
Abstract
The microaerobic synthesis of 3-hydroxybutyric acid by the Escherichia coli strain BOX3.1 ∆4 PL-atoB PL-tesB (MG1655 lacIQ, ∆ackA-pta, ∆poxB, ∆ldhA, ∆adhE, ∆fadE, PL-SDphi10-atoB, Ptrc-ideal-4-SDphi10-fadB, PL-SDphi10-tesB), which was previously directly engineered for the biosynthesis of the target compound from glucose through the reversed fatty acid β-oxidation pathway, was studied. A target product yield of 0.12 mol/mol was achieved. Inactivation of the nonspecific YciA thioesterase gene in the strain led to an increase in the yield of 3-hydroxybutyric acid to 0.15 mol/mol. For the optimization of biosynthesis of target product the strain MG∆4 PL-tesB (MG1655 ∆ackA-pta, ∆poxB, ∆ldhA, ∆adhE, PL-SDphi10-tesB) was engineered, and the genes encoding key enzymes of fatty acid β-oxidation were overexpressed in the strain from the plasmid pMW118m-atoB-fadB. The level of microaerobic synthesis of 3-hydroxybutyric acid by the strain MG∆4 PL-tesB (pMW118m-atoB-fadB) achieved in primary evaluation conditions reached 0.35 mol/mol. Inactivation in the strain of the gene of nonspecific thioesterase YciA led to only minor decrease in acetate byproduction. Further inactivation in the strain of gene encoding nonspecific thioesterase YdiI had virtually no effect on the level of synthesis of side products. Cultivation of the constructed strain MG∆4 PL-tesB ∆yciA (pMW118m-atoB-fadB) in bioreactor under the controlled conditions ensured achievement of a yield of 3‑hydroxybutyric acid amounting to 0.75 mol/mol.
Similar content being viewed by others

INTRODUCTION
The useful properties of many high value-added industrially important and physiologically active chemicals are caused by the presence of chiral centers in the corresponding molecules that determine the existence of optical isomers of chemically identical compounds. The production of optically pure enantiomers via stereoselective organic synthesis is often a challenging task that requires the use of expensive catalysts, harsh physical conditions, and aggressive solvents. The use of convenient chiral synthons as a starting material greatly simplifies the efficient synthesis of the target product.
In particular, enantiomers of 3-hydroxybutyric acid (3-HBA) can serve as a convenient starting material for the production of such important optically active compounds as drugs, pheromones, cosmetics, fragrances, and agrochemicals [1–4]. Normally, 3-HBA stereoisomers are not secreted by known microorganisms during the utilization of sugars. Nevertheless, their microbiological synthesis from cheap renewable raw materials can serve as an economically reasonable alternative to the energy- and resource-consuming [5] chemical method to obtain the corresponding compounds with upon the use of directly engineered, highly efficient microbial producing strains. Indeed, in recent years the feasibility of rational engineering of a such biotechnologically relevant bacterium as Escherichia coli for biosynthesis of both (R)- [6, 7] and (S)‑3-HBA [7, 8] from glucose has been demonstrated.
The corresponding biochemical pathway leading to the formation of the target compound consists of a sequential reactions that include the initial condensation of two precursor molecules, acetyl-CoA, into acetoacetyl-CoA, the further reduction of acetoacetyl-CoA into 3-hydroxybutyryl-CoA, and the final hydrolysis of the thioester bond of the latter with the formation of 3-HBA. These reactions are catalyzed by acetyl-CoA C-acetyltransferase (EC 2.3.1.9), NADPH-dependent (R)-3-hydroxyacyl-CoA dehydrogenase (EC 1.1.1.36) or NADH-dependent (S)-3-hydroxyacyl-CoA dehydrogenase (EC 1.1.1.35), and thioesterase (EC 3.1.2.20). Thus, the main reactions for the formation of the key chiral intermediate, 3-hydroxybutyryl-CoA, can be considered analogous to those of 3‑hydroxybutyrate biosynthesis, represented mainly in Ralstonia species, in the case of the (R)-stereoisomer, and of clostridial 1-butanol biosynthesis in the case of the (S)-stereoisomer. This rationalizes the choice of certain heterologous enzymes used to achieve the synthesis of a particular 3-HBA stereoisomer in recombinant strains, (R)-3-hydroxyacyl-CoA dehydrogenase PhaB from Ralstonia eutropha [6, 7] and (S)-3-hydroxyacyl-CoA dehydrogenase Hbd from Clostridium acetobutylicum [7, 8]. At the same time, the clostridial reactions of 1-butanol biosynthesis, from the biochemical point of view, are similar to the reversed reactions of the fatty acid β-oxidation (FABO) cycle, a biochemical pathway present in many microorganisms. The functional reversal of FABO was experimentally shown in a number of studies demonstrating the production from glucose or glycerol of various chain-length aliphatic carboxylic acids and alcohols, through this pathway, by recombinant E. coli strains [9–12]. Consequently, (S)-3-HBA can be synthesized by E. coli from glucose resulting from a partial one-turn reversal of FABO upon overexpression of a suitable thioesterase without the necessity of using any foreign genes.
Indeed, E. coli MG1655-derived strain BOX3.1 ∆4 PL-atoB PL-tesB, which is capable of enantioselective (S)-3-HBA biosynthesis from glucose through the reversed FABO, was previously successfully engineered [13]. In this strain, native AtoC and FadR-dependent regulatory regions of the atoB and fadB genes, which encode acetyl-CoA C-acetyltransferase and bifunctional (S)-3-hydroxyacyl-CoA dehydrogenase/enoyl-CoA hydratase (EC 1.1.1.35/EC 4.2.1.17), were replaced by artificial regulatory elements, comprising the effecient ribosome binding site of the φ10 gene from the T7 phage, as well as lambda phage PL or Ptrc-ideal-4 [14] promoters, respectively. In addition, the main pathways of mixed acid fermentation, which compete with reversed FABO for reduced equivalents and key precursor metabolites, pyruvate, and acetyl-CoA, were inactivated in the strain by deletions of ackA-pta, poxB, ldhA, and adhE genes. Multiple turns of the reversed FABO cycle in the strain were prevented by the deletion of the fadE gene, which encodes acyl-CoA dehydrogenase (EC 1.3.99.3), and the formation of 3-HBA from the direct CoA-precursor was ensured as a result of overexpression of the thioesterase II gene, tesB.
Nevertheless, despite the ability of the BOX3.1 ∆4 PL-atoB PL-tesB strain to stereoselectively synthesize (S)-3-HBA with a relatively high yield, which reaches 66% of the theoretical maximum under certain conditions, the observed secretion of acetic acid by the strain as the main by-product of glucose utilization suggested that further metabolic engineering of the strain or its reconstructed analogs would be beneficial for the synthesis of the target substance.
The goal of this work is to optimize the biosynthesis of (S)-3-hydroxybutyric acid from glucose by recombinant Escherichia coli strains through the reversed fatty acid β-oxidation pathway.
MATERIALS AND METHODS
Reagents. Restriction enzymes, Taq DNA polymerase, T4 DNA ligase (Thermo Scientific, Lithuania), Phusion high-fidelity DNA polymerase (Thermo Scientific, Finland), a Gibson assembly HiFi Builder kit and Q5 high-fidelity DNA polymerase (New England Biolabs, United States), and a QuikChange II Site-Directed Mutagenesis Kit (Agilent Technologies, United States) were used in the work. The polymerase chain reaction (PCR) products were purified via agarose gel electrophoresis and isolated with a QIAquick Gel Extraction Kit (Qiagen, United States). The oligonucleotides (Evrogen, Russia) are presented in Table 1. Components of the culture media, salts, and other reagents were produced by Panreac (Spain) and Sigma (United States).
Bacterial strains, plasmids, and media. The E. coli strain K-12 MG1655 (VKPM B-6195), the previously constructed E. coli strain MGΔ4 [15] (a derivative of the MG1655 strain, which lacks mixed acid fermentation pathways), and the previously constructed E. coli strain BOX3.1 ∆4 PL-atoB PL-tesB [13] with altered regulation of the expression of genes encoding the enzymes of aerobic fatty acid β-oxidation and thioesterase II (which also lacks mixed acid fermentation pathways) were used as the chassis for the construction of all strains obtained in this work. Table 2 presents the bacterial strains and plasmids used in the work. The reach LB, SOB, SOC media and minimal M9 medium [16] were used to cultivate the bacteria, if necessary, they were supplemented with ampicillin (100 μg/mL) or chloramphenicol (30 μg/mL).
Strain construction. The genes yciA and ydiI were inactivated in E. coli chromosome as described earlier [17].
Linear DNA fragments for the inactivation of target genes containing a chloramphenicol resistance marker (the cat gene) were obtained via PCR with primer pairs P1 and P2, P3 and P4, and the pMW118-(λattL-Cm-λattR) plasmid [18] as a template. The resulting DNA fragments were individually integrated into the chromosome of the E. coli MG1655 strain carrying the pKD46 helper plasmid. The correspondence between the assumed and experimentally obtained structures of the chromosomes of the selected strains with individually inactivated genes yciA and ydiI was confirmed via PCR analysis with pairs of locus-specific primers P5 and P6, P7, and P8.
The strains BOX3.1 Δ4 PL-atoB PL-tesB ΔyciA, MGΔ4 PL-tesB, MGΔ4 PL-tesB ΔyciA, and MGΔ4 PL-tesB ΔyciA ΔydiI were obtained via the introduction of individual modifications into the chromosomes of the target strains by P1-dependent transductions [16]. In the case of MGΔ4 PL-tesB strain, a previously obtained preparation of transducing phage carrying the corresponding genetic modification was used [13]. The marker flanked by the att sites of the lambda phage was removed from the chromosomes of the target strains using the pMWts-Int/Xis plasmid as described earlier [19]. The strains were transformed with the plasmids according to the standard procedure.
Plasmid construction. The pMW118m plasmid, a derivative of the low copy number pMW118 vector containing the genetic construct NotI-Ptrc-ideal-2-SDlacZ-SalI-XhoI-KpnI-TrrnB-SphI, was obtained via Gibson assembly. The linear DNA fragments with the required flank homology regions were obtained via PCR with pairs of specific primers. Linearized pMW118mod vector was obtained via PCR with the pMW118 plasmid used as a template and primers P9 and P10 complementary to its target regions. The DNA fragment containing the 5'-end homology to the flanking region of the linearized pMW118mod vector, NotI recognition site, Ptrc-idea-l-2 promoter (Olac-ideal-Ptrc/Olac-ideal) [14], SD of the lacZ gene, recognition sites SalI, XhoI, KpnI, and short 3'-end region of homology with the 5'-end region of the terminator of rrnB operon was obtained via PCR with primers P11 and P12, which have regions of mutual complementarity. The DNA fragment containing the recognition sites SalI, XhoI, KpnI, the terminator of E. coli rrnB operon, the SphI recognition site, and a sequence homologous to the flank region of the linearized pMW118mod vector was obtained via PCR with primers P13 and P14 and E. coli chromosomal DNA as a template.
The PCR fragments were purified and added to the reaction mixture of a NEB HiFi Builder kit containing exonuclease, high-fidelity DNA polymerase, and DNA ligase. To select the target, covalently closed, plasmid DNA, E. coli DH5alpha cells were transformed with the resulting reaction mixture, and the transformants were subsequently selected on the ampicillin-containing medium. The correspondence between the planned and experimentally obtained structures of the artificial genetic element NotI-Ptrc-ideal-2-SDlacZ-SalI-XhoI-KpnI-TrrnB-SphI in a series of plasmids isolated from the selected ApR transformants was confirmed via sequencing with primers P15 and P16.
The atoB and fadB genes were initially cloned in the pUC18 vector. For this purpose, the coding regions of the corresponding genes were amplified via PCR with primer pairs P17 and P18 in the case of the atoB gene and also with P19 and P20 and then P19 and P21 in the case of the fadB gene using chromosomal DNA of the E. coli MG1655 strain as a template. The primer design assumed that, as a result of such amplification, the coding regions of the genes would additionally contain recognition sites of BglII and BamHI at the flanks located immediately after the start and immediately before the stop codon, respectively, as well as the recognition sites of SalI at the 5'-ends and the recognition sites of XhoI and KpnI at the 3'-ends of the amplicons. The resulting DNA fragments were subsequently cloned in the pUC18 vector at SalI and KpnI sites and sequenced. The native recognition sites of BglII in the coding region of the fadB gene were further eliminated as a result of two-step, site-directed mutagenesis with the QuikChange II Site-Directed Mutagenesis Kit (Agilent Technologies, United States). For this, the plasmid with the cloned fadB gene was amplified via PCR with the locus-specific primers P22 and P23, digested with the DpnI, and subjected to a second round of PCR with primers P24 and P25. After the transformation of E. coli DH5alpha cells with the resulting reaction mixture, the subsequent selection of ApR clones, and the isolation of circular plasmid DNA from them, the latter were sequenced, and a plasmid containing the correct sequence of the fadB gene was selected. The atoB and fadB genes were subsequently recloned from the corresponding pUC-derived plasmids into the pMW118m-atoB-fadB plasmid under the control of the promoter Ptrc-ideal-2 and SDlacZ using AatII, SalI, and XhoI restriction sites.
Strain cultivation. Recombinant strains were grown overnight in M9 medium containing 2 g/L glucose at 37°C. For primary microaerobic cultivation, 5 mL of the resulting overnight cultures were diluted ten times by the addition of 45 mL of M9 medium containing 10 g/L glucose and 10 g/L yeast extract. The resulting cultures were incubated in 750-mL flasks closed with nonventilated caps on a rotary shaker at 250 rpm for 8 h at 37°C. The oxygen saturation of the medium was assessed in control flasks with the corresponding cultures upon incubation in the presence of resazurin. To induce the expression of genes under the control of LacI-dependent promoters Ptrc-ideal-2 and Ptrc-ideal-4, isopropyl-β-D-thiogalactoside (IPTG) was added to the culture medium to a final concentration of 1.0 mM 3 h after the start of incubation. When strains containing pMW118-derived plasmids were grown, 100 μg/mL ampicillin (Sintez, Russia) was added to the media.
Controlled microaerobic cultivation was carried out in a bioreactor as described earlier [13]. A combined medium containing (g/L): tryptone, 10; yeast extract, 5.0; Na2HPO4⋅12H2O, 15.1; KH2PO4, 3.0; NH4Cl, 1.0; NaCl, 0.5; CaCl2⋅2H2O, 0.015; MgSO4⋅7H2O, 0.5 and 5 mg/L thiamine was used. The aerobic phase of biomass accumulation was carried out for 7 h at 37°C and 850 rpm with an air flow of 0.5 L/min. The expression of genes under the control of LacI-dependent promoters was induced after 3 h of incubation. The microaerobic productive phase was initiated by the addition to the medium of glucose to a concentration of 25 g/L, a change in stirrer speed to 250 rpm, and a reduction of air flow to 0.1 L/min.
The cell suspensions collected for analysis were centrifuged at 10 000 g for 10 min. In the obtained supernatants, the concentrations of secreted metabolites and residual glucose were determined. All experiments were performed at least in triplicate.
Analytical methods. The concentrations of organic acids in culture liquids freed from the biomass by centrifugation were determined via high-performance liquid chromatography (HPLC) with a Waters HPLC system (United States). The Rezex ROA-Organic Acid H+ (8%) ion-exclusion column (Phenomenex, United States) with detection at a wavelength of 210 nm was used. An aqueous solution of sulfuric acid (2.5 mM) with a flow rate of 0.5 mL/min was used as the mobile phase. To measure the glucose concentration, the system was equipped with a Waters 2414 refractive index detector and the Spherisorb-NH2 column (Waters, United States). The mobile phase was an acetonitrile/water mixture (volume ratio 75/25) at a flow rate of 1.0 mL/min.
The identification and quantification of 3-HBA in the culture liquids was performed by chromatography-mass spectrometry according to previously developed procedures [13]. An Agilent 6890N gas chromatograph equipped with a 7683B autosampler and an Agilent 5975 mass selective detector and a DB-5MS capillary column (Agilent, United States) with a length of 30 m, an inner diameter of 0.25 mm, and a film thickness of 0.25 μm was used. Helium with a flow rate of 1.0 mL/min was used as the carrier gas. A 1-μL sample was injected into the evaporator in a split injection mode of 1 : 50. The evaporator temperature was 230°C. The following temperature program of the column oven was used: initial isotherm of 1 min at 60°C; subsequent gradient to 120°C at a rate of 3°C/min and then to 220°C at a rate of 20°C/min; final isotherm of 1 min at 220°C. Electron ionization (electron impact, 70 eV) was used in mass-selective detector mode with control of specified ions (117.10 m/z, 131.10 m/z, 147.10 m/z, 191.10 m/z). The ion-source temperature was set at 230°C. Sample preparation included extraction of the analyte from the culture liquid, evaporation of the extracts in a stream of nitrogen, and derivatization to obtain trimethylsilyl derivatives. The obtained data were processed with the Agilent MSD ChemStation Software. Library data (retention index and mass spectrum) were used to identify the target analyte. Quantitative analysis was performed with the appropriate standard.
The enantiomeric form of 3-HBA synthesized by the strains was determined via chiral gas chromatography with a flame ionization detector with an Agilent CP-Chirasil-Dex CB column (25 m length, 0.25 mm inner diameter, 0.25 µm film thickness). Preliminary derivatization of the analyte was carried out to obtain 3-acetoxybutyric acid methyl ester. Acetylated derivatives of commercially available acetylated methyl esters of (R)- and (S)-3-HBA (Sigma-Aldrich, United States) were used as standards. A Shimadzu GC-2010 gas chromatograph (Japan) equipped with an AOC-5000 autosampler was used. Helium at a flow rate of 1.0 mL/min was used as the carrier gas. The split injection mode was used (1 : 20, sample volume 1 μL). The injector temperature was 200°C, the column oven temperature was 110°C, and the detector temperature was 250°C. The data were processed with the GCsolution software (Shimadzu, Japan).
RESULTS AND DISCUSSION
The biosynthesis of 3-HBA from two molecules of acetyl-CoA through the reversed FABO requires the consumption of one reduced NADH equivalent. Glycolytic utilization of glucose leads to the formation of two pyruvate molecules and is accompanied by the generation of two NADH. The anaerobic conversion of pyruvate to acetyl-CoA by pyruvate formate lyase (EC 2.3.1.54) do not lead to the formation of NADH, while the action of pyruvate dehydrogenase (EC 1.2.4.1/2.3.1.12/1.8.1.4) under aeration is coupled with the formation of an additional NADH molecule per each formed acetyl-CoA. Consequently, in E. coli strains lacking the fermentation ability, the level of anaerobic NADH formation will exceed that required for efficient 3-HBA biosynthesis. In addition, it is known that E. coli cells with inactivated pta, ldhA, and adhE genes are incapable of anaerobic growth [20]. As a result, glycolysis in strains constructed to produce 3‑HBA will be suppressed during anaerobiosis, not only by the increased intracellular NADH pool but also by the excessive generation of ATP [21]. Conversely, the intracellular availability of NADH, which is required for the 3-HBA biosynthesis, would be limited in the presence of oxygen by the action of the respiratory electron-transport chain. Thus, neither completely anaerobic nor aerobic conditions can be considered optimal for 3-HBA biosynthesis from glucose via reversed FABO by the previously constructed BOX3.1 ∆4 PL-atoB PL-tesB strain.
Indeed, during the primary characterization, the BOX3.1 ∆4 PL-atoB PL-tesB strain did not synthesize significant amounts of 3-HBA from glucose aerobically, and the production of the target substance during anaerobiosis was rather low [13]. In this work, the biosynthetic potential of the strain was initially assessed under microaerobic cultivation. Under the respective conditions, the strain, depending on the induction of the fadB gene expression under the control of the LacI-dependent promoter, synthesized from glucose 2.9–4.9 mM 3-HBA with a molar yield of 0.07–0.12 mol/mol (Table 3). At the same time, the strain secreted the most of the consumed glucose in the form of pyruvic and acetic acids. The formation of 3-HBA by the strain observed in the absence of induced fadB expression, could be explained by a certain leakage of the Ptrc-ideal-4 promoter [14], while the secretion of pyruvic and acetic acids, which was littly dependent of the induction of 3-HBA synthesis, could be caused by a number of reasons. First of all, the lack of available intracellular NADH in the strain could lead to an increased secretion of these precursors in 3‑HBA synthesis. However, the accumulation of significant amounts of succinate and malate by the strain indicated that the cultivation conditions were sufficiently anoxic for the formation of the corresponding dicarboxylates as a result of the functioning of the reductive branch of the tricarboxylic acid cycle, which is usually active under anaerobic conditions. An increased level of acetate synthesis by the strain, with relatively low 3-HBA formation, could be a consequence of both the insufficient activity of FABO enzymes overexpressed in the strain and the result of the activity of nonspecific thioesterases other than the directly overexpressed thioesterase II (TesB). Indeed, E. coli cells naturally possess at least eight thioesterases capable of hydrolyzing the thioester bond of CoA-derivatives to form the corresponding carboxylates. Among them, YciA, YbgC, and YdiI thioesterases demonstrate a fairly broad substrate specificity for acyl derivatives of coenzyme A [10, 22, 23], while YciA shows maximum activity towards acetyl-CoA [10].
Thus, yciA gene was firstly inactivated to evaluate the efficiency of potential approaches to improve the 3-HBA synthesis in the BOX3.1 ∆4 PL-atoB PL-tesB strain. As a result of this modification, the 3-HBA yield and the level of the synthesis of the target compound by the derivative strain BOX3.1 Δ4 PL-atoB PL-tesB ΔyciA increased by almost 20%, while the yield and the level of acetate synthesis decreased by the same value (Table 3). However, the level of pyruvate secretion by the strain was unchanged, and acetate remained the main product secreted by the strain during glucose utilization. This fact indicated that namely the intracellular level of proteins catalyzing the FABO reactions apparently limited the production of 3-HBA by the constructed strain. Although the PL promoter of the lambda phage, which controls the expression of the atoB gene of acetyl-CoA C-acetyltransferase in the strain, is one of the strongest for E. coli and the Ptrc-ideal-4 promoter located before the gene of 3-hydroxyacyl-CoA dehydrogenase (fadB) is slightly inferior to it in strength [14], the chromosomal expression of single copies of the corresponding genes obviously could not ensure the enforced reversal of FABO in the strain and promote increased synthesis of the target compound.
To verify this hypothesis, atoB and fadB genes were cloned in the expression vector pMW118m-atoB-fadB under the control of the Ptrc-ideal-2 promoter and were initially expressed in a specially constructed MG ∆4 PL-tesB strain. Besides expression level of the corresponding genes encoding FABO enzymes, the respective MG ∆4 PL-tesB (pMW118m-atoB-fadB) strain was nearly isogenic to the BOX3.1 ∆4 PL-atoB PL-tesB strain. The differences consisted in the absence of the lacIQ mutation in the MG ∆4 strain and the presence of the intact fadE gene. However, without the fatty acids in the medium, the expression of fad-regulon genes, including fadE, is repressed in E. coli by the FadR transcriptional regulator, and the activity of the corresponding enzymes is absent in the cell [24]. Also, the leakage of the Ptrc-ideal-4 promoter observed in LacIQ strain BOX3.1 ∆4 PL-atoB PL-tesB did not allow to expect strict repression of similar in characteristics Ptrc-ideal-2 promoter [14] upon its location within the plasmid.
Indeed, during microaerobic glucose utilization, the strain MG ∆4 PL-tesB (pMW118m-atoB-fadB) secreted similar amounts of metabolites regardless of the presence of IPTG in the medium (Table 3). At the same time, the amount of 3-HBA synthesized by the strain MG ∆4 PL-tesB (pMW118m-atoB-fadB) and the yield of target compound increased by almost three times, up to 14.9 mM and 0.35 mol/mol, as compared with the corresponding parameters demonstrated by the BOX3.1 ∆4 PL-atoB PL-tesB strain upon induction (4.9 mM and 0.12 mol/mol). The 3-HBA synthesis by the strain rose primarily at the expense of a sharp decrease in pyruvate secretion, while the level of acetate formation decreased slightly. Thus, the increase in 3-HBA formation by the strain MG ∆4 PL-tesB (pMW118m-atoB-fadB) was the result of an intensification of the carbon flux through acetyl-CoA and the subsequent reactions of reversed FABO consuming the respective precursor. This was caused by the increased synthesis of the AtoB and FadB in the cell, which was achieved resulting from the plasmid expression of the respective genes. Nevertheless, the yield of 3-HBA synthesized by the strain MG ∆4 PL-tesB (pMW118m-atoB-fadB) was far from the theoretical maximum, 1 mol/mol, and acetate secretion remained notable.
Deletion of the gene encoding the main nonspecific thioesterase YciA, however, did not lead to a pronounced decrease in acetate secretion by the corresponding derivative strain MGΔ4 PL-tesB ΔyciA (pMW118m-atoB-fadB) (Table 3). It can be assumed that other nonspecific thioesterases were responsible for the hydrolysis of the acetyl-CoA thioester bond with the formation of acetate in the MGΔ4 PL-tesB ΔyciA (pMW118m-atoB-fadB). YdiI, first of all, can be considered such a thioesterase. It has a noticeably lower activity (0.036 μg/mg/min) towards acetyl-CoA than YciA (0.222 μg/mg/min), but comparable with that of TesB overexpressed in the strain (0.030 μg/mg/min) [10]. Therefore, the ydiI gene was inactivated in the strain MGΔ4 PL-tesB ΔyciA (pMW118m-atoB-fadB). During microaerobic glucose utilization, the resulting strain MGΔ4 PL-tesB ΔyciA ΔydiI (pMW118m-atoB-fadB) synthesized, regardless of the presence of IPTG in the medium, amounts of 3-HBA and acetate similar to those of the parent strain MGΔ4 PL-tesB ΔyciA (pMW118m-atoB-fadB) (Table 3). Thus, the acetate secretion by the strain MGΔ4 PL-tesB ΔyciA ΔydiI (pMW118m-atoB-fadB) was apparently caused by the side activity of overexpressed thioesterase II.
It is known that anaerobic utilization of fatty acids by E. coli cells via the FABO is impossible without the presence of an external electron acceptor in the medium [25]. Thus, despite the absence of direct evidence, it can be assumed that the activity of FABO enzymes, which require the participation of NADH/NAD+ cofactors, may, in some extent, be repressed by the high intracellular NADH pool. For this reason, maintenance of the optimal intracellular redox balance is obviously the key requirement for the efficient synthesis of target products through the reversed FABO. Upon the microaerobic cultivation in flasks, saturation of the medium with oxygen cannot be maintained at a constant level and, as noted above, the final conditions achieved during the incubation of the strains were nearly anaerobic. It was previously shown that the optimal conditions for 3-HBA biosynthesis from glucose by the BOX3.1 ∆4 PL-atoB PL-tesB strain were achieved upon conducting the productive phase in a bioreactor with maintenance of constant oxygen saturation of the medium at a relatively low level [13]; therefore, the biosynthetic characteristics of the strain MGΔ4 PL-tesB ΔyciA (pMW118m-atoB-fadB) were evaluated during the growth and 3-HBA biosynthesis in the bioreactor as described in Materials and Methods section.
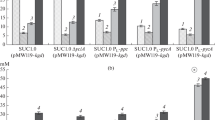
In the bioreactor, the strain MGΔ4 PL-tesB ΔyciA (pMW118m-atoB-fadB) synthesized to the end of incubation ~104 mM 3-HBA from glucose with a yield of 0.75 mol/mol (Fig. 1) and an enantiomeric excess of (S)-stereoisomer reaching > 99.5%. The strain secreted acetate to a final level of ~51.8 mM with a yield of ~0.38 mol/mol, while accumulating a relatively low pyruvate. Under similar conditions, the BOX3.1 ∆4 PL-atoB PL-tesB synthesized 3-HBA from glucose with a yield of 0.66 mol/mol and acetate by-production markedly higher than that of the strain MGΔ4 PL-tesB ΔyciA (pMW118m-atoB-fadB) [13].

Concentrations of metabolites (mM) secreted by the strain MGΔ4 PL-tesB ΔyciA (pMW118m-atoB-fadB) during microaerobic utilization of glucose in a bioreactor in the presence of IPTG: pyruvate (1), acetate (2), and 3-HBA (3).
CONCLUSIONS
Thus, the production of 3-HBA from glucose by directly engineered E. coli was optimized resulting from the reconstruction of the producing strain in combination with the biosynthesis of the target substance under the conditions favoring its efficient formation.
At the same time, the results of the study indicated that the biosynthetic characteristics of the obtained 3-HBA producing strain can be further improved by precise coordination in the cell of the activities of key enzymes responsible for the conversion of acetyl-CoA into the target product. Such coordination can be ensured as a result of fine tuning of the expression level of the corresponding genes. In addition, optimization of the cultivation conditions of the producing strain aimed to the achievement of a favorable intracellular redox balance can also contribute to the achievement of an increased level of glucose conversion to 3-HBA.
REFERENCES
Blacklock, T.J., Sohar, P., Butcher, J.W., Lamanec, T., and Grabowski, E.J.J., Org. Chem., 1993, vol. 58, no. 7, pp. 1672–1679.
Chiba, T. and Nakai, T., Chem. Lett., 1985, vol. 161, pp. 651–654.
Mori, K. and Sugai, T., Synthesis, 1982, vol. 9, pp. 752–753.
Mori, K. and Takikawa, H., Tetrahedron, 1990, vol. 46, pp. 4473–4486.
Spengler, J. and Albericio, F., Curr. Org. Synth., 2008, vol. 5, no. 2, pp. 151–161.
Liu, Q., Ouyang, S.P., Chung, A., Wu, Q., and Chen, G.Q., Appl. Microbiol. Biotechnol., 2007, vol. 76, no. 4, pp. 811–818.
Tseng, H.C., Martin, C.H., Nielsen, D.R., and Prather, K.L., Appl. Environ. Microbiol., 2009, vol. 75, no. 10, pp. 3137–3145.
Lee, S.H., Park, S.J., Lee, S.Y., and Hong, S.H., Appl. Microbiol. Biotechnol., 2008, vol. 79, no. 4, pp. 633–641.
Dellomonaco, C., Clomburg, J.M., Miller, E.N., and Gonzalez, R., Nature, 2011, vol. 476, no. 7360, pp. 355–359.
Clomburg, J.M., Vick, J.E., Blankschien, M.D., Rodriguez-Moyá, M., and Gonzalez, R., ACS Synth. Biol., 2012, vol. 1, pp. 541–554.
Gulevich, A.Y., Skorokhodova, A.Y., Sukhozhenko, A.V., Shakulov, R.S., and Debabov, V.G., Biotechnol. Lett., 2012, vol. 34, no. 3, pp. 463–469.
Gulevich, A.Yu., Skorokhodova, A.Yu., Stasenko, A.A., Shakulov, R.S., and Debabov, V.G., Appl. Biochem. Microbiol., 2016, vol. 52, no. 1, pp. 21–29.
Gulevich, A.Y., Skorokhodova, A.Y., Sukhozhenko, A.V., and Debabov, V.G., J. Biotechnol., 2017, vol. 244, pp. 16–24.
Skorokhodova, A.Yu., Zimenkov, D.V., Gulevich, A.Yu., Minaeva, N.A., Biryukova, I.V., and Mashko, S.V., Biotekhnologiya, 2006, no. 3, pp. 6–16.
Gulevich, A.Yu., Skorokhodova, A.Yu., Morzhakova, A.A., Antonova, S.V., Sukhozhenko, A.V., Shakulov, R.S., and Debabov, V.G., Appl. Biochem. Microbiol., 2012, vol. 48, no. 4, pp. 383–388.
Sambrook, J., Fritsch, E., and Maniatis, T., Molecular Cloning: A Laboratory Manual, 2nd ed., New York: Cold Spring Harbor Lab. Press, 1989.
Datsenko, K.A. and Wanner, B.L., Proc. Natl. Acad. Sci. U. S. A., 2000, vol. 97, no. 12, pp. P. 6640–6645.
Katashkina, Zh.I., Skorokhodova, A.Yu., Zimenkov, D.V., Gulevich, A.Yu., Minaeva, N.I., Doroshenko, V.G., Biryukova, I.V., and Mashko, S.V., Mol. Biol. (Moscow), 2005, vol. 39, no. 5, pp. 823–831.
Gulevich, A.Yu., Skorokhodova, A.Yu., Ermishev, V.Yu., Krylov, A.A., Minaeva, N.I., Polonskaya, Z.M., Zimenkov, D.V., Biryukova, I.V., and Mashko, S.V., Mol. Biol. (Moscow), 2009, vol. 43, no. 3, pp. 547–557.
Fischer, C.R., Tseng, H.C., Tai, M., Prather, K.L., and Stephanopoulos, G., Appl. Microbiol. Biotechnol., 2010, vol. 88, no. 1, pp. 265–275.
Koebmann, B.J., Westerhoff, H.V., Snoep, J.L., Nilsson, D., and Jensen, P.R., J. Bacteriol., 2002, vol. 184, no. 14, pp. 3909–3916.
Zhuang, Z., Song, F., Zhao, H., Li, L., Cao, J., Eisenstein, E., Herzberg, O., and Dunaway-Mariano, D., Biochemistry, 2008, vol. 47, no. 9, pp. 2789–2796.
Chen, M., Ma, X., Chen, X., Jiang, M., Song, H., and Guo, Z., J. Bacteriol., 2013, vol. 195, no. 12, pp. 2768–2775.
Fujita, Y., Matsuoka, H., and Hirooka, K., Mol. Microbiol., 2007, vol. 66, no. 4, pp. 829–389.
Campbell, J.W., Morgan-Kiss, R.M., and Cronan, J.E., Mol. Microbiol., 2003, vol. 47, no. 3, pp. 793–805.
ACKNOWLEDGMENTS
The authors are grateful to I.I. Gubaidullin from the NRC “Kurchatov Institute”-GosNIIGenetika for help with the cloning of the atoB and fadB genes, as well as the construction of the expression plasmid carrying these genes.
Funding
The work was supported by a grant from the Russian Foundation for Basic Research (project no. 18-29-08059).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interests. The authors declare that they have no conflict of interest.
Statement on Animal Welfare. This article does not contain any studies involving animals performed by any of the authors.
Statement of Compliance with Standards of Research Involving Humans as Subjects. This article does not contain any studies involving humans as subjects.
Additional information
Translated by D. Novikova
Rights and permissions
Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gulevich, A.Y., Skorokhodova, A.Y. & Debabov, V.G. Optimization of (S)-3-Hydroxybutyric Acid Biosynthesis from Glucose through the Reversed Fatty Acid β-Oxidation Pathway by Recombinant Escherichia coli Strains. Appl Biochem Microbiol 57, 161–169 (2021). https://doi.org/10.1134/S0003683821020046
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0003683821020046