
Abstract
We conducted year-round, monthly monitoring of the stable isotope composition of DIC and water in hypereutrophic Lake Kierskie, western Poland, along with isotope measures of calcite collected in sediment traps installed at 16 and 30 m water depth in the lake. Isotope data were supplemented by previously published data on physico-chemical variables in the lake water column. We sought to determine how carbon and oxygen isotopic disequilibria in calcite deposited in the lake’s laminated sediments vary seasonally, and what factors drive this variability. Deposition of calcite out of equilibrium with DIC and water was documented over the entire study period. For δ18O, the disequilibrium difference between successive months far exceeded the amplitude of the seasonal variability in the isotope composition of water. The biggest difference between the measured and calculated δ13Ccalcite and δ18Ocalcite values was observed during late autumn and winter sediment resuspension and redeposition (2.4‰ and 5.4‰, respectively). In the spring, δ13Ccalcite and δ18Ocalcite offsets from equilibria, 0.5‰ and 1.3‰, respectively, resulted from rapid precipitation of large calcite crystals. During summer, intense productivity and processes related to calcifying algae (“vital effects”) caused lower δ13C (0.5–1.8‰) and δ18O (2.8–2.9‰) in calcite. Differences between isotope values of calcite collected from the two water depths were small, and might have resulted from different settling velocities of small and large crystals, and/or preferential dissolution of smaller grains. We suggest that winter laminae should be excluded from isotope studies of varved sediments whenever possible, as they likely contain redeposited carbonate in which the isotope value is not indicative of conditions in the lake at the time of laminae formation. We also recommend supplementing isotope analysis of calcite in varved lake sediments with seasonally resolved analysis of carbonate content. It appears that major shifts in the proportion of carbonate deposited across seasons can cause notable changes in mean annual values of δ18Ocalcite and δ13Ccalcite, even if DIC and water isotopic compositions remain stable.
Similar content being viewed by others


Introduction
Stable carbon (δ13C) and oxygen (δ18O) isotope ratios in lake sediment carbonates are common proxies in paleolimnological studies, used to infer past dissolved inorganic carbon (DIC) and water isotopic compositions, respectively (Goslar et al. 1999; von Grafenstein et al. 2000; Mangili et al. 2007; Jonsson et al. 2010). The relationship between the isotope composition of carbonates and that of DIC and water, however, is not always straightforward, as multiple processes can cause deposition of carbonates in isotopic disequilibrium with ambient conditions (Fronval et al. 1995; Leng and Marshall 2004), many of which vary seasonally. For instance, kinetic isotope fractionation is most likely to occur during spring when calcite crystals form rapidly (Teranes et al. 1999a; Bluszcz et al. 2009), whereas factors related to organisms that control carbonate precipitation are more important during peak summer productivity (Bluszcz et al. 2009).
The seasonal variability of factors that cause deposition of carbonates out of equilibrium with DIC and water has not been considered in paleolimnological studies, though it almost certainly has an impact on interpreting carbonate isotope records in some lakes. In particular, such variability can play a role in disequilibrium if the proportion of carbonate deposited in different seasons throughout the annual cycle changes over time, and/or the difference between the isotopic composition of carbonates and ambient DIC and water varies between seasons. Detailed microfacies studies of varved sediments show that the proportions of carbonates deposited in different seasons of the annual cycle can change markedly over long temporal scales. For instance, Żarczyński et al. (2018) studied the 2000-year-long varved record from Lake Żabińskie (NE Poland) and found transitions between periods when calcite was deposited throughout the year to periods when calcite was restricted mainly to spring laminae, or occurred in similar amounts in both spring and summer laminae. It remains unclear, however, how the difference between the isotopic composition of carbonates and ambient conditions, i.e. DIC and water, varies between seasons. Few studies have reported on the topic of isotopic equilibrium or disequilibrium during calcite precipitation in lakes with varved sediments (Teranes et al. 1999b; Bluszcz et al. 2009; Ustrzycka et al. 2018). Precipitation of calcite out of isotopic equilibrium with ambient conditions is problematic in paleolimnological studies because it precludes reliable environmental inferences that rely on stable isotope measurements in sediments. Further actualistic studies will be required to better understand the conditions under which calcite precipitates and the processes that control carbon and oxygen stable isotope composition in CaCO3.
One lake in which modern conditions of sedimentation, including calcite precipitation, were recently investigated (Apolinarska et al. 2020), is Lake Kierskie, a hypereutrophic body of water located in western Poland. We conducted year-round monthly monitoring of the carbon and oxygen stable isotope composition of DIC and water, respectively, in Lake Kierskie, combined with isotopic studies of calcite collected in sediment traps installed in the lake at water depths of 16 and 30 m. Results were supplemented by previously published data on physico-chemical characteristics of the lake water (Apolinarska et al. 2020). Our aim was to document seasonal variability in carbon and oxygen isotopic disequilibria in calcite deposited in this highly eutrophic lake with laminated sediments, and identify the drivers of this variability. We used our results to address how seasonal variability in drivers of calcite disequilibrium with DIC and water affects paleolimnological studies. Lastly, we attempted to quantify the disequilibrium in calcite precipitates in the sediment.
Study site
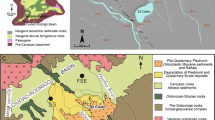
Lake Kierskie is located in the Poznań Lake District, on the northwest periphery of the city of Poznań (52°27′ N; 16°47′ E), at 72 m asl (Fig. 1). It was formed during recession of the Weichselian ice sheet from the area ca. 18.5 ka BP (Kozarski 1995). The postglacial morphology of the lake catchment (70.6 km2) is dominated by flat and undulating upland, as well as by a low-lying former glacial tunnel valley, within which the lake basin is located. Pleistocene glacial till and glacio-fluvial deposits are the dominant surface sediments in the area. Climate of the region is transitional, between the mild and humid Atlantic climate of western Europe and the continental climate of eastern Europe. The mean annual air temperature is 8 °C, with monthly averages between − 2.2 °C in January and 18 °C in July (Woś 1999). Annual precipitation between 1981 and 2015 ranged from 275 to 715 mm (Szyga-Pluta and Grześkowiak 2016).

Location of Lake Kierskie in Poland, and bathymetric map of Lake Kierskie with the location of the sediment traps marked with a white dot
Elongated Lake Kierskie is divided into a deeper (zmax = 35 m) southern basin and shallower (zmax = 20 m) northern basin (Fig. 1), and covers an area of 2.86 km2 (Fig. 1). The lake is fed by several inflows and is drained by a single outflow on its northeast end (Fig. 1). Kowalczak and Sosiński (1995) indicated that groundwater inflow contributes a substantial fraction of the lake hydrologic budget. Mean water residence time in Lake Kierskie is estimated to be 25–33 years (Jańczak and Sziwa 1995).
A recent (November 2015–October 2016) monthly sediment trap study in Lake Kierskie included monitoring of limnological variables comprising water clarity, hardness, alkalinity, nutrient and solute concentrations, trophic state, and phytoplankton community composition (Apolinarska et al. 2020). The study was motivated by recovery of a ~ 14-m-long core of discontinuously laminated sediments from the lake that was collected to explore the processes that lead to seasonal differences in the sediments deposited in Lake Kierskie. Sediment flux to the traps displayed a clear seasonal pattern related to physical and chemical characteristics of Lake Kierskie waters, the annual cycle of water-column mixing and stratification, and primary productivity, which directly or indirectly control the formation of the three major sediment constituents, i.e. organic matter (OM), calcite (CaCO3) and biogenic silica (SiO2biog). Intense SiO2biog sedimentation in early spring and heavy phytoplankton-related calcite precipitation during late spring and summer probably reflect the eutrophic to hypereutrophic conditions in the lake, indicated by Carlson’s (1977) Trophic State Index (maximum value = 60.6), which in turn reflects high phosphate concentrations, high summer chlorophyll-a concentrations and low Secchi disk transparency. High trophic status of Lake Kierskie is maintained by elevated nutrient concentrations in the lake input waters, i.e. high total nitrogen (TN) values of 2.79–6.71 mg l−1 and total phosphorus (TP) values of 0.12–0.87 mg l−1 (Grzonka et al. 2016), which drain cultivated lands, meadows and urbanised areas that today dominate the catchment (Apolinarska et al. 2020). In addition to nutrient loads delivered by inflows, the lake is commonly used for recreational purposes, and several beach resorts are located along the shores. More detailed characteristics of the lake and its catchment are presented in Apolinarska et al. (2020).
Materials and methods
Field sample collection
To identify the factors that control stable isotope values in calcite deposited in Lake Kierskie, we carried out a monitoring study of water-column characteristics and trophic status at roughly monthly intervals between November 2015 and October 2016, at the deepest site in the lake (35 m; GPS: N 52° 26′ 53.7″ E 16° 47′ 49.8″). Water samples for isotope analyses (δ13C of DIC and δ18O of water) were collected at 1 m water depth, placed into 10-ml glass septa test tubes and preserved with two drops of HgCl2 to prevent biological activity. Details regarding measurements of water transparency, temperature, dissolved oxygen concentration, conductivity, pH, ion concentrations, chlorophyll-a concentrations and phytoplankton analyses of lake waters were presented in Apolinarska et al. (2020).
Geochemical (Apolinarska et al. 2020) and isotopic (this study) analyses were carried out on sediment trap samples. Sediment traps were installed at 16 m water depth (two PVC tubes) and 30 m water depth (four PVC tubes) to track sedimenting particles in the water column and detect changes in the isotope composition of depositing calcite. Sediments were collected from plastic caps mounted at the lower end of 1-m-long, 6-cm-diameter PVC tubes. All water and sediment samples were kept refrigerated prior to laboratory analyses.
Laboratory work
Water analysis
Isotope analyses of DIC and water were conducted using a GasBench-II headspace autosampler online, with a Finnigan MAT 253 isotope ratio mass spectrometer (IRMS) in a continuous flow system. Accuracy of the δ13CDIC results was determined using three international carbonate standards, NBS 18, NBS 19 and LSVEC, which were measured along with each series of samples. δ13CDIC values are reported in standard δ-notation in per mil (‰) versus Vienna Pee Dee Belemnite (V-PDB). Accuracy of the δ18O results was established by measuring three international standards (GISP, W6444, W67400) for each series of measurements. δ18Owater values are reported in per mil relative to V-SMOW. Precision was tested by replicate analysis of laboratory standards and was found to be better than ± 0.2‰ for δ13CDIC and ± 0.25‰ for δ18Owater.
Sediment samples
Prior to isotope analyses, sediments collected in the two tubes at 16 m depth were combined. Likewise, sediments in the four tubes from 30 m depth were combined, thereby yielding a single sample per depth. Dried sediment samples were homogenised using a mortar and pestle. Stable isotope compositions of sediment calcite (δ13Ccalcite and δ18Ocalcite) were determined using a Kiel IV online carbonate preparation line connected to a Thermo Finnigan Delta + mass spectrometer. Calcite was dissolved using 100% phosphoric acid (density 1.9) at 75 °C. All values are reported as δ values, where δ = (Rsample/Rstandard − 1) × 1000, in per mil relative to V-PDB by assigning a δ13C value of 1.95‰ and a δ18O value of − 2.20‰ to NBS19. Reproducibility was checked by repeated measurement of NBS19 and was better than ± 0.03 and 0.07‰, for δ13C and δ18O, respectively.
Calculated δ13Ccalcite and δ18Ocalcite values
The ability of δ13C and δ18O values in calcite precipitated in Lake Kierskie to accurately record δ13C values of the DIC and δ18O values of the lake waters and water temperature (the two variables that control δ18Ocalcite), can be verified by determining the state of isotopic equilibrium during calcite precipitation.
Under equilibrium conditions, δ13Ccalcite should be 0.9‰ higher relative to δ13C in HCO3- (Romanek et al. 1992), which, in the pH range 7.6–8.9, observed in Lake Kierskie (Apolinarska et al. 2020), is the dominant dissolved carbon species (Wetzel 2001). Therefore, theoretical equilibrium δ13Ccalcite values were calculated by adding 0.9 to δ13C values in DIC (Fig. 2b).

a Carbon stable isotope values of DIC (δ13CDIC) and oxygen stable isotope values of water (δ18Owater) in samples collected at 1 m water depth. b δ13C and c δ18O values of calcite collected in the sediment traps installed at 16 and 30 m water depth (diamonds), and theoretical δ13C and δ18O values of calcite (triangles). C and O isotope values of water and DIC are presented as means of two months to better reflect the isotopic composition of DIC and water. d Summary of the dominant processes that affect sedimentation in Lake Kierskie. e CaCO3 flux to the sediment traps. f Concentration of HCO3− and CO32− in the epilimnion at 1 m water depth. g Lake water temperature (1 m water depth) shown along with temperature data from the meteorological station at Ławica airport, approximately 3 km SE from Lake Kierskie
The paleotemperature equation proposed by Kim and O'Neil (1997), commonly applied to calculate the equilibrium precipitation of lacustrine calcite from solution (Teranes et al. 1999a; Leng and Marshall 2004; Ustrzycka et al. 2018), was used to determine theoretical δ18Ocalcite values:
where T is the temperature in Kelvin (i.e. °C + 273.18), and α is the fractionation between calcite and water (18O/16O)calcite/(18O/16O)water.
The equation of Kim and O'Neil (1997) was re-expressed by Leng and Marshall (2004) in a more convenient form to solve for temperature:
where δc is δ18O of the sample calcite compared to the PDB international standard and δw is the δ18O of the sample water compared to the SMOW international standard.
To better reflect the conditions of calcite precipitation between the field sampling campaigns, mean values of water temperature and mean δ18Owater values measured at the beginning and end of each of the periods of sediment accumulation in the traps were used in the above equation.
Because of calcite dissolution in the water column of Lake Kierskie reported by Apolinarska et al. (2020), calculated δ13Ccalcite and δ18Ocalcite values were compared with δ13C and δ18O values measured in calcite trapped at 16 m (Fig. 2b, c). We assumed there had been less alteration of crystals and their isotope values in shallower-water traps (16 m), as the distance the calcite had settled was shorter, compared to carbonate in the deep-water trap.
Results
Stable isotope composition of lake surface waters
The δ13C of DIC and the δ18O of water in Lake Kierskie differ seasonally. DIC was 13C-depleted during the autumn and winter, with the lowest δ13CDIC value (− 9.05‰) measured in October 2016 (Fig. 2a). In spring and summer, 13C-enrichment was observed, and the highest δ13CDIC value (− 2.18‰) was recorded in June. The range of δ18Owater values was much smaller compared to that observed for δ13CDIC values (Fig. 2a). Lake waters became slightly 18O-enriched between early spring, i.e. in April when a minimum value of − 4.47‰ was recorded, and late summer (September), when a maximum value of − 3.44‰ was measured.
Stable isotope composition of calcite
Carbon and oxygen stable isotope values of calcite differed across seasons. Although values in calcites trapped at 16 and 30 m in the water column differed slightly, the seasonal trend of changes in the isotope values was independent of water depth. Average δ13C values in calcite deposited during the late autumn and winter were − 4.15‰ (16 m) and − 4.45‰ (30 m), respectively (Fig. 2f). Similar average δ13Ccalcite values were measured in calcite precipitated in the spring, − 4.30‰ (16 m) and − 4.38‰ (30 m). The most 13C-enriched calcite was collected in the summer, − 3.43‰ (16 m) and − 3.79‰ (30 m), however strong 13C depletion of the calcite was observed in late summer, with values of − 7.0 and − 6.53‰ at 16 and 30 m, respectively. Calcite samples with the lowest δ18O values were deposited during the late autumn and winter (Fig. 2c, − 7.77‰ at 16 m and − 7.88‰ at 30 m, on average), and in the summer (− 8.86‰ at 16 m and − 8.31‰ at 30 m, on average). The most 18O-enriched calcite was precipitated during spring (− 5.65‰ at 16 m and − 5.68‰ at 30 m, on average).
Carbon stable isotope values of calcite from the shallower trap were from 0.37‰ higher to 0.47‰ lower than values from the deeper trap (Fig. 2b). In most samples, the difference between δ18O values in calcite from 16 and 30 m was within ± 0.2‰ (Fig. 2c). Exception was 18O-enriched calcite (2.3‰) collected from the deeper trap in September.
Calculated versus measured δ13Ccalcite and δ18Ocalcite values
δ13C values in calcite deposited in the sediment traps during winter and autumn were ~ 2.4‰ higher compared to calculated equilibrium δ13Ccalcite values (Figs. 2b, 3). In early spring, 13C enrichment in the sediment trap calcite decreased to 0.5‰. In contrast, δ13C values in the calcite precipitated between mid-April and mid-September were between 0.5 and 4.2‰ lower than expected under equilibrium conditions (Figs. 2c, 3).

Theoretical vs. measured δ13C and δ18O values of calcite
Theoretical δ18Ocalcite values were in all months higher than measured δ18Ocalcite values (Figs. 2c, 3, 4). A large difference between measured and calculated δ18Ocalcite values was observed during late autumn (5.07‰) and winter (5.69‰) (Fig. 2c). In early spring, it decreased to 2.30‰ and during late spring was even lower, ranging between 1.28 and 1.66‰. During summer, the difference between calculated and measured δ18Ocalcite values increased to 2.84–2.93‰, and was the highest (6.27‰) in the late summer samples (Fig. 2c).

Theoretical and measured δ18O values of calcite versus epilimnetic water temperatures (mean of values measured at the start and end of each sedimenting period). δ18O values of calcite precipitated during particular seasons are marked with different symbols
Discussion
Calcite isotopic disequilibrium with DIC and water—seasonal variability and its drivers
Previously recognised, season-specific disequilibria were usually restricted to spring (Teranes et al. 1999a; Bluszcz et al. 2009). In Lake Kierskie, deposition of calcite out of isotopic equilibrium with ambient conditions was reported throughout the entire monitoring period, with differences in magnitude and causes of the disequilibrium observed among seasons (Figs. 2b, c, 3).
The biggest difference between measured and calculated δ18Ocalcite values was observed during late autumn (5.07‰) and winter (5.69‰) (Figs. 2c, 4), similar to what was reported for Baldeggersee (Teranes et al. 1999a) and Sacrower See (Bluszcz et al. 2009). Calcite deposited during autumn and winter was also out of isotopic equilibrium with ambient DIC, ~ 2.4‰ enriched in 13C compared to theoretical δ13Ccalcite values. Carbonates collected at that time originated exclusively from resuspension and redeposition of previously deposited sediments (Fig. 2d; Apolinarska et al. 2020), therefore δ13C and δ18O values of the calcite were not controlled by ambient conditions in late autumn and winter. Instead, δ13C and δ18O values of the redeposited calcite reflect the δ13C and δ18O values of calcite precipitated in the previous seasons.
Calcite deposited during the early spring (17 March–16 April 2016) is a mixture of material resuspended and redeposited during spring water mixing, and newly precipitated biogenic calcite from active biological assimilation of CO2 (Apolinarska et al. 2020). Compared to late autumn and winter, this mixture displays a smaller difference between the measured and calculated δ13C (~ 2.4‰ during autumn and winter versus 0.5‰ in spring) and δ18O (5.07–5.69‰ during autumn and winter versus 2.30‰ in spring) values in calcite (Figs. 2b, c, 3, 4).
Enhanced precipitation of large calcite crystals started later (Fig. 2d), after phosphate ions were assimilated by the primary producers and their concentration in the epilimnion decreased (Apolinarska et al. 2020). During periods of rapid CaCO3 precipitation, mass-dependent processes that occur contribute to kinetic isotope effects, leading to preferential incorporation of light 12C and 16O isotopes into calcite, and therefore lower δ13Ccalcite and δ18Ocalcite values (Fronval et al. 1995; Bluszcz et al. 2009; Teranes et al. 1999a). Such an effect was observed in many biogenically mediated, fast-growing carbonates (McConnaughey 1989 and references therein). In Lake Kierskie, large calcite crystals that precipitated between 15 April and 16 June from CaCO3-supersaturated waters, were out of carbon (1.10–1.27‰ depleted in 13C) and oxygen (1.28–1.66‰ depleted in 18O) isotopic equilibrium with water (Fig. 2b, c, d). Similar, though smaller disequilibria were reported from Lake Areso (Fronval et al. 1995) and Baldegersee (Teranes et al. 1999a).
Along with reduced rates of CaCO3 precipitation during summer, the difference between calculated and measured δ18O values of the calcite should also be much smaller. Such results have been reported from several lakes (Fronval et al. 1995; Filippi et al. 1999; Teranes et al. 1999a; Bluszcz et al. 2009). In contrast to expectations, however, in Lake Kierskie the difference between calculated and measured δ18O values of calcite increased from 1.28 to 1.66‰ in spring, to 2.84–2.93‰ between 16 June and 24 August, and 6.27‰ between 24 August and 13 September (3.98‰ at 30 m) (Fig. 2c). Carbon stable isotope values of the calcite were 0.54 to 1.81‰ depleted in 13C relative to δ13C of theoretical calcite (Fig. 2b). Exceptionally strong depletion of 13C, by 4.21‰, was observed in late summer (Fig. 2b).
Determination of the causes of isotopic disequilibria is difficult. Two likely explanations, however, are provided below. First, greater disequilibrium may result from changes in water pH and the resultant shift in the proportions of major carbon forms in the water (Fig. 2g). At higher pH, when hydroxylation of CO2 (CO2 + OH− ↔ CO32−) becomes dominant over hydration of CO2 (CO2 + H2O ↔ H+ + HCO3−), carbonate species are 18O-depleted (Usdowski et al. 1991; Usdowski and Hoefs 1993), resulting in lower δ18O values in calcite and greater disequilibrium. Such a relation was reported in Baldeggersee and Soppensee, where δ18O values in authigenic calcite declined in response to elevated CO32− concentrations in surface waters (Teranes et al. 1999a). Although correlation between the extent of δ18O disequilibrium (Δδ18Omeasured-calculated) and concentration of CO32− in the epilimnion was not observed in Lake Kierskie (Fig. 5), local pH values, in the microenvironment close to picoplankton cell walls, which serve as nuclei for CaCO3 precipitation (Dittrich and Obst 2004), can be very high. In addition, evidence suggests that in conditions of CO2 shortage, generated during intense photosynthesis, some phytoplankton groups that dominate in alkaline systems, including some green algae, cyanobacteria and diatoms, can use bicarbonate (HCO3−), which results in release of OH− from cells (Borowitzka 1989; Park et al. 2008), causing further increases in pH. Since biogenically induced calcite precipitation occurs during relatively short-lived (a few days), discrete productivity peaks (Teranes et al. 1999a), detailed characteristics of CaCO3 precipitation would require monitoring of the ambient conditions during those peaks.

Relation between the magnitude of δ18O disequilibrium (Δδ18Omeas-calc) and the amount of CO32− in lake waters
Isotopic disequilibria in biogenic calcite precipitated during summer can also be explained by the “vital effects” of calcifying algae. For instance, Bluszcz et al. (2009) argued that this explained the more negative δ18O values in calcite precipitated by the green alga Phacotus lenticularis (Ehrenberg) Diesing in Lake Sacrower See, Germany. Species morphologies and the sizes of calcite crystals precipitated by different phytoplankton groups indicate that different taxa cause distinct precipitation processes (Dittrich and Obst 2004; Dittrich et al. 2004). In Lake Kierskie, the summer phytoplankton assemblage was composed mainly of chlorophytes, cryptophytes and dinoflagellates, whereas the diatom bloom occurred during spring (Apolinarska et al. 2020). Some of the identified phytoplankton taxa are known to be associated with calcite precipitation, e.g. picocyanobacteria, filamentous cyanobacteria, the diatom Stephanodiscus hantzschii Grunow and chlorophytes (Phacotus lendneri Chodat and representatives of the genus Chlamydomonas) (Stabel 1986; Kosamu and Obst 2009; Lenz et al. 2018; Karami et al. 2019), however the “vital effects” of particular species are not known and are beyond the scope of this study.
The drivers of calcite isotopic disequilibrium with DIC and water during all but winter and early spring seasons, when redeposition took place, are at least indirectly related to the high trophic state of Lake Kierskie. In particular, precipitation of large calcite crystals during late spring, caused by high phosphorus concentration, and shifts in the relative proportions of carbon species, triggered by intense photosynthesis during summer, contribute to the disequilibrium. Consequently, changes in primary productivity likely contribute to shifts in isotope values of calcite precipitated in lakes. Indeed, this has been confirmed by several previous studies. For instance, in Lake Areso (Fronval et al. 1995) a 2–2.5‰ decrease in δ18O values was observed between calcite precipitated in preindustrial and modern times. In Lake Ontario, calcite became 2.3‰ 18O-depleted between 1940 and 1987 (Schelske and Hodell 1991), whereas δ18O values in calcite from Lake Greifen decreased by 2–3‰ between the 1950s and mid-1970s (McKenzie 1985; McKenzie and Hollander 1993). In all these lakes, changes in isotopic values of carbonates were accompanied by marked increases in trophic state.
Precipitating calcite can be subject to several processes as it settles to the lake bottom, but those processes were not as important in Lake Kierskie as the processes listed above, in causing isotopic disequilibrium. This is illustrated by generally small differences between δ13C and δ18O values in calcite samples from 16 and 30 m water depth (Fig. 2b, c). However, CaCO3 dissolution in the hypolimnion of Lake Kierskie reported by Apolinarska et al. (2020) may play a role in slight, but systematic 13C and 18O depletion in calcite collected from the deeper sediment trap. Such depletion is explained by selective dissolution of smaller calcite crystals as they settle to the lake bottom (Ramisch et al. 1999), and consequent dominance of larger ones in the sediment record. As the larger calcite crystals are more depleted in 13C compared to the smaller ones (Teranes et al. 1999a, b), lower δ13C values are observed in the calcite collected from the deep sediment trap. One could also invoke differences in settling velocities between small and large crystals to explain differences between δ13C and δ18O compositions at different depths. Nevertheless, considering that reported settling velocities of calcite crystals range between 3 and 55 m d−1 (Rossknecht 1980; Teranes et al. 1999a), the one month interval between sediment trap sampling dates in Lake Kierskie would likely reduce any differences in isotope values.
What is the effect on paleolimnological inferences of seasonal variability in drivers of calcite disequilibrium with DIC and water?
Our data show that seasonal variation in the extent of calcite isotopic disequilibrium with ambient conditions can be considerable in Lake Kierskie. For instance, with respect to δ18O values, seasonal variability of disequilibrium is several times greater than the amplitude of yearly differences in δ18O values of water (Fig. 2a, c). This implies that major shifts in the amount of carbonate deposited in particular seasons can affect the annual mean values of bulk calcite δ18O and δ13C, even without noticeable changes in DIC and water isotopic compositions. Major shifts in the timing of calcite deposition are likely to occur on long time scales (Żarczyński et al. 2018). Therefore, we argue that seasonal variation in the magnitude of calcite isotopic disequilibria with ambient conditions can have important implications for interpretation of calcite δ18O and δ13C records from lake sediment cores.
We suggest that the best way to obtain calcite δ18O and δ13C records from varved lake sediment sequences that reliably reflect past DIC and water isotopic composition, is to focus on analysis of laminae that represent a single season. This, however, requires that seasonal laminae be thick enough to be separated during sampling for carbonate isotopic analysis, as for instance, in Lake Mondsee, which has a seasonally resolved record of δ18Ocarb and δ13Ccarb (Kämpf et al. 2019). Winter laminae are the most likely to contain redeposited carbonates (Apolinarska et al. 2020; Roeser et al. 2021), and the isotopic composition of such carbonate is thus not indicative of ambient conditions at the time of lamina formation. Therefore, we advise that winter laminae be excluded from analysis, even if sampling of discrete spring or summer laminae is not feasible.
The thin nature of laminae in many varved sequences precludes obtaining seasonally resolved calcite δ18O and δ13C records. Consequently, most inferences are based on isotopic analysis of bulk carbonate samples that represent several years of deposition (Ralska-Jasiewiczowa et al. 2003; Lauterbach et al. 2011; Słowiński et al. 2017). For such records, we recommend isotopic analysis of bulk calcite samples, complemented by at least semi-quantitative analysis of carbonate content at seasonal resolution, by the means of, for instance, thin section observations or X-ray fluorescence core scanning (Żarczyński et al. 2018; Bonk et al. 2015). We suggest that past fluctuations in DIC and water isotopic composition are not always the main reason for isotopic variations in sediment calcite, especially if the latter occurred concurrently with major shifts in the proportion of carbonates deposited in particular seasons.
Conclusions
Year-round monthly monitoring of the carbon and oxygen stable isotope compositions of the dissolved inorganic carbon and lake water, respectively, and δ18O and δ13C records in calcite trapped at 16 and 30 m water depth in Lake Kierskie, western Poland, enabled identification of persistent isotopic disequilibria between authigenic calcite and ambient conditions. The magnitude and causes of such disequilibria in this highly eutrophic lake, however, differed between seasons.
The largest-magnitude isotopic disequilibria were found in calcite trapped during autumn and winter. Calcium carbonate that precipitates at that time originates exclusively from resuspension and redeposition of previously deposited sediments, and thus δ13C and δ18O values of the calcite are not controlled by ambient conditions at the time of redeposition. Therefore, whenever possible, winter laminae should be excluded from isotope studies of varved sediments.
In spring, rapid precipitation of large calcite crystals led to kinetic isotope effects and preferential incorporation of light 12C and 16O isotopes into CaCO3. As a result, carbonate δ13C and δ18O values were 1.10–1.27‰ and 1.28–1.66‰ lower, respectively, than theoretical calcite formed under equilibrium conditions. In contrast to the findings of other studies where offsets from isotopic equilibria were restricted to spring, the disequilibrium precipitation of CaCO3 in Lake Kierskie continued into summer. Although the causes of the 13C (0.54–1.81‰) and 18O (2.84–2.93‰) depletion in calcite relative to values expected in isotopic equilibrium is difficult to determine, we argue that the isotopic disequilibria observed during summer can be explained by changes in the proportions of major dissolved carbon forms that occur, along with a change in water-column pH. We also suggest that the processes related to calcifying algae, so-called “vital effects,” including differences in cell sizes and rates of photosynthesis among phytoplankton species, and the ability to use HCO3− in conditions of CO2 shortage during periods of intense photosynthesis, can be important. These explanations, however, require further investigation.
Differences in isotope values in calcite trapped at 16 and 30 m in the water column in Lake Kierskie were small and resulted mainly from selective dissolution of the smaller calcite crystals in the water column. The proportional increase in the share of 13C-depleted larger crystals was reflected in decreased δ13C calcite values with greater water depth.
The disequilibria observed between expected and measured C and O isotope values of authigenic calcite in Lake Kierskie can, to some extent, be associated with the high trophic state of the lake, i.e. high phosphorus concentration in water promotes precipitation of large calcite crystals in spring, whereas intense productivity (in response to high trophy) leads to a shift in carbon species and greater dominance of species that can use HCO3−, thereby affecting δ13C and δ18O values of precipitated calcite. This finding is particularly relevant for paleolimnological studies, because it revealed that isotope values in sediment records may be affected by isotopic disequilibria. Given this potential problem, we suggest that care be taken whenever lower δ18O values in deposited calcite are accompanied by other sedimentary evidence for increased lake trophic state.
We also recommend that isotopic analysis of calcite in varved lake sediment sequences be supplemented with seasonally resolved measures of sediment carbonate content. We suggest that past DIC and water isotopic composition may not have been the major controls on sediment calcite isotopic values, if the latter occurred along with major shifts in the proportion of carbonates deposited in particular seasons.
The present study provided new insights into the application of stable isotope analyses of carbonates in paleolimnological studies. It also highlighted the need for further investigations. The data from Lake Kierskie are based on one year of monthly observations, which should be extended to test whether the seasonal disequilibrium precipitation of calcite is persistent and replicable. Because multiple factors influence stable isotope values in calcite, future monitoring studies should include lakes of different characteristics, which will enable better understanding of the processes related to disequilibrium precipitation of carbonate sediments.
References
Apolinarska K, Pleskot K, Pełechata A, Migdałek M, Siepak M, Pełechaty M (2020) The recent deposition of laminated sediments in highly eutrophic Lake Kierskie, western Poland: 1 year pilot study of limnological monitoring and sediment traps. J Paleolimnol 63:283–304
Bluszcz P, Luke A, Ohlendorf C, Zolitschka B (2009) Seasonal dynamics of stable isotopes and element ratios in authigenic calcites during their precipitation and dissolution, Sacrower See (northeastern Germany). J Limnol 68:257–273
Bonk A, Tylmann W, Benjamin A, Enters D, Grosjean M (2015) Modern limnology and varve-formation processes in Lake Żabińskie, northeastern Poland: comprehensive process studies as a key to understand the sediment record. J Limnol 74:358–370
Borowitzka MA (1989) Carbonate calcification in algae—initiation and control. In: Mann S, Webb J, Williams RJP (eds) Biomineralization. VCH Verlagsgesellschaft, Weinheim, pp 116–135
Carlson RE (1977) A trophic state index for lakes. Limnol Oceanogr 22:361–369
Dittrich M, Obst M (2004) Are picoplankton responsible for calcite precipitation in lakes? Ambio 33:559–564
Dittrich M, Kurz P, Wehrli B (2004) The role of autotrophic picocyanobacteria in calcite precipitation in an oligotrophic lake. Geomicrobiol J 21:45–53
Filippi ML, Lambert P, Hunziker J, Kübler B, Bernasconi S (1999) Climatic and anthropogenic influence on the stable isotope record from bulk carbonates and ostracodes in Lake Neuchâ, Switzerland, during the last two millennia. J Paleolimnol 21:19–34
Fronval T, Bo Jensen N, Buchardt B (1995) Oxygen isotope disequilibrium precipitation of calcite in Lake Arresø, Denmark. Geology 23:463–466
Goslar T, Bałaga K, Arnold M, Tisnerat N, Starnawska E, Kuźniarski M, Chróst L, Walanus A, Wiȩckowski K (1999) Climate-related variations in the composition of the Late Glacial and early Holocene sediments of Lake Perespilno (eastern Poland). Quaternary Sci Rev 18:899–911
Grzonka B, Dondajewska R, Fabiś M, Grześkowiak A, Żak J, Domek P (2016) Sprawozdanie z monitoringu jakości wód Jeziora Kierskiego oraz jego dopływów i odpływu. Environment and Water Consulting, Poznań, p 49
Jańczak J, Sziwa R (1995) Charakterystyka morfometryczno-hydrologiczna jezior i zbiorników wodnych Poznania. In: Kaniecki A, Rotnicka J (eds) Wody powierzchniowe Poznania Problemy wodne obszarów miejskich. Wydawnictwo Sorus, Poznań, pp 344–355
Jonsson CE, Andersson S, Rosqvist GC, Leng MJ (2010) Reconstructing past atmospheric circulation changes using oxygen isotopes in lake sediments from Sweden. Clim Past 6:49–62
Kämpf L, Plessen B, Lauterbach S, Nantke C, Meyer H, Chapligin B, Brauer A (2019) Stable oxygen and carbon isotopes of carbonates in lake sediments as a paleoflood proxy. Geology 48:3–7
Karami F, Balci N, Guven B (2019) A modeling approach for calcium carbonate precipitation in a hypersaline environment: a case study from a shallow, alkaline lake. Ecol Complex 39:100774
Kim S-T, O’Neil JR (1997) Equilibrium and nonequilibrium oxygen isotope effects in synthetic carbonates. Geochim Cosmochim Acta 61:3461–3475
Kosamu IBM, Obst M (2009) The influence of picocyanobacterial photosynthesis on calcite precipitation. Int J Environ Sci Te 6:557–562
Kowalczak P, Sosiński W (1995) Hydrogeologia zlewni Jeziora Kierskiego a problem ochrony wód. In: Kaniecki A, Rotnicka J (eds) Wody powierzchniowe Poznania Problemy wodne obszarów miejskich. Wydawnictwo Sorus, Poznań
Kozarski S (1995) Deglacjacja północno-zachodniej Polski: warunki środowiska i transformacja geosystemu (~ 20–10 ka BP). Continuo, Wrocław
Lauterbach S, Brauer A, Andersen N, Danielopol DL, Dulski P, Hüls M, Milecka K, Namiotko T, Plessen B, Grafenstein UV, Participants D (2011) Multi-proxy evidence for early to mid-Holocene environmental and climatic changes in northeastern Poland. Boreas 40:57–72
Leng MJ, Marshall JD (2004) Palaeoclimate interpretation of stable isotope data from lake sediment archives. Quaternary Sci Rev 23:811–831
Lenz S, Uta G, Juergen G, Michael S, Maren L, Uta R (2018) Calcite production by the calcifying green alga Phacotus lenticularis. J Limnol 77:1792
Mangili C, Brauer A, Plessen B, Moscariello A (2007) Centennial-scale oscillations in oxygen and carbon isotopes of endogenic calcite from a 15,500 varve year record of the Piànico interglacial. Quaternary Sci Rev 26:1725–1735
McConnaughey T (1989) 13C and 18O isotopic disequilibrium in biological carbonates: I. Patterns. Geochim Cosmochim Acta 53:151–162
McKenzie JA (1985) Carbon isotopes and productivity in the lacustrine and marine environment. In: Stumm W (ed) Chemical processes in lakes. Wiley, New York, pp 99–118
McKenzie JA, Hollander DJ (1993) Oxygen isotope record in recent carbonate sediments from Lake Greifen, Switzerland (1750–1986): application of continental isotopic indicator for evaluation of changes in climate and atmospheric circulation patterns. In: Swart PK, Lohmann KC, McKenzie JA, Savin S (eds) Climate change in continental isotopic records, AGU geophysical monography. American Geophysical Union, New York, pp 101–112
Park RA, Clough JS, Wellman MC (2008) AQUATOX: Modelling environmental fate and ecological effects in aquatic ecosystems. Ecol Model 213:1–15
Ralska-Jasiewiczowa M, Goslar T, Różański K, Wacnik A, Czernik J, Chróst L (2003) Very fast environmental changes at the Pleistocene/Holocene boundary, recorded in laminated sediments of Lake Gościaż, Poland. Palaeogeogr Palaeoecol 193:225–247
Ramisch F, Dittrich M, Mattenberger C, Wehrli B, Wüest A (1999) Calcite dissolution in two deep eutrophic lakes. Geochim Cosmochim Acta 63:3349–3356
Roeser P, Dräger N, Brykała D, Ott F, Pinkerneil S, Gierszewski P, Lindemann C, Plessen B, Brademann B, Kaszubski M, Fojutowski M, Schwab MJ, Słowiński M, Błaszkiewicz M, Brauer A (2021) Advances in understanding calcite varve formation: new insights from a dual lake monitoring approach in the southern Baltic lowlands. Boreas. https://doi.org/10.1111/bor.12506
Romanek CS, Grossman EL, Morse JW (1992) Carbon isotope fractionation in synthetic aragonite and calcite: effects of temperature and precipitation rate. Geochim Cosmochim Acta 56:419–430
Rossknecht H (1980) Phosphatelimination durch autochthone Calcitfallung im Boddensee–Obersee. Arch Hydrobiol 88:328–344
Schelske CL, Hodell DA (1991) Recent changes in productivity and climate of Lake Ontario detected by isotopic analysis of sediments. Limnol Oceanogr 36:961–975
Słowiński M, Zawiska I, Ott F, Noryśkiewicz AM, Plessen B, Apolinarska K, Rzodkiewicz M, Michczyńska DJ, Wulf S, Skubała P, Kordowski J, Błaszkiewicz M, Brauer A (2017) Differential proxy responses to late Allerød and early Younger Dryas climatic change recorded in varved sediments of the Trzechowskie palaeolake in Northern Poland. Quaternary Sci Rev 158:94–106
Stabel HH (1986) Calcite precipitation in Lake Constance: chemical equilibrium, sedimentation, and nucleation by algae. Limnol Oceanogr 31:1081–1094
Szyga-Pluta K, Grześkowiak K (2016) Warunki pluwialne w Poznaniu w latach 1981–2015. Bad Fizjogr Ser A 67:239–256
Teranes JL, McKenzie JA, Bernasconi SM, Lotter AF, Sturm M (1999a) A study of oxygen isotopic fractionation during bio-induced calcite precipitation in eutrophic Baldeggersee, Switzerland. Geochim Cosmochim Acta 63:1981–1989
Teranes JL, McKenzie JA, Lotter AF, Sturm M (1999b) Stable isotope response to lake eutrophication: calibration of a high-resolution lacustrine sequence from Baldeggersee, Switzerland. Limnol Oceanogr 44:320–333
Usdowski E, Hoefs J (1993) Oxygen isotope exchange between carbonic acid, bicarbonate, carbonate, and water: a re-examination of the data of McCrea (1950) and an expression for the overall partitioning of oxygen isotopes between the carbonate species and water. Geochim Cosmochim Acta 57:3815–3818
Usdowski E, Michaelis J, Bottcher ME, Hoefs J (1991) Factors for the oxygen isotope equilibrium fractionation between aqueous and gaseous CO2, carbonic acid, bicarbonate, carbonate, and water (19 °C). Z Phys Chem 170:237–249
Ustrzycka A, Piotrowska N, Bonk A, Filipiak J, Tylmann W (2018) Isotopic fingerprints of the Lake Żabińskie (NE Poland) hydrological system on contemporary carbonates precipitated in the lake. Isot Environ Health Stud 54:225–243
von Grafenstein U, Eicher U, Erlenkeuser H, Ruch P, Schwander J, Ammann B (2000) Isotope signature of the Younger Dryas and two minor oscillations at Gerzensee (Switzerland): palaeoclimatic and palaeolimnologic interpretation based on bulk and biogenic carbonates. Palaeogeogr Palaeoecol 159:215–229
Wetzel RG (2001) Limnology. Lake and river ecosystems. Academic Press, San Diego
Woś A (1999) Klimat Polski. Wydawnictwo Naukowe PWN, Warszawa
Żarczyński M, Tylmann W, Goslar T (2018) Multiple varve chronologies for the last 2000 years from the sediments of Lake Żabińskie (northeastern Poland)—comparison of strategies for varve counting and uncertainty estimations. Quaternary Geochronol 47:107–119
Acknowledgements
We are grateful to the Kaskada Hotel and Recreation Resort owner and employees for assistance and rental of equipment. Two anonymous reviewers are acknowledged for comments on the previous version of the manuscript. The study was financed with funds from the Institute of Geology, Faculty of Geographical and Geological Sciences, and Department of Hydrobiology, Faculty of Biology, Adam Mickiewicz University, Poznań.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Apolinarska, K., Pleskot, K., Pełechata, A. et al. Seasonal deposition of authigenic calcite out of isotopic equilibrium with DIC and water, and implications for paleolimnological studies. J Paleolimnol 66, 41–53 (2021). https://doi.org/10.1007/s10933-021-00184-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10933-021-00184-y