
Abstract
Recently unique spans of genetic data are produced by researchers, there is a trend in genetic exploration using machine learning integrated analysis and virtual combination of adaptive data into the solution of classification problems. Detection of ailments and infections at early stage is of key concern and a huge challenge for researchers in the field of machine learning classification and bioinformatics. Considerate genes contributing to diseases are of huge dispute to a lot of researchers. This study reviews various works on Dimensionality reduction techniques for reducing sets of features that groups data effectively with less computational processing time and classification methods that contributes to the advances of RNA-Sequencing approach.
Similar content being viewed by others


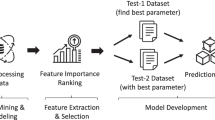
Introduction
Bioinformatics studies are based on major roles played by DNA (Deoxyribonucleic acids), RNA (Ribonucleic acids), genetic and genomic relating issues. Genomics have produced enormous amount of data that consist of extensive bit produced in the structure of protein–protein association and structure of 3-D [2]. Bioinformatics data needs to be stored in efficient ways, due to the major difficulty of its tools crashing when large data are stored in it [1]. Microarray technologies are microscopic slides containing thousands of prearranged series of samples of DNA, RNA, genes, protein, or tissues among others that represents most of the human genome [5]. A lot of works has been done in microarray to help in identifying human diseases [3]. By classifying microarray data into normal form of samples, it is probable to identify and find treatment for particular diseases [4].
Microarray based gene expression has been extensively used all through the last decade, in recent times RNA-Seq have substitute microarrays as the technology of preference in quantifying gene expression data such as cancer, pattern recognition, among other diseases, due to its valued advantages such as less noisy data, identifying innovative transcripts which does not require prearranged transcripts of concentration [6]. Over the past decades, initiatives have been put in place to support the scale up of anti-malaria interventions in Africa [13]. A major factor of malaria vector is Anopheles Gambiae with its broadest distribution in West Africa. Anopheles is the key vector of malaria with an emerging model for molecular and genetic studies of mosquito parasites interactions. There are forms of type of species responsible for majority of malaria transmission across [22]. Data analysis for the gene expression analysis is in advances.
RNA-Seq is an effective technique for gene expression profiling of organisms that utilizes the capabilities of Next Generation Sequencing (NGS) technologies. RNA-Seq is a capable means of having variety of applications such as; determining novel transcripts, detecting and quantifying the joint isoforms, informative sequence variation, synthesis detection, gene expression based classification to identify the significant transcripts, distinguishing biological samples and forecasting the results from large scale gene expression data which can be generated in a single run [7]. The gigantic quantity of data produced over the period of time has developed in many folds with trillions of datasets through several medium of information storage. Dimension reduction is a vital responsibility which engage huge sum of data proposed to be useful and non-redundant, making promising the consequent learning and generalization steps leading to enhanced human interpretations [1].
High dimensionality results into curse-of-dimensionality and heavy computation problems; frequently, dimensionality reduction techniques are applicable to machine learning technologies to ease relating issues. As dimensionality increases in data, the rate of computation in conventional dimensionality reduction approaches develops exponentially, and this makes computation prohibitively difficult. Increasing dimensionality approaches will map a lower and reduced dimensional feature space onto a higher-dimensional feature space and develop the linear separability [17]. Dimensionality reduction is a technique that is supportive, vital and essential; it trims, distinguishes and exemplifies datasets by converting a high dimensional dataset into a lower dimensional dataset by influencing the significant variables that prompts the unique data [8]. Classifying the sample of RNA-seq data thus can be used to identify cancer or not.
Currently machine learning technology has one of the greatest and promising approaches, they have been proposed and applied in a diverse of research works, such as recognition, language translation, bioinformatics and others [55]. RNA-seq skills are speedily developing, and its major challenges is addressing the huge amount of procedural noises that can determine about half percentage of cell disparities in expression sizes [56]. The dependability of capacities for RNA-Seq technology is labeled its sensitivity and accuracy. Both sensitivity and accuracy differ generally amongst RNA-seq technologies [56].
Studying the procedural differences and effects in the RNA-Seq data, this study reveals the ability to learn methods to be used and needed for improving and correcting biological analyses. These applications are beneficial as innovative technologies are developed with undiscovered preconceptions.
In this study several classifier algorithms with techniques of dimension reduction which are often applied during investigation of high dimensional data, to denoise and streamline data for several benefits that are helpful to the better performance of RNA-Seq data are reviewed, this study presents an assessment on diverse dimensionality reduction and classification approaches to improve the performance of RNA-Seq and propose models to accomplish improved performance by presenting promising prospects in predictions, in addition suggest approaches for RNA-seq data that will provide a research source for real-world RNA-based diagnosis and gene detection applications.
RNA-Seq
RNA sequencing is one of the key options in evaluating expression levels [9]. RNA-Seq is capable of executing lacking previous understanding of the sequences of importance and permits diversity of applications such as; assessment of nucleotide variations, reforming of transcriptome, assessment of methylation prototype, among others. RNA-seq technology contains several advantages that surpasses the microarray technology [10], for example; the elevated intensity of data reproducibility throughout the flow-cells, reducing the number of procedural replicates for a research. RNA-seq identifies and enumerates the expression of useful related proteins with comparable but distinguishable amino acid sequence, predetermined by miscellaneous genes (isoforms) [11]. The rate of next-generation sequencing research has fall significantly in the facet of high-throughput sequencing methodologies. However, a clear understanding about the qualitative and quantitative analysis of RNA-Seq has not yet been achieved, especially when compared to older methodologies such as microarray technology [12].
RNA-Seq is an exceptionally expression analysis technology helpful for several concerning precise states [15], with the advancing fame of RNA-Seq technology, numerous software and channels have been put in place for gene expression analysis from these data [16]. The invasion of high-dimensional and noisy data into the biological sciences has to taunt out the ability of lower-dimensional data structure to be crucial. Numerous examples have been provided recently of how lower-dimensional structure is capable of providing better insight in the biology world, helping as an understanding and visualization tools. Biological data has experienced an inundated high-dimensional and noisy data to a never before extent. Interesting structures can be uncovered using dimensionality reduction techniques in high-dimensional RNA-Seq data, little insight about a biological and technical considerations that possibly will explain the uncovered arrangement are given [14], there is no compromise regarding which methodology is most suitable to ensure the strength of results in terms of robustness and accuracy.
RNA-Seq study involves the phases of preprocessing, positioning, and quantification. There are several approaches to achieve these phases, although there is no best suitable procedure [63]. RNA-Seq data are available for downloading in several repository such as the NCBI (https://www.ncbi.nlm.nih.gov/sra), among others. RNA-Seq consists of next generating sequence (NGS) technology with millions of DNA fragments of novel transcript detection, they contain non-coding RNA detection and splice detection, with good-quality and non-degraded RNA of small amount required depletion [61].
A review of dimensionality reduction technologies
There has been a lot of research on medical diagnosis of malaria and other ailments in literature, and a whole lot of reports have been made on accuracies of classifications [61].
High dimensional data are challenging, they contain high computational cost and memory usage [18]. In machine learning, dimensionality reduction techniques consist of; Feature Selection and Feature Extraction.
The feature selection technique discovers the relevant features from the original dataset using objective measures, to reduce the number of features and to remove the irrelevant, redundant and noises from high-dimensional data [20]. The feature extraction technique is used to obtain the most relevant information from the original data and represent that information in a lower dimensionality space; it selects a new set of features and transforms the features into a linear or nonlinear combination of original features [21]. Dimensionality reduction techniques is an important factor which is used to reduce the features of the original data without the loss of information, it can be used remotely or in combination to improve performances such as accuracy among other parameters.
Dimensionality reduction is very beneficial and vital when evaluating high dimensional data [57], it is an essential analytic factor for RNA-seq data analysis. Appropriate dimensionality reduction algorithms can facilitate effective evaluations and classification performance of different approaches in relation to their capability to improve features of the innovative expression in terms of their performance metrics such as accuracy, sensitivity, specificity, recall, robustness, computational scalability, computational cost, among others [58].
Feature selection dimensionality reduction approach
Feature selection is widely used in data preprocessing for machine learning technology, it is fundamentally used for reducing data by getting rid of irrelevant and redundant features in a data [19]. Feature selection is a dimensionality reduction technique that improve the clarity of information the benefits a precise data, trims down time of training the learning algorithms, improves prediction performance and enhances visualization of data. Feature selection consists of three relevant variable selection categories; Filter methods, wrapper methods and embedded methods [18]. Diverse learning algorithms perform proficiently and provide better precise results when data holds non-redundant and significant attributes. Datasets have huge amount of irrelevant and redundant features, there is need for a proficient feature selection technique to extract relevant features. Feature selection methods are important for selecting informative genes proceeding to classification of RNA-Seq data for prediction and diagnosis of diseases, to improve the classification accuracy.
There are several feature selection techniques applied to malaria vectors among other ailments, such as typhoid, tuberculosis, diarrhea, measles, among others. Filter methods, wrapper methods, ensemble methods and embedded methods are the popularly used feature selection techniques.
In recent years, most authors are focusing on hybrid approaches used for feature selection. Before any model is applied to the data, it is always better to remove noisy and inconsistent data to get more accurate results in less time. Reducing the dimensionality of a dataset is of paramount importance in real world applications. Moreover, if most important features are selected, the complexity decreases exponentially. Several feature selections approaches have been applied to ailment datasets in recent year to explore valuable information, the utilization of feature selection methods is done on clinical databases for the prediction of numerous chronic diseases such as diabetes, heart disease, strokes, hypertension, among others. Various learning algorithms work efficiently and give more accurate results if the data contains more significant and non-redundant attributes. As the medical datasets contains large number of redundant and irrelevant features, an efficient feature selection technique is needed to extract interesting features relevant to the disease. A feature selection approach for the classification of cancer microarray gene expression dataset model, presented by [42], their paper, used data on microarray gene expression level to determine marker genes that are relevant to a type of cancer. They investigated a distance-based feature selection method for two-group classification problem. In order to select marker genes, the Bhattacharyya distance is implemented to measure the dissimilarity in gene expression levels between groups. They used the support vector machine to make classification with use of the selected marker genes. The performance of marker gene selection and classification are illustrated in both simulation studies and two real data analysis.
Filter based feature selection method
Filter based feature selection is based on certain evaluation principle [25]; it is a non-dependent approach, giving various performance on prediction. Filter based approach provides rapid and proficient results on execution. As a result, they are ideal for big databases. They perform efficiently with huge databases, computationally less expensive and efficient. Filter approach provides execution of results very fast with high-quality overview and it is low computational complexity [24]. They pick subsets of features by using relevant model learning algorithm and independent of whichever classifier, it ranks features on assessment condition basis. They depend on the fundamental uniqueness of data and their variable selection procedure requires execution formerly by means of this approach, they use statistical procedures for conveying scores to features due to their robustness against over-fitting compared to other methods and procedures.
The drawback of these approaches is that they pay no attention to the classification interaction, feature dependencies and failure in picking the most ‘‘useful” features [23]. Filter approach also have several drawbacks because it pays no consideration on the classifier’s interaction, features are not considered, and neglects several features that are not functional by themselves but can be valuable when combined with others. Filter algorithms are evaluated on different criteria; distance, information, dependency and consistency [25]. Filter based feature selection consists of the following algorithms that are relatively beneficial to a better performance of RNA-Seq [19]; ANOVA (Analysis of Variance), T-test feature selection, Information gain (IG), Fisher score, Chi-squared test, Correlation-based Feature Selection (CBFS), among others.
Wrapper based feature selection method
Wrapper based feature selection picks features by giving suitable consideration to the usage of knowledge algorithm. The major benefit over filter methods is locating the major constructive features and optimal selection of features is carried out for the learning algorithm [25]. Wrapper based feature selection approach explores the best subset in a feature by taking into account the learning algorithm to be used, it utilizes a precise classifier to evaluate the selected features quality, the classifier runs severally to assess the features quality, based on accuracy of a model, scoring is assigned. A wrapper-based method performs optimal selection of features and calculates the estimated accuracy for each feature using the bias of the induction algorithm to select features of the learning algorithm [27]. Wrapper method considers reliance along with features; it has an enhanced presentation in terms of predictive metrics, improved classifier relations, and optimizes the classifier performance and gives more precise results in comparison to filter methods [26]. However, it has a difficulty of utilizing an additional learning algorithm which result needs of executing the algorithms over again, it has more computational complexity, larger time of implementation, over-fitting dataset, huge computational resources, expensive than filter methods computationally, lacks generality and large datasets are less scalable. Wrapper based methods are complex and results to over-fitting on small training datasets. Methods that can be applied to RNA-Seq are; Simulated annealing, Sequential forward selection (SFS), Genetic Algorithms (GA), Recursive feature elimination (RFE) method, backward elimination Method, among others [19].
Embedded feature selection method
Embedded feature selection approach is usually guided by the learning process search known as the nested subset method [28]. It measures the ‘‘usefulness” of feature subsets and feature selection is carried out as a training process [17]. They work specifically for optimizing the performance of the learning algorithm. This makes use of data available and generates solutions faster. Search is guided by learning process in this approach by carrying out training process, the benefits of filter and wrapper approaches are aggregated and precise to the learning machines. Feature subsets usefulness is measured by using the supervised learning algorithms and optimizes the performance of a learning algorithm. They are inexpensive computationally and less prone to over-fitting. They are better classifiers with its dependencies between features capturing effectively, available data usage and providing faster solutions. Moreover, the computational complexity is better [29]. Its major limitation is taking dependent classification decisions, hence affecting the selected features by the hypothesis that the varying classifiers [30]. They are specific, with poor generality, considerate selection of relevant features for classifier usage and computationally costly. Embedded feature selection methods include; Decision Trees “ID3 (Iterative Dichotomiser 3), C4.5/5.0 algorithms”, CART (Classification and Regression Trees) and Random forest algorithm, Support vector machines- Recursive Feature Elimination (SVM-RFE) approach, Least Absolute Shrinkage and Selection Operator (LASSO) method, Elastic Net, Ridge Regression, Artificial neural networks, Weighted Naïve Bayes, Sequential Forward, Selection(SFS), Feature selection using the weighted vector of SVM, among others. Table 1 below shows some feature selection algorithms and their respective characteristics.
Feature extraction dimensionality reduction approach
Feature extraction is used in obtaining new latent optimal component features from a given dataset by transforming the data into a reduced complexity form of features, it gives a simple data representation of each variable in a feature subspace as a combination of linear input variables. Feature extraction is a more general method. Various methods exist, such as; Principal component analysis (PCA), Non-Linear principal component analysis, Kernel-PCA, independent component analysis, among others [18]. The most popular and widely used feature extraction approach is the PCA, introduced by Karl; it consists of an orthogonal transformation to convert samples belonging to correlated variables into samples of linearly uncorrelated variables. It can project the data from the original space into a lower dimensional space in an unsupervised manner (Table 2). Table 2 shows some feature extraction algorithms and their respective characteristics.
Related works
A comparative analysis of dimensionality reduction techniques on microarray gene expression data was carried out by authors [35], to assess the performance of the PCA, Kernel PCA (K-PCA), Locally Linear Embedding (LLE), Isomap, Diffusion Maps, Laplacian Eigenmaps and Maximum Variance Unfolding, in terms of visualization of microarray data.
In 2014, Xintao et al., [34] worked on dimensionality reduction model for high dimensional data. The experiment shows that the proposed dimension reduction techniques can be effective in analyzing and modeling the atmospheric corrosion data. The feature selection method shows that feature subset is optimal; feature extraction method reserves the original structure, discriminate information, and the integrity of data, etc. Their paper proposed dimensionality reduction solution that can solve the high-dimensional sample data problem.
Authors in [15] worked on dimensionality reduction of RNA-Seq Data. They worked on tissue type with principal components; they devised a principled way of choosing which latent components to consider when exploring the data. They used RNA-seq data taken from the brain and showed some of the most biologically informative Principal Components are high-dimensional. They used CSUMI (Component Selection Using Mutual Information) to explore how technical artifacts affect the global structure of the data, validating previous results and demonstrating how their method can be viewed as a verification framework for detecting undiscovered biases in emerging technologies, and compared CSUMI to two correlation-based approaches, showing theirs outperforms both.
2015, Sean, Jian, Jadwiga and Bonnie [62] worked on learning dimensionality reduction and RNA-Seq, they presented a mixed method called the component selection using mutual information (CSUMI)—that practices a mutual information concepts to translate the outcomes of PCA. CSUMI was applied to GTEx RNA-seq data. Their method exposed the unseen association among principal components (PCs) and methodological bases of variation across data samples. They worked on tissue natures and how it disturbs PCs, they developed a method for selecting which PCs to study when discovering the data. They applied the procedure to a brain RNA-seq data showed how informative PCs are higher-dimensional. They further used CSUMI to determine how technical artifacts disturb the comprehensive construction of the data. In conclusion, they relate CSUMI to correlation-based methods.
In 2015, Emma and Christopher, [36] worked on dimensionality reduction models for zero-inflated single cell gene expression analysis. They developed Zero Inflated Factor Analysis (ZIFA), which explicitly models the dropout characteristics, and shows that it improved the performance on simulated and biological datasets. They tested the relative performance of ZIFA against PCA, Probabilistic PCA (PPCA), Factor analysis (FA) and, where appropriate, non-linear techniques including Stochastic Neighbor Embedding (t-SNE), Isomap, and Multidimensional Scaling (MDS).
Authors in [3] worked on a feature selection based on One-Way-ANOVA for microarray data classification, by combining Analysis of Variance (ANOVA) for feature selection; to diminish high data dimensionality of feature space and SVM algorithms technique for classification; to reduce computational complexity and effectiveness. Noises and computational burden arising from redundant and irrelevant features are eliminated. It reduces gene expression data, which can drop the cancer testing cost significantly. The proposed approach selects most informative subset of features for classification to obtain a high-performance accuracy, sensitivity, specificity and precision.
A feature extraction comparative analysis for the classification of colon cancer in microarray dataset was carried out in [8]. The study demonstrates the effectiveness of feature extraction as a dimensionality reduction process, and investigates the most efficient approach that can be used to enhance classification of microarray. Principal Component Analysis (PCA) and Partial Least Square (PLS), an unsupervised and supervised technique respectively are considered, Support Vector Machine (SVM) classifier was carried out. The overall result shows that PLS algorithm provides an improved performance of about 95.2% accuracy compared to PCA algorithms.
In 2016, Michael et al. [33] worked on PCA and reported low intrinsic dimensionality on microarray data gene expression. They reevaluate their approach and showed linear intrinsic dimensionality of higher global map. They analyzed furthermore whereby PCA fails to distinguish relevant biologically information and point out methods that can overcome these limitations. Their results refine understanding of the gene expression spaces structures and shows that PCA critically depends on the effect size of the biological signal as well as on the fraction of samples containing this signal.
In 2017, Zhengyan, Li, and Chi, [37] classified lung adenocarcinoma and Squamous cell carcinoma using RNA-Seq Data. They used gene expression profile to discriminate NSCLC (Non-Small Cell Lung Cancer) patient’s subtype. We leveraged RNA-Seq data from The Cancer Genome Atlas (TCGA) and randomly split the data into training and testing subsets. To construct classifiers based on the training data, we considered three methods: Logistic Regression on Principal Components (PCR), logistic regression with LASSO shrinkage (LASSO), and Kth Nearest Neighbors (KNN). Performances of classifiers were evaluated and compared based on the testing data. Results: All gene expression-based classifiers show high accuracy in discriminating LUSC (Lung Squamos Cell) and LUAD (Lung Adenocarcinoma). The classifier obtained by LASSO has the smallest overall misclassification rate of 3.42% (95% CI: 3.25–3.60%) when using 0.5 as the cutoff value for the predicted probability of belonging to a subtype, followed by classifiers obtained by PCR (4.36%, 95% CI: 4.23–4.49%) and KNN (8.70%, 95% CI: 8.57–8.83%). The LASSO classifier also has the highest average for the receiver operating characteristic curve (AUC) value of 0.993, compared to PCR (0.987) and KNN (0.965). There results suggest that mRNA (Messenger Ribonucleic Acids-Sequencing) expressions are highly informative for classifying NSCLC subtypes and may potentially be used to assist clinical diagnosis.
In 2017, Arowolo et al. [38] worked on microarray dataset using hybrid dimensionality reduction model. Dimensionality reduction technique was combined, to address the highly correlated data problems and to select significant variables of features. One-Way-ANOVA was used for feature selection to get an optimal number of genes; Principal Component Analysis (PCA) and Partial Least Squares (PLS) were used separately as feature extraction methods, to reduce the selected features. Support Vector Machine (SVM) was used as a classification method on colon cancer dataset. Combining feature selection and feature extraction gave an efficient dimensional space. Redundant and irrelevant features were removed and an accuracy of about 98% was achieved.
In 2018, Jiucheng et al. [39] worked on selections of microarray genes using a supervised local linear embedding and correlation coefficient for classification. The feature genes selection has gained great significance in biology. This study proposed a supervised locally linear embedding and Spearman’s rank correlation coefficient feature selection method, based on the linear embedding and correlation coefficient algorithms. Supervised locally linear embedding used the class label information and improved the performance of the classification. Spearman’s rank correlation coefficient removes the co-expression genes. The experiment results were obtained on four public tumor microarray datasets.
In 2018, Byungjin, Ji, and Duhee, [40] worked on Single-cell RNA (Sc-RNA) sequencing technologies and bioinformatics pipelines. Technical challenges in single-cell isolation and library preparation on computational analysis pipelines available for analyzing scRNA-seq data was their major focus. Improvements of molecular and cell biology available in bioinformatics tools that will greatly facilitate both the basic science and medical applications of these sequencing technologies were carried out.
2018, a transcriptomic study of malaria was investigated on a systemic host–pathogen interaction, [61]. This study considered malaria as an example for the assessment of transcriptomic general host–pathogen relations in persons, abundance of the direct host–pathogen communication happens inside the blood, a voluntarily tested section of the body. They explained lessons learned from transcriptomic trainings of malaria and how it guides studies of host–pathogen relations in other transmittable diseases. They suggested that the probable of transcriptomic trainings to advance the understanding of malaria as an ailment remains partway unexploited because of limitations in learning strategy rather than as a significance of scientific limitations. Additional developments involve combination of transcriptomic information with diagnostic methods from extra scientific corrections, with epidemiology and mathematical modeling.
2019, Leihong, Xiangwen and Joshua [54] worked on an HetEnc deep learning predictive model for biological dataset, they proposed a novel deep learning-based approach that helps in separations of information domain. In their study, they assessed HetEnc on a two-platform dataset, the HetEnc holds the probable to increase multi-platforms.
2019, Li and Quon [56] worked on detection of model patterns in mitigating noises technically in large genomic data. They showed that procedural disparity in sequential datasets can be alleviated by evaluating feature discovery designs alone and disregarding feature quantification capacities. Their outcome embraces datasets having low discovery noise comparative to quantification noise. They determined state-of-the-art performance of discovery pattern models.
2019, Chieh and Ziv [60] developed a continuous state HMM in modelling time series ScRNA-Seq, they defined the CSHMM and provided learning and inference algorithms that allows the method in determining both structural branching process and assignment of cells. They analyzed cells datasets and showed the method accurately inferred topology of branching correctly.
In 2019, Shiquan, Jiaqiang, Ying and Xiang [58] worked on robustness, accuracy and scalability of dimensionality reduction approaches for RNA-Seq analysis, they provided a relative evaluation using dimensionality reduction approaches for RNA-Seq trainings. They compared 18-dimension reduction approaches on 30 openly obtainable RNAseq datasets covering several sample scopes and sequencing techniques. They assessed the performance of diverse approaches in terms of ability of recovering features of the innovative expression matrix, clustering of cells and reconstruction of lineage in terms of accuracy and robustness. They assessed the computational scalability of diverse methods in terms of their cost of computation.
Classification
Classification is the method of envisaging the class of certain data points. Classes are occasionally termed as targets or labels or groups. Classification analytical demonstration is approximating a mapping function (f) from input variables (X) to discrete output variables (y). For example, gene expression analysis can be known as a classification problem. This is a binary classification where there are classes. A classifier exploits training data to recognize how specified input variables relate to the class. When the classifier is trained precisely, it can be used to detect an unknown ailment. Classification is a supervised learning approach where the labels are with the input data. There are several applications in classification in various fields such as in credit endorsement, target marketing, medical diagnosis, among others [66].
Dataset samples belong to classes such as malignant dataset or non-malignant dataset, the goal is to classify these samples and produce classified samples based on its measurements in RNA-Seq. Classifier training of high-dimensional data sets is a great challenge that has received varieties of attention from the research community. A standard way of addressing the challenges is majorly done by using pre-processors and applications of classification algorithm that controls complexity model through regularization [32]. Machine learning is an approach that scientifically addresses some questions such as, how systems can be programmed to automatically learn and to improve with experience. Learning in this context is not considered a real learning process but recognizing complex patterns and make intelligence decisions based on data. Machine learning develops algorithms that discover knowledge from specific data and experience, based on computational principles. Classification aims to develop rule decisions that discriminate between samples of different classes based on the gene expression profile. Discovery of significant classification rules to accomplish the classification task is suitable for bio-medical research. Some of the widely used classifiers are Decision Tree, Neural Network (NN), Bat Algorithm, Artificial bee colony (ABC), Particle swarm optimization (PSO), K-NN, Support Vector Machine (SVM), among others [31], Convolution Neural Network (CNN) [59].
Multi-layer Perceptron (MLP)
MLP is a feedforward neural network error backpropagation useful in numerous grounds owing to its prevailing and constant learning algorithm [64]. Neural network studies the training samples by regulating the synaptic weight rendering to the error amount on the output layer. The back-propagation algorithm has a limited apprising the synaptic weights and preconceptions, and effective for calculating all the fractional results of the cost function with respect to these permitted parameters. A perceptron is a submissive design classifier. Adopting [64], the rule weight-update in backpropagation algorithm is well-defined as follows:
where w is the weight update achieved through the nth reiteration over the foremost loop of the algorithm, η is a positive constant called the learning rate, δ is the error term associated with j, and 0 ≤ α < 1 is a momentum constant.
Support vector machine (SVM)
SVM have become a popular classification method, and has been widely used to classify gene expression data measured on RNA-Seq Data [65]. SVM can be functional without alteration even when p > n. adopting the [65] model:
Hyperplane is defined as
Evaluation measures
Executing malaria vector data analysis in data mining system by utilizing classification algorithms requires, getting the evaluation measures requires the output results of a classification confusion matrix, which comprises of the metrics used in evaluating the classified models, where the model predicts the classes and outcomes (True Positive TP, True Negative TN, False Positive FP, and False Negative) [41]:
Accuracy
The closeness of a measured value to the standard or known value is termed as accuracy. It is otherwise stated as a weighted arithmetic means of precision and the recall.
Sensitivity
The true positive rate is also called as sensitivity is defined as the fraction of positives which are appropriately recognized.
where TP denotes the number of true positives that correctly classified the diagnostic test and FN denotes the number of false negatives that incorrectly classified the normal diagnostic test region.
Specificity
A true negative is called as specificity which evaluates the fraction of negatives that are appropriately recognized.
where, TN represents the number of true negatives that are properly classified in a normal portion and FP represents the number of false positives that incorrectly classifies the diagnostic regions.
Precision
The precision is the portion of the retrieved document that is related to the query and it called for the positive predictive rate (PPR).
Recall
The recall is termed as sensitivity, which is the proportion of the portion of recovered and relevant instances.
TP, TN, FP, FN
The four parameters deliberate such as TP (True Positive), TN (True Negative), FP (False Positive), and FN (False Negative) are utilized to classify the normal labels in a given the Datasets.
Conclusion
In this study, various dimensionality reduction and classification techniques are investigated to know how effectively they can be used to gain higher learning performance of algorithms that ultimately improves the predictive accuracy of various classifiers. Among dimensionality reduction algorithms, some algorithms involve in removal of irrelevant or redundant features while others involve in removal of both types of features. These algorithms develop small feature subset consisting of same types of features as in the original feature set or derive some new features from original features depending on the need. Classification of RNA-Seq is an emerging research area in the field of bioinformatics, applying dimensionality reduction techniques projects a better performance of the technology, therefore exploiting this study will bring about novels that will enrich the classification of RNA-Seq for diseases.
Availability of data and materials
Not applicable.
Abbreviations
- RNA-Seq::
-
Ribonucleic Acids-Sequencing
- DNA::
-
Deoxyribonucleic acids
- NGS::
-
Next Generation Sequencing
- 3-D::
-
Three Dimensional
- ANOVA::
-
Analysis of Variance
- IG::
-
Information Gain
- CBFS::
-
Correlated Based Feature Selection
- SFS::
-
Sequential Forward Selection
- GA::
-
Genetic Algorithm
- RFE::
-
Recursive Feature Elimination
- ID3::
-
Iterative Dichotomies 3
- CART::
-
Classification and Regression Trees
- SVM-RFE::
-
Support vector machines- Recursive Feature Elimination
- LASSO::
-
Least absolute shrinkage and selection operator
- PCA::
-
Principal Component Analysis
- ICA::
-
Independent Component Analysis
- PLS::
-
Partial Least Square
- K-PCA::
-
Kernel Principal Component Analysis
- LLE::
-
Locally Linear Embedding
- CSUMI::
-
Component Selection Using Mutual Information
- ZIFA::
-
Zero Inflated Factor Analysis
- PPCA::
-
Probabilistic PCA
- FA::
-
Factor Analysis
- SNE::
-
Stochastic Neighbor Embedding
- MDS::
-
Multidimensional Scaling
- SVM::
-
Support Vector Machine
- NSCLC::
-
Non-Small Cell Lung Cancer
- TCGA::
-
The Cancer Genome Atlas
- PCR::
-
Principal Component Regression
- MLP::
-
Multi-layer Perceptron
- K-NN::
-
Kth Nearest Neighbor
- mRNA::
-
Messenger Ribonucleic Acids-Sequencing
- LUSC::
-
Lung Squamos Cell
- LUAD::
-
Lung Adenocarcinoma
- Sc-RNA::
-
Single-cell Ribonucleic Acids
- NN::
-
Neural Network
- ABC::
-
Artificial bee colony
- PSO::
-
Particle swarm optimization
- TP::
-
True Positive
- TN::
-
True Negative
- FP::
-
False Positive
- FN::
-
False Negative
- PPR::
-
Positive Predictive Rate
References
Prathusha P, Jyothi S. Feature extraction methods: a review. Int J Innov Res Sci Eng Technol. 2017;6(12):22558–77.
Usman MA, Shahzad A, Javed F. Using PCA and Factor Analysis for Dimensionality Reduction of Bio-informatics Data. Int J Adv Comp Sci Appl. 2017;8(5):415–26.
Arowolo MO, Abdulsalam SO, Saheed YK, Salawu MD. A Feature Selection Based on One-Way-Anova for Microarray Data Classification. Al-Hikmah J Pure Appl Sci. 2016;3:30–5.
Sheela T, Lalitha R. An approach to reduce the large feature space of microarray gene expression data by gene clustering for efficient sample classification. Int J Comp Appl. 2018. https://doi.org/10.26808/rs.ca.i8v3.01.
Joseph MD, Madhavi D. Analysis of cancer classification of gene expression data a scientometric review. Int J Pure Appl Math. 2018;119(12):1–10.
Zararsız G, Dincer G, Selcuk K, Vahap E, Gozde EZ, Izzet PD, Ahmet O. A Comprehensive Simulation Study on Classification of RNASeq Data. PLOS Opened J. 2017. https://doi.org/10.1371/journal.pone.0182507.
Witten DM. Classification and Clustering of Sequencing Data Using a Poisson Model. Ann Application Stat. 2011;5(4):2493–518.
Arowolo, M.O., Isiaka, R.M., Abdulsalam, S.O., Saheed, Y.K., and Gbolagade, K.A. (2017).A Comparative Analysis of Feature Extraction Methods for Classifying Colon Cancer Microarray Data. Eur Allian Innov Endor Trans Scalable Information Systems. Vol. 4, No. 14, pp. 1–6.
Costa-Silva J, Domingues D, Lopes FM. RNA-Seq differential expression analysis: An extended review and a software tool. PLoS ONE. 2017;12(12):1–12. https://doi.org/10.1371/journal.pone.0190152.
Ana C, Pedro M, Sonia T, David G, Alejandra C, Andrew M, Michał WS, Daniel JG, Laura LE, Xuegong Z, Ali M. Survey of Best Practices for RNA-seq Data Analysis. Genome Biol. 2016;17(13):1–10. https://doi.org/10.1186/s13059-016-0881-8.
Agarwal A, Koppstein D, Rozowsky J, Sboner A, Habegger L, Hillier LW. Comparison and calibration of transcriptome data from RNA-Seq and tiling arrays. BMC Genomics. 2010;11(1):1–11.
Kratz A, Carninci P. The devil in the Details of RNA-seq. Nature Biotechnol. 2014;32(9):882–4.
Mariangela B, Eric O, William AD, Monica B, Yaw A, Guaofa Z, Joshua H, Ming L, Jiabao X, Andrew G, Joseph F, Guiyun Y. RNA-Seq analyses of changes in the anopheles Gambiae transcriptome associated with resistance to Pyrethroids in Kenya. Parasit Vectors. 2015. https://doi.org/10.1186/s13071-015-1083-z.
Sean S, Jian P, Jadwiga B, Bonnie B. Discovering what dimensionality reduction really tells us about RNA-Seq data. J Comp Biol. 2015. https://doi.org/10.1089/cmb.2015.0085.
Zhang ZH, Jhaveri DJ, Marshall VM, Bauer DC, Edson J, Narayanan RK. A Comparative Study of Techniques for Differential Expression Analysis on RNA-Seq Data. PloS ONE. 2014;9(8).
Oshlack A, Robinson MD, Young MD. From RNA-seq reads to differential expression results. Genome Biol. 2010;11(12):1–8.
Zena MH, Duncan FG. A review of feature selection and feature extraction methods applied on microarray data. Hindawi, Adv Bioinform. 2015;1:1–13. https://doi.org/10.1155/2015/198363.
Priyanka J, Dharmender K. A review on dimensionality reduction techniques. Int J Comput Appl. 2017;173(2):42–7.
Divya J, Vijendra S. Feature selection and classification systems for chronic disease prediction: A review. Egyptian Inform J. 2018. https://doi.org/10.1016/j.eij.2018.03.002.
Nadir OFE, Othman I, Ahmed HO. A novel feature selection based on one-way ANOVA F-Test for E-mail spam classification. Res J Appl Sci Eng Technol. 2014;7(3):625–38.
Arul VK, Elavarasan UN. A Survey on Dimensionality Reduction Technique. Int J Emerg Trends Technol Comput Sci (IJETTCS). 2014;3(6):36–42.
Jiang X, Peery A, Hall AB, Sharma A, Chen XG, Waterhouse RM, Komissarov A. Genome analysis of a major urban malaria vector mosquito. Anopheles Stephensi. 2014. https://doi.org/10.1186/s13059-014-0459-3.
Lavanya C, Nandihini M, Niranjana R, Gunavathi C. Classification of Microarray Data Based On Feature Selection Method. International Conference on Engineering Technology and Science. Int J Innov Res Sci Eng Technol. 2014;3(1): 1261–1264.
Yu L, Liu H. Feature selection for high-dimensional data: a fast correlationbased filter solution. ICML. 2003;3:856–63.
Kumar V, Minz S. Feature selection. SmartCR. 2014;4(3):211–29.
Maldonado S, Weber R. A wrapper method for feature selection using support vector machines. J Infom Sci. 2009;179(13):8–17.
Tang J, Alelyani S, Liu H. Feature selection for classification: a review. Data Classification: Algorithm Applications. 2014;37.
Eswari T, Sampath P, Lavanya S. Predictive methodology for diabetic data analysis in big data. Procedia Computing Science. 2015;50:203–8.
Xiao Z, Dellandrea E, Dou W, Chen L. ESFS: A New Embedded Feature Selection Method Based on SFS. Rapports de recherché; 2008.
Peng Y, Wu Z, Jiang J. A novel feature selection approach for biomedical data classification. J Biomed Inform. 2010;43(1):15–23.
Sumathi A, Santhoshkumar S, Sakthivel NK. Development of an efficient data mining classifier with microarray data set for gene selection and classification. J Theor Appl Inf Technol. 2012;35(2):209–14.
Emad MM, Enas MFE, Khaled TW. Survey on different methods for classifying gene expression using microarray approach. Int J Comput Appl. 2016;150(1):12–22.
Michael L, Franz M, Martin Z, Andreas S. Principal components analysis and the reported low intrinsic dimensionality of gene expression microarray data. Sci Rep. 2016;6:1–11. https://doi.org/10.1038/srep25696.
Xintao Q, Dongmei F, Zhenduo F. An efficient dimensionality reduction approach for small-sample size and high-dimensional data modeling. J Comput. 2014;9(3):576–83.
Christoph B, Hans K, Christian R, Xiaoyi J. Comparative study of unsupervised dimension reduction techniques for the visualization of microarray gene expression data. BMC Bioinformatics. 2010;11(1):1–11.
Emma P, Christopher Y. ZIFA: Dimensionality reduction for zero-inflated single-cell gene expression analysis. Genome Biol. 2015;16(1):1–10.
Zhengyan H, Chi W. Classifying Lung Adenocarcinoma and Squamous Cell Carcinoma using RNA-Seq Data. Cancer studies and molecular medicine. Open J. 2017;3(2):27–31. https://doi.org/10.17140/CSMMOJ-3-120.
Arowolo MO, Sulaiman OA, Isiaka RM, Gbolagade KA. A Hybrid Dimensionality reduction model for classification of microarray dataset. Int J Inform Technol Comput Sci. 2017;11:57–63. https://doi.org/10.5815/ijitcs.2017.11.06.
Jiucheng X, Huiyu M, Yun W, Fangzhou H. Feature genes selection using supervised locally linear embedding and correlation coefficient for microarray classification. Comput Math Methods Med. 2018. https://doi.org/10.1155/2018/5490513.
Byungjin H, Ji HL, Duhee B. Single-cell RNA sequencing technologies and bioinformatic pipelines. Exp Mol Med. 2018;50(8):96–104. https://doi.org/10.1038/s12276-018-0071-8.
Balamurugan M, Nancy A, Vijaykumar S. Alzheimer’s Disease Diagnosis by Using Dimensionality Reduction Based on KNN Classifier. Biomed Pharmacol J. 2017;10(4):1823–30.
Wenyan Z, Xuewen L. Feature selection for cancer classification using microarray gene expression data. Biostat Biometr Open Access J. 2017;1(2):1–7.
Pavithra D, Lakshmanan B. Feature selection and classification in gene expression cancer data. International Conference on Computational Intelligence in Data Science. IEEE. 2017, pp. 1–6
Kumara M, Rath NK, Swain A, Rath SK. Feature selection and classification of microarray data using MapReduce based ANOVA and KNearest neighbor. Procedia Comput Sci. 2015;54:301–10.
Uysal AK, Gunal S. A novel probabilistic feature selection method for text classification. Knowledge Based System. 2012;36(6):226–35.
Arul VK, and Elavarasan N. A survey on dimensionality reduction technique. Int J Emerg Trends Technol Comput Sci. 3(6):36–41.
Nalband S, Sundar A, Prince A, Agarwal A. Feature selection and classification methodology for the detection of kneejoint disorders. Comput Methods Programs Biomed. 2016;127:10–22.
Sivapriya TR, Banu N, Kamal AR. Hybrid Feature Reduction and Selection for Enhanced Classification of High Dimensional Medical Data IEEE International Conference on Computational Intelligence and Computing Research. 2013, pp. 327–30.
Guyon I. Gene selection for cancer classification using support vector machines. Machine Learn. 2002;46(1):389–422. https://doi.org/10.1023/A:1012487302797].
Joaquim PD, Hugo A, Luis ACR. A weighted principal component analysis and its application to gene expression data. IEEE/ACM Trans Comput Biol Bioinform. 2011;8(1):246–52. https://doi.org/10.1109/TCBB.2009.61.
Jin L, Yong X, Ying LG. Semi-supervised Feature Extraction for RNA-Seq Data Analysis. Conference: International Conference on Intelligent Computing, 2015.
Lucas A. 2013. “Package ‘amap’,”, http://cran.r-project.org/web/packages/amap/vignettes/amap.pdf.
Ching ST, Wai ST, Mohd SM, Weng HC, Safaai D, Zuraini AS. A review of feature extraction software for microarray gene expression data. Hindawi Publishing Corporation Biomend Research International. 2014;2014:1–16.
Leihong W, Xiangwen L, Joshua X. HetEnc: A Deep Learning Predictive Model for Multi-Type Biological Dataset. BMC Genomics. 2019;20(638):1–19. https://doi.org/10.1186/s12864-019-5997-2.
Cohen JB, Simi M, Campagne F. 2018. Genotype Tensors: Efficient Neural Network Genotype Callers. bioRxiv; 2018. p. 338780.
Li R, Quon G. scBFA: modeling detection patterns to mitigate technical noise in large-scale single-cell genomics data. Genome Biol. 2019;20(193):1–12. https://doi.org/10.1186/s13059-019-1806-0.
Lan HN, Susan H. Ten quick tips for effective dimensionality reduction. PLoS Comput Biol. 2019. https://doi.org/10.1371/journal.pcbi.1006907.
Shiquan S, Jiaqiang Z, Ying M, Xiang Z. Accuracy, robustness and scalability of dimensionality reduction methods for single cell RNASeq analysis. BioRxiv. 2019. doi:https://doi.org/10.1101/641142.
Huynh P, Nguyen V, Do T. Novel hybrid DCNN-SVM model for classifying RNA-Seq gene expression data. J Inform Telecommun. 2019;3(4):533–47. https://doi.org/10.1080/24751839.2019.1660845.
Chieh L, Ziv B. Continuous-State HMMS for Modeling Time-Series Single-Cell RNA-Seq Data. Bioinform Oxford Academic. 2019;35(22):4707–15. https://doi.org/10.1093/bioinformatics/btz296.
Hyun J, Athina G, Thomas DO, Michael L, Lachlan JC, David JC, Aubrey JC. Transcriptomic studies of malaria: a paradigm for investigation of systemic host-pathogen interactions. Microbiol Mol Biol Rev. 2018;82(2):1–17.
Sean S, Jian P, Jadwiga B, Bonnie B. Discovering what dimensionality reduction really tells us about RNA-Seq data. J Comput Biol Res Articles. 2015;22(8):715–28.
Conesa, A. (2016). A survey of Best Practices for RNA-seq Data Analysis. Genome Biology, 2016. Vol. 17, No. 1, pp. 13–23.
Mehdi P, Jack YY, Mary QY, Youping D. A comparative study of different machine learning methods on microarray gene expression data. BMC Genomics. 2016;9(13):1–13. https://doi.org/10.1186/1471-2164-9-S1-S13.
Kean MT, Ashley P, Daniela W. Statistical analysis of next generation sequencing data, frontiers in probability and the statistical sciences. Springer International Publishing Switzerland, 2014. pp. 219–246
Ayon D. Machine learning algorithms: a review. Int J Comput Sci Inform Technol. 2016;7(3):1174–9.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
Not applicable.
Corresponding author
Ethics declarations
Competing interests
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Arowolo, M.O., Adebiyi, M.O., Aremu, C. et al. A survey of dimension reduction and classification methods for RNA-Seq data on malaria vector. J Big Data 8, 50 (2021). https://doi.org/10.1186/s40537-021-00441-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40537-021-00441-x