
Abstract
Silicon sulfide (SiS2) was synthesized from Si, SiC, Si3N4, and a eutectic Al-Si liquid. An Ar-CS2 gas mixture, after bubbling through liquid CS2, was passed over pulverized Si, SiC, Si3N4, or a eutectic Al-Si alloy liquid. White, needle-shaped SiS2 was precipitated as a single phase on the colder downstream surface when Si powder in a SiO2 liner was heated over 1273 K. This deposition mechanism involves a reaction between Si and CS2 to form SiS2, with the vaporization of SiS(g) and deposition as SiS2(s) from SiS(g) and S2(g). Quartz was unaffected by the reaction gases. Notably, Si, SiC, Si3N4, and the eutectic Al-Si liquid were found to be suitable sources for SiS2 synthesis, although this was suppressed owing to Al2S3 formation on the latter.
Similar content being viewed by others
Introduction
Metallic sulfides with layered morphologies, such as SiS2, are employed in all-solid rechargeable battery electrolytes.[1] The two-dimensional polymeric chain structure of SiS2 makes it suitable for Li intercalation.[2,3,4,5] Unlike some sulfides, SiS2 is not a naturally occurring mineral and must be synthesized. This study focuses on SiS2 synthesis from powdered Si, SiC, and Si2N3, as well as eutectic Al-Si liquid. The objectives of the study were as follows: (1) to determine which of the Si sources, if any, would form SiS2; (2) to determine the reaction processing conditions for SiS2 formation; and (3) to characterize the SiS2 and associated products. The proposed process offers the benefit of providing a means for recycling common Si waste. This sulfide synthesis approach is applicable to catalyst formation or laterite upgrading.
The standard synthesis process for SiS2 employs the reaction of Al2S3 with SiO2 at temperatures above 1523 K.[2,3,4,5,6,7,8,9,10,11] Direct reaction between S and Si is difficult because of the high melting point of Si, the high vapor pressure of S, and easy sublimation of SiS2. Although some sulfides can be formed by slow heating of encapsulated metal and S powder followed by a high-temperature soak lasting weeks or months, this is too slow for commercial production. In this study, the potentially faster and lower-temperature reaction of the selected Si sources with CS2 vapor entrained in Ar was investigated.
Literature
Many researchers[12,13,14,15,16,17,18,19,20,21,22,23] have investigated the reaction of metal oxides with CS2 to form metal sulfides via the following reaction:
at temperatures as low as 1273 K. Even very stable rare-earth metal oxides can be converted to the corresponding sulfides or oxysulfides via this route.[6] This method has been suggested for use in nuclear fuel processing, laser applications, and thermoelectric devices.[12,13,14,15,16,17,18,19,20,21,22,23,24,25] Furthermore, CS2 is more reducing than H2S and has been demonstrated as a means of refining the oxides and nitrides of Ti, V, and Sc.[26,27,28,29,30] Many investigators[12,13,14,15,16,17,18,19,20,21,22,23, 26, 30,31,32,33,34] have used a CS2 bubbler to produce CS2 gas for subsequent reactions and to limit the gas circuit for safety. The selected CS2 bath temperature was close to room temperature, between 263 K and 313 K. Previous papers published by the authors[26, 30] reported the use of a bath at higher temperatures.
The structure of SiS2 is orthorhombic (a = 0.561 nm, b = 0.547 nm, c = 0.9583 nm[3]), and the condensed form of SiS2 is a white, needle-like crystal with a melting point of 1363 K.[35,36,37,38] Notably, SiS2 exhibits a boiling phenomenon between 1373 K and 1473 K.[37, 39] Since its melting and boiling points are close to each other, it undergoes significant sublimation of SiS2 below melting point.[3, 5, 7,8,9,10,11, 35, 40] This phenomenon suggests that SiS2 might be a good candidate for vapor refining; however, condensation must occur rapidly because SiS is also known to deposit from the transport gas during cooling.[3, 4, 6,7,8] Any SiS2 product must be isolated from the ambient atmosphere to prevent deliquescence and reaction with water vapor to form H2S and SiO2.[4, 6,7,8, 40] Therefore, the product must be stored in a dry and inert atmosphere.
Literature published between 1880 and 1930[6, 11, 30, 35, 41] showed that SiS2 was formed from reactions with CS2 in china clay reactors, indicating that Al2O3 and SiO2 reacted with CS2. However, this reaction could not be confirmed by the preliminary experiments in this study nor by the thermochemical computations[11, 35, 36, 42, 43] described later. Therefore, SiO2 was not selected as a Si source herein; instead, Si, SiC, Si3N4, and eutectic Al-Si were used. The binary Al-Si system exhibits eutectic behavior at Al-12.8 wt pct Si and at 850 K.[44] Since Al-Si alloy is a basic Al alloy, Si recycling via selective sulfurization is desirable. The SiS2 formation reactions are as follows:
Carbon contamination in SiS2(s) does not lead to significant problems in Li battery applications.
The reaction with Al2S3 and SiO2 has been used to synthesize SiS2 since 1935.[3, 5, 7, 9, 10, 39] Byerley and Teo reported that Al2S3 and SiO2 reacted in a N2 atmosphere at 1523 K, and that SiS2 sublimated in vacuum at 1523 K to produce high-purity SiS2.[9, 10] The sublimation of SiS2 has been reported by Haas,[41] as well as by Schumb and Bernard,[45] to be slow. Byerley and Teo,[9, 10] and Emons and Theisen[46] determined the partial pressures of sulfides and S2 by Knudsen cell measurements, and obtained results in good agreement with others,[42, 47] as shown in Figure 1; however, there were differences in the delineation of the pertinent reactions.

Partial pressures of gases in Si-S binary system
The extrapolation to 1 atm SiS(g) of the reaction
approaches the boiling point of SiS2 (1403 K), as estimated from the JANAF tables.[37] The boiling process of SiS2 necessarily includes the sublimation of SiS, as written in Reaction [3].[9, 10, 38, 46, 47] Since the early 1930s, the evaporation of sulfides has been the subject of numerous investigations,[11, 37, 41,42,43] and the existence of two sulfides, namely SiS2 and SiS, has been recognized by 1965.[4, 10, 41]
The properties of solid SiS have not been investigated because of its strong deliquescence and subsequent disproportionation. Moreover, SiS gas has been commonly reported to form a black or gray deposits after the reactions of Si with S, H2S, or other sulfides, such as CuS, FeS, ZnS, and Al2S3.[7, 8, 10, 31, 45, 48] Additionally, SiS shows yellow, orange-red, dark brown, reddish black, and black colors because of its instability at room temperature in an ambient atmosphere.[9, 10, 41, 45]
In 1939, Barrow and Jevons[31] reported that SiS could be formed from Si and gaseous CS2, although SiS and SiS2 could not be distinguished by spectroscopic analysis of distilled SiS. Schumb and Bernard[45] reported that SiS was formed by heating a mixture of pure SiS2 and excess Si at 1123 K, and that SiS was not formed by the reaction between Si and S2 gas at 1 atm. Byerley and Teo[10] reported the decomposition of pure SiS2(s) in the Knudsen cell to SiS(g) and S2(g) (Figure 1). The partial pressures of the products of Reaction [4] were approximately 100 times lower than those of the products of Reaction [3].
The vapor pressure compiled in HSC Chemistry[36] for Reaction [5]
is only 6.5 x 10−4 atm at the reported boiling point of SiS2.[35, 37] Furthermore, the vapor pressure of SiS exceeds 1 atm at the melting point of SiS2[36]; therefore, SiS(g) predominates. The employed database[36] cites JANAF thermodynamic tables,[37] the calorimetric data[7, 11, 43, 48] from which are favored. The contradictory reports of vapor pressure measurements have been the topic of numerous discussions[11, 37, 41,42,43, 49] and the need for accurate thermochemical data persists. Herein, we use the experimental reports from the evaporation studies[8,9,10, 41, 46, 47, 50] and the database summarized in HSC Chemistry,[36] as shown in Figure 1.
Crystalline SiS has not been reported and solid SiS is formed as an amorphous structure only from condensing gaseous SiS,[3, 9, 10, 31, 41, 45, 50] i.e.,
whereby SiS decomposes to Si and SiS2 after annealing without crystallization.[10, 33, 34, 41, 42, 45] This suggests that SiS is thermodynamically unstable and that SiS may not be formed via Reaction [2a].
The vaporization of SiS2 is strongly related to the chemical reactions of the SiS system, and a possible mechanism involves the proportional reaction of Eq. [3]. The results of this approach have been shown to match that of the compositional analysis of gas mixtures by mass spectroscopy, according to Coppens et al.[49]
Yokoyama32) patented the synthesis of SiS2 by the reaction of SiC and CS2 based on Reaction [2b]. He reported that only non-volatile carbon remained after Reaction [2b] and that SiS2, not SiS, was evaporated. Notably, SiS2 is condensed at the cold part of the reactor. In Yokoyama’s study, 0.9 kg of SiS2 was produced at 1073 K in 1 hour from 2.1 kg of CS2 and 0.8 kg of SiC mixed with the clay. Such a fast reaction at relatively low temperatures is an attractive way to form SiS2, but neither a detailed mechanism nor thermal conditions were presented, and no additional data have been reported subsequently. Therefore, the fundamental information on CS2 gas sulfurization is the focus of this study to provide an effective means of stable synthesis of SiS2, and perhaps, SiS.
TiO2, TiO, TiN, and TiC have reportedly been sulfurized at 1273 K.[15,16,17, 22, 26] Trivalent TiN and 4-valent TiC were sulfurized more quickly than divalent TiO.[26, 27]
Industrial quantities of CS2 liquid are available for commercial processing and thermochemical data are available for CS2 gas sulfurization; thus, it is possible to analyze the chemical processes involved in SiS2 synthesis.
Thermochemical
Figure 2 shows the standard Gibbs free energy changes (ΔGo) for the reactions to form SiS2 from Si, SiC, and Si3N4.[36] Since they are negative at all temperatures, the reactions from Si, SiC, and Si3N4 are favorable when the reactants are in their standard states, which are the pure condensed phases and ideal gases at unit activity.

Standard Gibbs free energy changes involved in CS2 gas sulfurization
The binary Al-Si system shows a simple eutectic melting behavior at Al-12.8 wt pct Si and 850 K.[44] The dilution of Si as the mixture reduces the thermochemical activity of Si, and a slight negative deviation from the ideal mixing behavior is known in Al-Si binary system.[44, 51] Although the modification due to less activity depends on temperature and Si concentration, it consequently increases ΔG from ΔGo for Reaction [2a] shown in Figure 2. At 1473 K and the eutectic composition, for example, the activity of Si relative to pure solid Si is 0.067,[44] and ΔG for Reaction [2a] increases from ΔGo by +33 kJ, although ΔG is sufficiently negative to drive Reaction [2a].
Reaction [7] from SiO2 is significantly positive at all temperatures, confirming that SiO2 is unreactive, thereby making it suitable for reactor construction.
The positive deviation is much greater if CO2 is the C-bearing product. In this study, SiO2 was used as a non-reactive material in CS2 gas flow. The thermal decomposition of CS2 gas via high-temperature reaction, i.e.,
is also insignificant if partial pressures of both CS2 and S2 are near 1 atm. However, the positive value of ΔGo is not large enough to completely obviate the partial decomposition of CS2; thus, it should be considered.
The thermal decomposition of CS2 via Reaction [8] occurs partially. The Gibbs free energy change for Reaction [8] is +19.1 kJ/mol at 1273 K[36] for the reference states of pure graphite and gases with unit activity, giving an equilibrium constant of 0.167. This means that the S2 pressure will be 16.7 pct of the CS2 pressure under the reasonable assumption of ideal gas behavior. A typical CS2 partial pressure in the Ar feed stream is evaluated as the equilibrium vapor pressure at the CS2 bath temperature. Figure 3 shows the vapor pressures of CS2 at the bath temperature and the reaction temperature, i.e., PCS2(Tbath) and PCS2(T), respectively. The figure also shows the partial pressure of S2, PS2(T), at the reaction temperature T, via the thermal decomposition of CS2 when carbon is at unit activity and the Ar pressure is the same at both temperatures. For example, the partial pressure of S2 at 1273 K is as high as 0.028 atm, corresponding to PCS2 = 0.196 atm at 273 K.

Partial pressures of Ar, CS2, and S2 gases at temperatures of evaporation and reaction
The various possible reactions in the C–S–Si system complicate the sorting out process of the significant reactions from the insignificant ones. Herein, the reactions involved in the experiments are analyzed from a thermodynamic perspective.
Experimental
Single-crystal semiconductor-grade lump Si (>99.9999 pct) was ground and sieved into -100 mesh powder. Powdered SiC (>99 pct) and Si3N4 (>99 pct) were obtained from Kojundo Chemical Lab. Co., Ltd. Their particle sizes were as large as 2–3 μm. The Al-12.6 wt pct Si eutectic was alloyed in air using pure Al (99.995 pct) from Nippon Light Metals Co. Ltd and an Al-25 wt pct Si alloy from Hirano Seizaemon Syouten Co. Ltd. The cast ingot was cut into small pieces after the oxidized surface was removed. Liquid CS2 (>99.5 pct) was obtained from Fujifilm Wako Pure Chemical Corp.
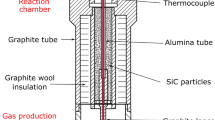
Figure 4 shows the experimental setup. Pure Ar gas (>99.9998 pct) was bubbled at a constant rate into a bottle containing 50 mL of liquid CS2 kept in a water bath at a constant temperature (± 0.2 K). The CS2 vapor pressure generated an adequate Ar-CS2 gas mixture.[36] The CS2 mass was recorded as sulfurization proceeded; it was found to vary linearly with time[26, 27, 30] and was found to be sufficient for the sulfurization of 0.5 g Si over 1 hour. The CS2 feed rate was controlled by a constant flow of Ar and by varying the CS2 temperature. The linear decrease in mass with reaction time indicated that the bubbler produced a Ar-CS2 gas mixture of constant composition and that adequate heat was being supplied to the liquid to satisfy the enthalpy of CS2 liquid vaporization.

Experimental setup for the sulfurization process
A typical CS2 feed rate at 273 K was 11.5 mL/min (8.6 x 10−6 mol/s), which was achieved with an Ar flow rate of 10 mL/min. The CS2 bath temperature was selected to be either 273 K or 283 K.
A 40-mm-ID 1-m-long dense alumina (>99.5 pct) tube was set in the 0.5-m-long electric furnace. The unreacted exhaust gas was captured in a 1.0 M NaOH solution.[26, 27, 30, 33, 34, 39, 48, 52] Alternatively, CS2 could have been recovered by using a gas chiller. The temperature of the sample was monitored and controlled at the external surface of the alumina tube. The temperature profile inside the alumina tube was measured precisely in a stream of Ar gas before the sulfurization process, such that the sample temperature in the hot zone was accurately known.
Samples were held in an Al2O3 (>99.95 pct) boat (14 mm wide, 100 mm long) for runs 1–4 and 13–15 or in a transparent 16-mm-ID quartz tube that was 100, 150, or 300 mm long for runs 5–10. The samples for runs 5–10 were placed upstream, in the hot region of the quartz tube. The cold end of the tube was downstream and was maintained at a controlled temperature, as shown in Figure 4. The 150-mm-long tube consisted of two pieces, the upper and lower halves, to easily retrieve the condensates. The quartz tube with the sample powder was evacuated and then heated in Ar gas. The gas mixture of Ar and CS2 was then introduced after reaching the desired temperature. Both heating and cooling were conducted under pure Ar flow at a constant flow rate (20 mL/min). The sample mass was recorded after cooling to quantify the sulfurization degree.
The resulting phases were identified by X-ray diffraction (XRD) analysis using Philips X’Pert Pro (Cu Kα). The morphology of the specimens was determined by using a JEOL JSM 6510LA scanning electron microscope (SEM) equipped with an energy-dispersive X-ray spectrometer (EDS). The C and S concentrations were determined via infrared spectroscopy analysis using LECO CS-600 and oxygen concentration was measured by the inert-gas fusion method using a LECO TC-600 analyzer.
After the initial Si run series with Si, all subsequent runs employed a transparent quartz tube inserted inside the Al2O3 tube, as shown in Figure 4, to prevent possible reactions with Al2O3.
When the samples were cooled to room temperature after each run and were still in Ar atmosphere, both ends of the quartz tube were sealed with the rubber caps and placed in a glovebox filled with dry, oxygen-free Ar gas. The tube consisted of three 100-mm-long pieces to facilitate precipitate recovery. The Si source was placed at the hot end of the tube, such that it was positioned in the hottest zone in the alumina tube. The liquid CS2 was typically maintained at 293 K under a constant Ar flow rate of 10 mL/min.
Results
The results from reacting CS2 to sulfurize Si, SiC, Al-Si eutectic alloy, and Si3N4 are presented for each Si source.
Sulfurization of Si powder
The sulfurization of Si was initially performed in an Al2O3 boat and then in a SiO2 liner to avoid potential reactions with Al2O3. The experimental conditions are listed in Table I.
Synthesis in Al2O3
White powder, identified as SiO2, was found to lightly coat the Si particles as the conclusion of run 1, as shown in Figure 5(a). This was probably the result of the unintentional moist air hydrolysis of SiS2 or SiS.[7, 11, 33,34,35, 41, 45] A thin glossy gray layer, similar to tar, was found adhered to the alumina boat after runs 2–4. The XRD patterns showed this to be primarily α-Al2O3, possibly with some amorphous SiS.[9, 10, 41, 45] Most of the residue remained as unreacted Si, even after 4 h. Single-phase SiS2 could not be synthesized at the initial position of the Si powder in the Al2O3 boat. Several thermochemical reactions occur between Si and CS2 gas.

Appearance of the samples (runs 1-4) in the alumina boats after sulfurization for 4 hours at various temperatures
Significant amounts of white, needle-like SiS2 precipitates were found in the lower-temperature region of the Al2O3 tube. The size of this precipitation became more longer and the larger amount of precipitates were found as the Si powder was heated to higher temperatures.
Synthesis in SiO2
Figure 6 shows a black product, which remained only near the hot end, while the white, needle-like precipitates were formed 100 mm from the furnace center, where the temperature was ~900 K. This result agrees with those of runs 1–4 that make use of an Al2O3 boat. Farther downstream, dark red precipitates were gradually transitioned to yellow.

Appearance of SiO2 tube after sulfurization; temperature profile and the precipitates in the tube, where the Si powder was heated at 1273 K for 4 hours (run 8)
No precipitates were found in the few millimeter-long gap between the inner SiO2 and the outer Al2O3 tubes. Furthermore, no product was precipitated upstream from the initial sample position, meaning that the flow of reaction gas carried or generated the condensate downstream. The white needles or sometimes thin and long plates grew in the upstream and inward radial directions, as shown in Figure 7.

White precipitates on the inner surface of SiO2 half tubes from (a) run 10 (1273 K, 2 hours) and (b) run 7 (1273 K, 8 hours)
Using the upper- and lower-half tubes of silica, the quantity of the white product on the inner surface of the half tubes was recorded for several runs. The amounts were identical in both halves, and the precipitates had similar morphologies. Gravity had no discernable effect and no trace of liquid was found after runs 7 and 10. Therefore, the white precipitates were deposited from the gas onto the cold substrate and were not hydrolyzed.
Figure 8 shows the XRD spectra for selected sample locations for run 8 (1273 K, 4 hours). Notably, SiS2 was absent at location (a), the initial position where the Si powder was placed. Near the center of the quartz tube, at location (b), the white deposit was identified as SiS2. Most of the initial Si powder was converted to an amorphous phase, while a lesser amount was transformed to SiC. The small amount of the black or gray material between the initial Si position and the position of SiS2 deposition could not be identified through XRD analysis. This material was very thin and highly hygroscopic, thereby suggesting that it was an amorphous SiS precipitate. The yellow deposit downstream from SiS2 deposition was identified as S8, which is the stable form of solid sulfur. The broad halo pattern in XRD indicated the existence of either amorphous S or amorphous SiS. Notably, S exhibits a yellow or brownish color when it solidifies from liquid, depending on its purity and thickness.

XRD patterns of the specimens taken from the various parts of the tube in run 8
Figure 9 shows the elemental analysis of the white precipitates produced in 2 hours at 1273 K using an Ar-CS2 mixture generated at 273 K. The sample prepared in run 10 was identified as SiS2 by XRD analysis; however, more than 4 hours at 1273 K was necessary to determine the Si/S ratio of the phases.

Elemental analysis of sulfur and carbon in the white precipitates obtained at 1273 K
Sulfurization of SiC
Fine SiC powders (2–3 μm) were heated to 873 K for 3 days and to 1073 K for 1.5 day. Thereafter, Ar gas was bubbled at a rate of 10 mL/min through liquid CS2 at 293 K. The color, size, and XRD patterns of these samples were those of pure SiC and unchanged, as shown in Figures 10(b) and (c). There were no deposits in the furnace even after a very long reaction time at these low temperatures.

XRD patterns of SiC powder after heating in Ar-CS2 gas mixture at various temperatures
SiC powder was exposed to the mixed gas at 1273 K for 1 day. The appearance of the tube was identical with that when pure Si was used. Needle-shaped crystals of SiS2 precipitated downstream on the quartz wall. The position of deposition, as well as the XRD spectra, was also identical as with the use of Si powder (Figure 10(e)).
Sulfurization of liquid Al–Si eutectic
Two 3.5-g samples of Al-13 wt pct Si alloy were, in separate runs, set in an Al2O3 boat and reacted for 1 day in a mixture formed by bubbling 10 mL/min of Ar through liquid CS2 at 293 K. The Al–Si alloy was not stirred. One sample was heated to 873 K and the other to 1273 K. Both had solidified surfaces after cooling, indicating melting.
At 873 K, no deposition of needle-shaped crystals was found in the colder regions of the furnace. The sample heated at 1273 K, however, formed needle-shaped crystals downstream. The XRD results shown in Figure 11 confirmed the presence of single-phase SiS2 without Al contamination. However, the metallic luster of the Al–Si alloy was lost and sulfurized to black after solidification at 1273 K. Notably, the product on the alloy surface was identified to be α-Al2S3 via XRD analysis.

XRD patterns of Al-Si alloys, where the reaction time was set commonly to 1 day
Sulfurization of Si3N4
Figure 12 shows the result when the same Ar-CS2 gas mixture was generated by bubbling 10 mL/min Ar through CS2 liquid at 293 K, after passing over the Si3N4 powder in the transparent quartz at 1273 K for 1 day. White, needle-shaped crystals precipitated on both the upper and lower halves of the tube in the cold region. These were identified by XRD as single-phase SiS2. The Si3N4 powder was placed more widely than the previous samples, but only the powder at the hottest region changed its color, as shown in Figure 12. The region between the hottest area of Si3N4 and the SiS2 deposition area was slightly covered with black particles, which could not be identified by XRD because of their small mass and reactivity. The black–gray particles were found only at the lower half of the tube.

Appearance of sulfurized sample and white precipitates on the inner surface of SiO2 tube
Figure 13 shows the XRD patterns of the samples heated for 1 day at temperatures lower than 1273 K. There were neither precipitates of SiS2 crystals nor phase change of Si3N4 at 873 K, 973 K, and 1073 K observed. When the sample was heated to 1173 K, a small amount of SiS2 precipitates, as shown in Figure 13(f), was observed. Furthermore, SiS2 crystals were formed at the lowest temperature (1173 K) among the four materials studied when Si3N4 was used.

XRD patterns of Si3N4 samples, where reaction time was set commonly to 1 day and liquid CS2 was kept at 293 K under Ar flow rate of 10 mL/min. (a): starting material, Si3N4, (b) through (e): after sulfurization at 873 K, 973 K, 1073 K, and 1173 K, respectively, (f) white powder precipitated by sulfurization at 1173 K
Discussion
Sulfurization of Si powder
In Al2O3
The expected direct Reaction [2a] did not occur in runs 1–4 at the hottest regions in the furnace. Reaction [9] at the hot zone was expected to form amorphous carbon and gaseous SiS, followed by its partial deposition as amorphous SiS, as shown in Reaction [10].
The formation of SiS(g) may preferentially occur through Reaction [9] at the high-temperature zone. The vaporized SiS(g) may then be carried by the gas flow to decompose and condense as SiS at the lower-temperature zone[10, 46] via Reaction [10]. The formation of crystalline SiS was neither reported in the literature nor detected in this work.
Gaseous SiS may decompose to SiS2(s) and Si(s) as the reverse of Reaction [4]. However, the co-deposition of Si(s) with SiS2(s) was not detected. A possible route to form SiS2(s) from the gas phase remained only in Reaction [11], i.e., a route via SiS(g).
SiS once deposited is transformed to SiS2 by exposed to S2(g), and it again evaporates as SiS2(g). The black or gray deposition with a strong hygroscopic property in addition to the odor of H2S upon exposure to air can be tentatively identified as amorphous SiS. The coexistence of S2(g) is key to SiS2 crystals by Reaction [11] because the parallel evaporation Reaction [3] does not form the S2(g) required by Reaction [11].
Another possible reaction for SiS(g) deposition is that with CS2(g) as:
Nevertheless, carbon deposition was not observed; thus, Reaction [12] was dismissed as a viable path to produce SiS2. Although SiS2 vaporization was independently observed in this work, its pressure was thermodynamically 1000 times lower than that of SiS vaporization, as shown in Figure 1.
In SiO2
The needle-like samples shown in Figure 14(a) were analyzed by elemental mapping. They exhibited a homogeneous distribution of O, Si, and S. The trace amounts of C were probably from the carbon tape used for SEM sample preparation. Carbon was not detected in the crystalline grains. The O profile showed that the crystal surface contained a considerable amount of oxygen, which was expected as the specimens were heavily oxidized. However, this does not correspond to the XRD data that show the SiS2 phase. The XRD profiles of this specimen were first measured in air and again after keeping in vacuum for 5 days, where EDS mapping revealed that a SiS2 phase had transformed into a mixture of crystalline and amorphous phases, probably SiO2. Because of the extreme sensitivity to hydrolysis, the surface may have decomposed into a very thin and amorphous SiO2 layer during handling. The characteristic H2S smell effusing from the sample supported the hydrolyzation hypothesis. The intensity of S, as shown in Figure 14(c), seems very low because of its low-intensity energy profile and the overlapping profiles with the other elements. The detected signal positions of S agreed with the locations of Si. Therefore, neither free C nor free S from the CS2 gas is attached to the SiS2 surface. This is consistent with the element analysis shown in Figure 9.

SEM image (a) and its element mapping of (b) silicon, (c) sulfur, (d) oxygen, and (e) carbon, of the specimen at run 8
The specimens were slightly oxidized during the transfer for analysis, and the precipitate volume was very small for short-time sulfurization, thereby preventing the separation of the needle-shaped SiS2 crystals from the surrounding tar-like amorphous material adhering as a thin sheet to the silica tube. As shown in Figure 9, a highly purified SiS2 was recovered from the central part of the tube at a longer sulfurization time. A small amount of carbon in the samples may have been present because of the sampling method. This phenomenon is consistent with SiS2 vapor deposition. The morphology and microstructure of the precipitated SiS2 samples are shown in Figure 14. The white precipitates resembled needles in macroscopic view, although they were assemblies of long square pillars. This directional growth is consistent with the orthorhombic SiS2 structure.
Sublimation of SiS
The vapor transport behavior of SiS2 was studied in the absence of CS2 by placing 1 g of SiS2 in the hot end of the SiO2 tube to observe its transport to the colder regions of the reactor. The vapor pressures of the solid SiS2, defined according to equation,[5] are 1.22 × 10−4 and 1.05 × 10−7 atm at 1273 K and 1000 K, respectively, while ΔH oSubl is 273.2 kJ/mol.[36] The sample was held at 1273 K for 4 hours in run 8N while pure Ar gas, devoid of CS2, was passed through the furnace at a 20 mL/min flow rate. The linear velocity of the Ar flow was as minimal as 1.15 × 10−3 m/s.
Figure 15 shows a photograph of the cooled quartz tubes after the heating cycle. White, needle-shaped SiS2 crystals were deposited downstream. The deposition position was approximately 110 to 140 mm from the initial position of SiS2, which corresponded to 1023 K to 1173 K based on the profile shown in Figure 6. This is approximately 100 degrees higher than the deposition of SiS2 derived from the reaction of CS2 with the selected Si sources. This 100 K higher SiS2 condensation temperature indicated that the vapor pressure of the evaporation-derived SiS2 was less than those obtained from the CS2 reactions.

Photo of the quartz tube after heating SiS2 in an Ar atmosphere at 1273 K (run 8N)
A small quantity of white precipitate was also found on the lower half of the upstream tube, and it was symmetrical with the downstream deposit. This symmetry indicated that SiS2 diffusion was fast compared to bulk flow. This upstream deposition was not observed in Ar-CS2 sulfurization. The sublimation-generated SiS2 is thought to have caused accelerated back diffusion and upstream deposition, whereas CS2-derived deposition ensues downstream deposition only. Furthermore, the orange–black deposits are unrelated to SiS2 vapor transport but associated with reactions involving CS2. The mechanisms of evaporation and deposition are different in these two cases.
These observations show that although SiS2 vapor transport occurs without CS2, the reaction of CS2 with Si sources will generate SiS2 at a rate that is three orders of magnitude slower than the SiS2 vaporization rate.
Reaction Mechanism
Figure 16 shows a suggested reaction path schematic. The CS2 gas evaporates from the liquid into the Ar carrier gas at approximately room temperature. This gas passes into the high-temperature region and reacts with Si. Because neither SiS(s) nor SiS2(s) was observed on the Si surface, their precipitations were neglected by combining Eqs. [6] and [9] to give:

A schematic of sulfurization mechanism when Ar/CS2 gas was used
The resulting C was observed to be amorphous. The C is proposed to react with Si to generate SiC, although the reaction is believed to be slow.
Carbon contamination in SiS2(s) does not cause significant problems in Li battery applications.
This reaction sequence exhausts S2(g), as shown in column 1 of Figure 16. The second column shows that a simultaneous reaction between Si and CS2 gas generates SiS(g) followed by subsequent condensation in the colder regions of the reactor. The formation of SiS2(s) on the Si surface and its vaporization as SiS(g) are feasible as the side reaction, as shown in the third column. The far-right path shows the condensation of S8 due to cooling from the S2(g) originating from the thermal decomposition of CS2, as well as the decomposition of SiS2 to SiS.
Figure 17 presents an illustration of the gas pressures at the vaporization[9] and the successive deposition[4] from Si + CS2, referring to Figure 1. These two reactions (upper and middle lines in Figure 17) are irreversible under gas flow. The evaporated SiS(g) at T2, as shown by the upper curve (red), is condensed as SiS2(s) by reacting with S2(g), as shown by the middle curve (black). A large amount of SiS(g) at 1273 K, for example, is carried to the lower-temperature area, whereas SiS(g) decomposes and condenses to solid SiS2 (middle curve). Based on the measurements of equilibrium vapor pressure among SiS2, SiS, and S2,[10, 41, 46] as shown in Figure 1, once SiS(s) is formed, it cannot exist at the higher temperature area because of the high vapor pressure of SiS(s). Under the exposure of S2(g) and by absorbing S2(g), SiS(g) deposits as SiS2(s). When this pressure is estimated to be as high as 10−6 atm, the temperature is evaluated as 951 K. Without the coexistence of S2(g), the pressure of SiS2(g) is low, as reported by Rosenqvist and Tungesvik.[8] The vaporization of pure SiS2(s) was independently examined and the deposit was found at 1073 K. (lower dashed curve in Figure 17). Therefore, the deposition temperature from SiS2(g) is 122 K different from the former mechanism via SiS(g).

Temperature dependencies of pressures related to the reactions
Figure 1 quantitatively shows the vapor pressure of solid SiS2 by Reaction [5].[36] Its vapor pressure ranges from 3.5 × 10−3 atm at 1473 K to 1.4 × 10−5 atm at 1173 K. Vapor transport rate in an Ar carrier gas is directly related to the product of the partial pressure of the transport species and the Ar flow rate. The low transport rate from SiS2(s) also agrees with the relatively low pressure of SiS2 in the Ar carrier gas.
The vapor pressures of SiS and S2 are higher than that of SiS2, as shown in Figure 1 if pure solid SiS2 exists at a constant temperature. This indicates the preferential evaporation as SiS(g) and S2(g). The sharp temperature dependencies of the reactions related to SiS(g) suggest that the deposition from SiS(g) as SiS2(s) preferentially occurs in the lower-temperature regions.
Sulfurization of SiC
After the runs for SiC, SiS2 precipitated at 1273 K. However, a significant fraction of the SiC powder remained unreacted at 1273 K, contrary to a patent claiming good recovery at 1073 K for 1 hour.[32] In the runs for Si, a small amount of SiC is one of the intermediates from Si to SiS2 as SiC is converted into SiS2 by the additional sulfurization under the same gas flow conditions.
The deposition position was also identical as obtained with the use of Si powder, the same as the XRD spectra (Figure 10(e)). The formation of SiS2 from SiC is favorable, but a minimum temperature of 1273 K is required for the SiC starting material.
Potential Diagram
Carbon deposits on the unreacted Si particles via Reaction [9]. This may produce SiC, as shown in Figure 16. However, SiC can form SiS2, as shown in Figure 10. Therefore, the total stability of the Si–S–C ternary system should be considered. Both the reactivity between the Si and CS2 gas, as well as that between the SiC and CS2 gas, can be summarized by the potential diagram shown in Figure 18, which is based on the employed database.[36] Although the thermodynamic database may need refinement, it provides a potential diagram that approximates the phase stability at 1273 K. The standard state activity of C, aC, is pure solid graphite, which is very similar to the activity of amorphous C deposits. Additionally, the S pressure cannot exceed 1 atm because the system is constrained to remain at atmospheric pressure by the bubblers. Similarly, the partial pressure of the CS2 gas depends on the bath temperature, T1, which is also less than 1 atm.

Potential diagram of Si–S–C system at 1273 K and equilibrium pressures of CS2 gas evaporated at T1
The potential diagram shown in Figure 18 was constructed based on the following conditions: The cross-hatched region is the feasible operating area to produce the reactive Ar-CS2 gas mixture; An equilibrium point lies in this region, especially on the line of aC = 1 in the stable SiS2 region if stable graphite coexists. Activities of C less than unity represent that the gas is not saturated with C; thus, the locations where aC < 1 denote conditions of no carbon deposition and the region where aC > 1 indicates the possible existence of amorphous C.
The region of solid SiS only exists at aC << 1; thus, it cannot exist in the presence of pure C. High partial pressure of S2 is beneficial for CS2 gas sulfurization, and such a condition is envisaged from the thermal decomposition of CS2. The red line of 2.56 × 10−4 atm S2 pressure was computed from the liquid CS2 being at only 200 K. The residual C from the decomposed CS2 gas did not contaminate the SiS2 product owing to the SiC coexistence. Higher S2 partial pressure conditions favor SiS2 formation as a single-phase without SiC contamination.
Sulfurization of liquid Al–Si eutectic
Because the melting point of Al2S3 was 1373 K,[36] the lighter and unsublimated solid Al2S3 floated on the surface of the liquid alloy. The Al2O3 crucible was unaffected by the environment, but the metallic Al alloy was readily converted into Al2S3 at 1273 K. The SiO2 crucible was not used in this experiment to avoid the confusion on the reaction between Al2S3 and SiO2.[9, 10]
The formation of Al2S3 consumes CS2 gas, and the covering of the liquid surface by Al2S3 may slow down the gas reaction between the Al–Si melt and CS2. However, quantitative analyses, such as phase equilibrium[39] and reaction kinetics, were not conducted because of the hygroscopic nature of SiS2.
Sulfurization of Si3N4
Notably, Si3N4 reacted with CS2 to form SiS2, whereas SiO2 did not. The formation of heavy gas, such as SiS, is presumed to be the cause of the formation of the black–gray particles found at the lower half of the SiO2 tube.
Conclusion
In this study, SiS2 was vapor-deposited using transparent silica tubes when Si, SiC, Si3N4, or Al–Si eutectic alloy was heated in a mixture of Ar and CS2(g) above 1273 K. Notably, SiS2 was also formed at 1173 K from Si3N4. The needle-shaped SiS2 crystals were precipitated at the downstream of gas flow at temperatures between 1073 K and 1173 K. They were semi-transparently white with minimal C contamination, as well as exhibited strong hygroscopic reactions in the open air and were easily oxidized to SiO2.
Consequently, a reaction mechanism is proposed consisting of the following: (1) evaporation of SiS gas after the reaction between Si and CS2, which is triggered by SiS2(s) formation and (2) successive deposition of SiS2(s) from SiS(g) at the low-temperature zone. The vaporization of the S2(g) by-product plays an important role in the deposition of SiS(g) as SiS2(s). A small amount of C contaminated the sample at the early stage proposed because of the thermal decomposition of the CS2 gas. At the later stage, C was removed by the excess amount of S2(g). A highly pure SiS2(s) can be obtained at 1273 K from low-quality Si sources using high partial pressure of S in CS2 gas.
References
M. Murayama, R. Kanno, M. Irie, S. Ito, T. Hata, N. Sonoyama, and Y. Kawamoto: J. Sol. Stat. Chem., 2002, vol. 168(1), pp. 140–48. https://doi.org/10.1006/jssc.2002.9701
W. Büssen, H. Fisher, E. Gruner, Naturwissenshaften, 1935, vol. 23, p.740. https://doi.org/10.1007/bf01498156
E. Zintl and K. Loosen: Z. Phys. Chem. (Leipzig) A, 1935, vol. 174(3), pp. 301–11.
C.T. Prewitt and H.S. Young: Science, 1965, vol. 149(3683), pp. 535–37. https://doi.org/10.1126/science.149.3683.535
5) Johannes Peters, Bernt Krebs, Acta Crystallogr. B Struct. Crystallogr. Cryst. Chem., 1982, vol. B38, pp. 1270-1272. https://doi.org/10.1107/s0567740882005469
J.W. Mellor: Comprehensive Treatise on Inorganic and Theoretical Chemistry, vol.VI, C (Part II), §60. Si, Silicides, Longmans, Green and Co. Ltd., London, 1930, pp. 985–90.
7) P. Rocquet, M.F. Ancey-Moret, Mémoires Présentés a la Société Chimique, 1954, vol. 7, pp. 1038-1043.
8) Terkel Rosenqvist, Karl Tungesvik, Trans Faraday Soc., 1971, vol. 67, pp. 2945-2951. https://doi.org/10.1039/tf9716702945
9) J. Byerley, W.K. Teo, Metall. Trans., 1973, vol. 4, pp. 419-422. https://doi.org/10.1007/bf02648693
J. Byerley and W.K. Teo: J. Inorg. Nucl. Chem., 1973, vol. 35(7), pp. 2195–2205. https://doi.org/10.1016/0022-1902(73)80282-9
I. Tomaszkiewicz, G.A. Hope, and P.A.G. O’Hare: J. Chem. Thermodynamics, 1997, vol. 29(9), pp. 1031–45. https://doi.org/10.1006/jcht.1997.0221
M. Skrobian, N. Sato, M. Saito, and T. Fujino: J. Alloys Compd., 1994, vol. 210(1-2), pp. 291–97. https://doi.org/10.1016/0925-8388(94)90152-x
13) Milan Skrobian, Nobuaki Sato, Takeo Fujino, Thermochim. Acta, 1995, vol. 249, pp. 211-219. https://doi.org/10.1016/0040-6031(95)90696-7
14) Milan Skrobian, Nobuaki Sato, Kohta Yamada, Takeo Fujino, Thermochim. Acta, 1995, vol. 255, pp. 201-209. https://doi.org/10.1016/0040-6031(94)02150-m
M. Saeki, M. Onoda, and Y. Yajima: J. Sol. State Chem., 1996, vol. 121(2), pp. 451–56. https://doi.org/10.1006/jssc.1996.0062
S. Hirai, K. Shimakage, Y. Saitou, T. Nishimura, Y. Uemura, M. Mitomo, and L. Brewer: J. Am. Ceram. Soc., 1998, vol. 81(1), pp. 145–51. https://doi.org/10.1111/j.1151-2916.1998.tb02306.x
J. Cuya, N. Sato, K. Yamamoto, A. Muramatsu, K. Aoki, and Y. Taga: Thermochim. Acta, 2004, vol. 410(1-2), pp. 27–34. https://doi.org/10.1016/s0040-6031(03)00366-6
18) S. Komatsuzaki, K. Takase, C.F. Smura, Y. Takahashi, Y. Takano, K. Sekizawa, S.J. Clarke, J. Alloys Compd., 2006, vol. 408-412, pp. 586-588. https://doi.org/10.1016/j.jallcom.2004.12.058
N. Sato, G. Shinohara, A. Kirishima, and O. Tochiyama: J. Alloys Compd., 2008, vol. 451(1-2), pp. 669–72. https://doi.org/10.1016/j.jallcom.2007.04.100
H. Yuan, J. Zhang, R. Yu, and Q. Su: J. Rare earths, 2013, vol. 31(3), pp. 327–30. https://doi.org/10.1016/s1002-0721(12)60280-4
O.M. Saad, T. Kuzuya, S. Hirai, and M. Ohta: Mater. Trans., 2010, vol. 51(12), pp. 2289–93. https://doi.org/10.2320/matertrans.m2010157
22) Michihiro Ohta, Shuhei Satoh, Toshihiro Kuzuya, Shinji Hirai, Masaru Kunii, Atsushi Yamamoto, Acta Mater., 2012, vol. 60, pp. 7232-40. https://doi.org/10.1016/j.actamat.2012.09.035
M. Ohta, P. Jood, M. Murata, C-H. Lee, A. Yamamoto, and H. Obara: Adv. Energy Mater., 2019, vol. 9(23), 1801304. https://doi.org/10.1002/aenm.201801304
24) Emmanuel Guilmeau, Antoine Maignan, Chunlei Wan, Kunihito Koumoto, Phys. Chem. Chem. Phys., 2015, vol. 17, pp. 24541-24555. https://doi.org/10.1039/c5cp01795e
25) Yuta Kikuchi, Yohan Bouyrie, Michihiro Ohta, Koichiro Suekuni, Makoto Aihara, Toshiro Takabatake, J. Mater. Chem. A, 2016, vol. 4, pp.15207-15214. https://doi.org/10.1039/c6ta05945g
E. Ahmadi, Y. Yashima, R.O. Suzuki, and S.A. Rezan: Metall. Mater. Trans. B, 2018, vol. 49(4), pp. 1808–21. https://doi.org/10.1007/s11663-018-1278-8
E. Ahmadi and R.O. Suzuki: Metall. Mater. Trans. B, 2020, vol. 51(1), pp. 140–48. https://doi.org/10.1007/s11663-019-01730-w
N. Suzuki, M. Tanaka, H. Noguchi, S. Natsui, T. Kikuchi, and R.O. Suzuki: Mater. Trans., 2017, vol. 58(3), pp. 367–70. https://doi.org/10.2320/matertrans.mk201613
T. Matsuzaki, S. Natsui, T. Kikuchi, and R.O. Suzuki: Mater. Trans., 2017, vol. 58(3), pp. 371–76. https://doi.org/10.2320/matertrans.m2016305
30) Takumi Kaneko, Yuta Yashima, Eltefat Ahmadi, Shungo Natsui, Ryosuke O. Suzuki, J. Solid State Chem., 2020, vol. 285, 121268. https://doi.org/10.1016/j.jssc.2020.121268
R.F. Barrow and W. Jevons: Proc. R. Soc. London A, 1938, vol. 169(936), pp. 45–65. https://doi.org/10.1098/rspa.1938.0194
R. Yokoyama: Japanese patent, S26-3067 filed on 1951.6.13.
M. Egashira, H. Katsuki, M. Mori, H. Kaneko, W. Kurahashi, and S. Kawasumi: Yogyo Kyokai Shi (Bull. Jpn. Ceram. Soc.), 1985, vol. 93(9), pp. 535–40. https://doi.org/10.2109/jcersj1950.93.1081_535
M. Egashira, Y. Ohyama, and H. Katsuki: J. Ceram. Soc. Jpn., 1988, vol. 96(1110), pp. 171–75. https://doi.org/10.2109/jcersj.96.171
D.I. Bletskan: J. Ovonic Research, 2005, vol. 1(5), pp. 47–52. http://www.chalcogen.ro/Bletskan.pdf
A. Roine: HSC Chemistry, 8.0.8, OutoTec Technologies, Pori, Finland, 2014.
NIST-JANAF thermochemical tables, 4th ed. Part.II, Cr-Zr, (Journal of physical and chemical reference data, Monograph: No. 9), ed. by Malcolm W. Chase, Jr., National Institute of Standards and Technology, Gaithersburg, MD USA, 1998, p.1867, p.1871 and p.1872.
E. Tiede and H. Thimann: Ber., 1926, 59 II B p.1703. Quoted in Ref. 47.
E.J. Kohlmeyer and H.W. Retzlaff: Z. anorg. Chem., 1950, vol. 261(3-4), pp. 248–60. https://doi.org/10.1002/zaac.19502610314
40) Chen Chen, Xiaotian Zhang, Lakshmi Krishna, Chito Kendrick, Shun-Li Shang, Eric Toberer, Zi-Kui Liu, Adele Tamboli, Joan M. Redwing, J. Crystal Growth, 2016, vol. 452, pp. 151-157. https://doi.org/10.1016/j.jcrysgro.2015.12.005
A. Haas: Angew. Chem., 1965, vol. 77, pp. 1066–75; Angew. Chem. Int. Ed. Engl., 1965, vol. 4(12), pp. 1014-1023. https://doi.org/10.1002/anie.196510141
P.A.G. O’Hare: J. Chem. Thermodyn., 1987, vol. 19(7), pp. 675–701. https://doi.org/10.1016/0021-9614(87)90090-5
M.S. Chandrasekharaiah and J.L. Margrave: J. Phys. Chem. Ref. Data, 1994, vol. 23, pp. 499–507. https://doi.org/10.1063/1.555960
J. L. Murray, A. J. McAlister, Bulletin of Alloy Phase Diagrams, 1984, vol. 5, pp. 74-84. https://doi.org/10.1007/bf02868729
W.C. Schumb and W.J. Bernard: J. Am. Chem. Soc., 1955, vol. 77(4), pp. 904–05. https://doi.org/10.1021/ja01609a029
46) H.-H. Emons, L. Theisen, Monat. Chemie, 1972, vol. 103, pp. 62-71. https://doi.org/10.1007/bf00912928
Handbook of Chemistry and Physics, 39th ed., Chemical Rubber Publ. Co., 1957–58, p. 598 quoted in Ref. 8.
H. Gabriel and C. Alvarez-Tostado: J. Am. Chem. Soc., 1952, vol. 74(1), pp. 262–64. https://doi.org/10.1021/ja01121a513
49) P. Coppens, S. Smoes and J. Drowart, Trans. Faraday Soc., 1967, vol. 63, pp. 2140-2148. https://doi.org/10.1039/tf9676302140
50) R.J. Fruehan, E.T. Turkdogan, Metall. Trans., 1971, vol. 2, pp. 895-902. https://doi.org/10.1007/bf02662752
51) M. Bonnet, J. Rogez, R. Castanet, Thermochimica Acta, 1989, vol. 155, pp. 39-56.
H. Yoshida, M. Matsubara, and S. Yoshida: Kagaku Kougaku Ronbunshu, 1992, vol. 18(3), pp. 281–87. https://doi.org/10.1252/kakoronbunshu.18.281
Acknowledgments
The authors thank the technical supports of SEM observation by Mr. N. Miyazaki. A part of this work was conducted at Laboratory of XPS analysis, Joint-use facilities, Hokkaido University, and supported by “Nanotechnology Platform” Program of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan. This work was also financially supported by Grant-in-Aid from MEXT (KAKENHI) under contacts Nr. 17H0343409 and 18F1805409 and from Japan Mining Industries Association. One of the authors (E.A.) acknowledges a fellowship from JSPS postdoctoral program (P18054).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Manuscript submitted April 5, 2020; accepted January 29, 2021.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Suzuki, R.O., Yashima, Y., Kaneko, T. et al. Synthesis of Silicon Sulfide by Using CS2 Gas. Metall Mater Trans B 52, 1379–1391 (2021). https://doi.org/10.1007/s11663-021-02103-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11663-021-02103-y