reporting AEs. Nasopharyngitis was the most reported AE and GI events were the second most reported AE. Only four subjects discontinued because of AEs, one in each group, and three of which were GI related. In this 52-week study, oral semaglutide was well tolerated and with a safety profile consistent with GLP-1-based therapy. The most common AEs for oral semaglutide were constipation and mild-to-moderate nausea, which diminished over time. The proportion of people who discontinued treatment because of AEs was 2–4% for people treated with oral semaglutide [19].
3.3.12 PIONEER 10
An open-label, 52-week randomized study completed in Japan, PIONEER 10, compared the safety, tolerability, and efficacy of oral semaglutide 3 mg, 7 mg, and 14 mg to subcutaneous dulaglutide 0.75 mg. Four hundred and fifty-eight subjects at least 20 years of age with T2DM inadequately controlled with one oral antihyperglycemic medication were randomized (2:2:2:1) to oral semaglutide 3 mg, 7 mg, or 14 mg once daily and dulaglutide 0.75 mg once weekly. Subjects treated with oral semaglutide 14 mg experienced a statistically significantly reduction, 1.8%, compared with 1.3% dulaglutide with 0.75 mg after 52 weeks while reductions for subjects taking oral semaglutide 3 mg and 7 mg were 0.7% and 1.4%, respectively. Glycosylated hemoglobin A1c below 6.5% was achieved by 21%, 43%, and 58% of people receiving treatment with oral semaglutide 3 mg, 7 mg, and 14 mg, respectively, compared with 41% of people treated with dulaglutide 0.75 mg. In addition to the HbA1c reduction, subjects receiving oral semaglutide 14 mg also showed a statistically significantly greater reduction in BW from baseline at week 52, with a reduction of 1.9 kg compared with a weight gain of 1.1 kg with dulaglutide (subjects treated with oral semaglutide 3 mg and 7 mg experienced a weight gain of 0.1 kg and weight reduction of 1.0 kg, respectively)
3.3.12.1 Safety
A comparable number of AEs were observed with oral semaglutide compared to dulaglutide 0.75 mg with 31%, 39%, 54%, and 40% of subjects experiencing GI AEs with oral semaglutide 3 mg, 7 mg, 14 mg, and dulaglutide respectively. The most frequently reported GI events were constipation and nausea and the percentage of subjects who discontinued treatment because of AEs was similar between groups with 3–6% of subjects treated with oral semaglutide compared to 3% of subjects treated with dulaglutide [37].
The PIONEER 11 and PIONEER 12 studies began recruiting subjects in 2019 and are currently open for enrollment in China. PIONEER 11 is evaluating the efficacy and safety of oral semaglutide in subjects with T2DM with an HbA1c of 7–10% who have been treated with only diet and exercise with a primary endpoint of change in Hb1A1c [38]. The PIONEER 12 study is evaluating the efficacy and safety of oral semaglutide compared to sitagliptin in subjects with T2DM with an HbA1c of 7–10.5% treated with metformin [39].
4 Conclusions
Overall, oral semaglutide demonstrated safety and noninferiority when compared to subcutaneous semaglutide as well as the other GLP-1RAs across its clinical studies. Oral semaglutide is novel in that it is the first oral GLP-1RA to provide patients with an alternative to the subcutaneous GLP-1RAs currently available. Many patients with T2DM are hesitant to move to treatment with injectable medications [14] but cannot achieve target HbA1c levels with previously available oral medications alone. Oral semaglutide is likely to allow many patients to continue with oral treatment who would otherwise require escalation of therapy using either an injectable GLP-1RA or insulin. Oral semaglutide is expected to produce an incremental benefit vs alternative T2DM treatments in terms of MACEs prevented; however, the CVD benefit is currently being evaluated in the SOUL trial [17, 20, 21, 26, 34].
References
Services USDoHaH. National diabetes statistics report, 2020. Estimates of diabetes and its burden in the United States: Center for Disease Control and Prevention; 2020. https://www.cdc.gov/diabetes/data/statistics-report/index.html. Accessed 3 Aug 2020
Saisho Y. β-cell dysfunction: its critical role in prevention and management of type 2 diabetes. World J Diabetes. 2015;6(1):109–24.
Kolluru GK, Bir SC, Kevil CG. Endothelial dysfunction and diabetes: effects on angiogenesis, vascular remodeling, and wound healing. Int J Vasc Med. 2012;2012:918267.
American Diabetes Association. Pharmacologic approaches to glycemic treatment: standards of medical care in diabetes: 2020. Diabetes Care. 2020;43(Suppl. 1):S98-110.
American Diabetes Association. Obesity management for the treatment of type 2 diabetes: standards of medical care in diabetes: 2020. Diabetes Care. 2020;43(Suppl. 1):S89-97.
Aroda V, Rosenstock J, Terauchi Y, et al. PIONEER 1: randomized clinical trial of the efficacy and safety of oral semaglutide monotherapy in comparision with placebo in patients with type 2 diabetes. Diabetes Care. 2019;42(9):1724–32.
Shah M, Vella A. Effects of GLP-1 on appetite and weight. Rev Endocr Metab Disord. 2014;15(3):181–7.
Rosenstock J, Allison D, Birkenfeld AL, et al. Effect of additional oral semaglutide vs sitagliptin on glycated hemoglobin in adults with type 2 diabetes uncontrolled with metformin alone or with sulfonylurea: the PIONEER 3 randomized clinical trial. JAMA. 2019;321(15):1466–80.
Dhindsa DS, Mehta A, Sandesara PB, Thobani A, Brandt S, Sperling LS. Strategies for appropriate selection of SGLT2-i vs. GLP1-RA in persons with diabetes and cardiovascular disease. Curr Cardiol Rep. 2019;21(9):100.
Pratley R, Amod A, Hoff ST, et al. Oral semaglutide versus subcutaneous liraglutide and placebo in type 2 diabetes (PIONEER 4): a randomised, double-blind, phase 3a trial. Lancet. 2019;394(10192):39–50.
Trujillo JM, Nuffer W, Ellis SL. GLP-1 receptor agonists: a review of head-to-head clinical studies. Ther Adv Endocrinol Metab. 2015;6(1):19–28.
Granhall C, Sondergaard FL, Thomsen M, Anderson TW. Pharmacokinetics, safety and tolerability of oral semaglutide in subjects with renal impairment. Clin Pharmacokinet. 2018;57(12):1571–80.
Pratley R, Aroda V, Lingvay I, et al. Semaglutide versus dulaglutide once weekly in patients with type 2 diabetes (SUSTAIN 7): a randomised, open-label, phase 3b trial. Lancet Diabetes Endocrinol. 2018;6:275–86.
Bucheit JD, Pamulapati LG, Carter N, Malloy K, Dixon DL, Sisson EM. Oral semaglutide: a review of the first oral glucagon-like peptide-1 receptor agonist. Diabetes Technol Ther. 2020;22(1):10–8.
Baekdal TA, Breitschaft A, Navarria A, Hansen CW. A randomized study investigating the effect of omeprazole on the pharmacokinetics of oral semaglutide. Expert Opin Drug Metab Toxicol. 2018;14(8):869–77.
Ahmann A, Capehorn M, Charpentier G, et al. Efficacy and safety of once-weekly semaglutide versus exenatide ER in subjects with type 2 diabetes (SUSTAIN 3): a 56-week, open-label, randomized clinical trial. Diabetes Care. 2018;41(2):258–66.
Marso SP, Bain SC, Consoli A, et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2016;375:1834–44.
Mosenzon O, Blicher TM, Rosenlund S, et al. Efficacy and safety of oral semaglutide in patients with type 2 diabetes and moderate renal impairment (PIONEER 5): a placebo-controlled, randomised, phase 3a trial. Lancet Diabetes Endocrinol. 2019;7(7):515–27.
Yamada Y, Katagiri H, Hamamoto Y, et al. Dose-response, efficacy, and safety of oral semaglutide monotherapy in Japanese patients with type 2 diabetes (PIONEER 9): a 52-week, phase 2/3a, randomised, controlled trial. Lancet Diabetes Endocrinol. 2020;8(5):377–91.
Bucheit J, Pamulapati LG, Carter N, Malloy K, Dixon DL, Sisson EM. Oral semaglutide: a review of the first oral glucagon-like peptide-1 receptor agonist. Diabetes Technol Ther. 2020;1:10–8.
Anderson SL, Beutel TR, Trujillo JM. Oral semaglutide in type 2 diabetes. J Diabetes Complications. 2020;34(4):107520.
Pais R, Gribble FM, Reimann F. Stimulation of incretin secreting cells. Ther Adv Endocrinol Metab. 2016;7(1):24–42.
Holst JJ, Visboll T, Deacon CF. The incretin system and its role in type 2 diabetes mellitus. Mol Cell Endocrinol. 2009;297:127–36.
Muller TD, Finan B, Bloom SR, et al. Glucagon-like peptide 1 (GLP-1). Mol Metab. 2019;30:72–130.
Bodnaruc AM, Prud’homme D, Blanchet R, Giroux I. Nutritional modulation of endogenous glucagon-like peptide-1 secretion: a review. Nutr Metab (Lond). 2016;13:92.
Sposito AC, Berwanger O, de Carvalho LSF, Kerr SF. GLP-1RAs in type 2 diabetes: mechanisms that underlie cardiovascular effects and overview of cardiovascular outcome data. Cardiovasc Diabetol. 2018;17(1):157.
Marathe CS, Rayner CK, Jones KL, Horowitz M. Effects of GLP-1 and incretin-based therapies on gastrointestinal motor function. Exp Diabetes Res. 2011;2011:279530.
Buckley ST, Baekdal TA, Vegge A, et al. Transcellular stomach absorption of a derivatized glucagon-like peptide-1 receptor agonist. Sci Transl Med. 2018;10(467):eaar7047.
Davies M, Pieber TR, Hartoft-Nielsen ML, Hansen OKH, Jabbour S, Rosenstock J. Effect of oral semaglutide compared with placebo and subcutaneous semaglutide on glycemic control in patients with type 2 diabetes: a randomized clinical trial. JAMA. 2017;318(15):1460–70.
Ismail R, Csoka I. Novel strategies in the oral delivery of antidiabetic peptide drugs: insulin, GLP 1 and its analogs. Eur J Pharm Biopharm. 2017;115:257–67.
Granhall C, Donsmark M, Blicher TM, et al. Safety and pharmacokinetics of single and multiple ascending doses of the novel oral human GLP-1 analogue, oral semaglutide, in healthy subjects and subjects with type 2 diabetes. Clin Pharmacokinet. 2018;58(6):781–91.
Baekdal T, Thomsen M, Kupcova V, Hansen CW, Anderson TW. Pharmacokinetics, safety, and tolerability of oral semaglutide in subjects with hepatic impairment. J Clin Pharmacol. 2018;58(10):1314–23.
Rodbard HW, Rosenstock J, Canani L, et al. Oral semaglutide versus empagliflozin in patients with type 2 diabetes uncontrolled on metformin: the PIONEER 2 trial. Diabetes Care. 2019;42(12):2272–81.
Husain M, Birkenfeld AL, Donsmark M, et al. Oral semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2019;381(9):841–51.
Pieber TR, Bode B, Mertens A, et al. Efficacy and safety of oral semaglutide with flexible dose adjustment versus sitagliptin in type 2 diabetes (PIONEER 7): a multicentre, open-label, randomised, phase 3a trial. Lancet Diabetes Endocrinol. 2019;7(7):528–39.
Zinman B, Aroda VR, Buse JB, et al. Efficacy, safety, and tolerability of oral semaglutide versus placebo added to insulin with or without metformin in patients with type 2 diabetes: the PIONEER 8 trial. Diabetes Care. 2019;42:2262–71.
ClinicalTrials.gov. PIONEER 10 trial. Available from: https://clinicaltrials.gov/ct2/show/NCT03015220. Accessed Oct 2020.
ClinicalTrials.gov. PIONEER 11 trial. Available from: https://clinicaltrials.gov/ct2/show/NCT04109547. Accessed Oct 2020.
ClinicalTrials.gov. PIONEER 12 trial. Available from: https://clinicaltrials.gov/ct2/show/NCT04017832. Accessed Oct 2020.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No sources of funding were received for the preparation of this article.
Conflict of interest
Mae Sheikh-Ali has received a speaker honorarium from Novo Nordisk, Eli Lilly, AstraZeneca, Janssen, and Merck. David Sutton has received a speaker honorarium from Novo Nordisk, Eli Lilly, Boehringer Ingelheim, Zealand Pharma, AbbVie, Abbot, and Amarin. Stephanie Niman, Jennifer Hardy, Rebecca F. Goldfaden, Jessica Reid, and Rushab Choksi have no conflicts of interest that are directly relevant to the content of this article.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Code availability
Not applicable.
Author contributions
All authors contributed to the review. RG had the idea for the article, SN and JR performed the literature search and data analysis, and all authors drafted and/or critically revised the review. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Niman, S., Hardy, J., Goldfaden, R.F. et al. A Review on the Efficacy and Safety of Oral Semaglutide. Drugs R D 21, 133–148 (2021). https://doi.org/10.1007/s40268-021-00341-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-021-00341-8