
Copy Number Variations of Glycoside Hydrolase 45 Genes in Bursaphelenchus xylophilus and Their Impact on the Pathogenesis of Pine Wilt Disease

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling and Inoculation Test
2.2. Identification and Quantification of Multi-Copy GH45 Genes
2.3. Detection of Copy Number Variations
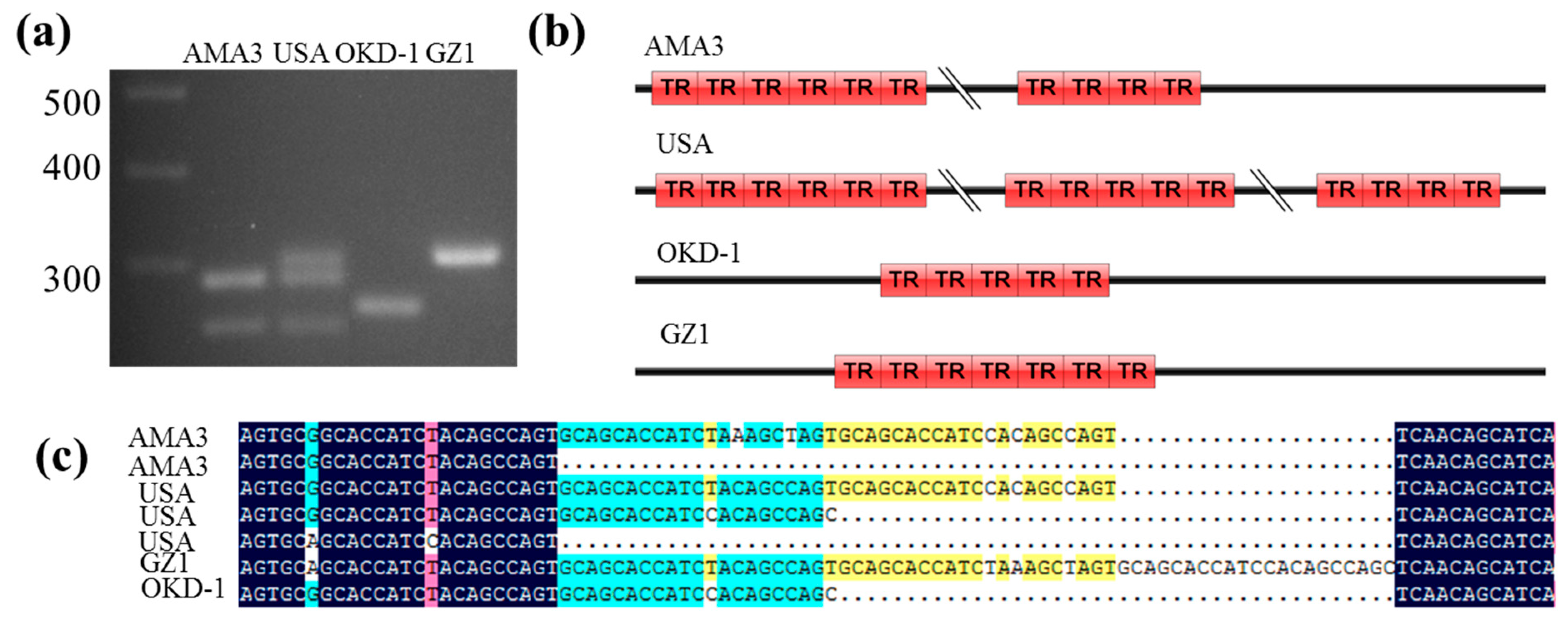
3. Results
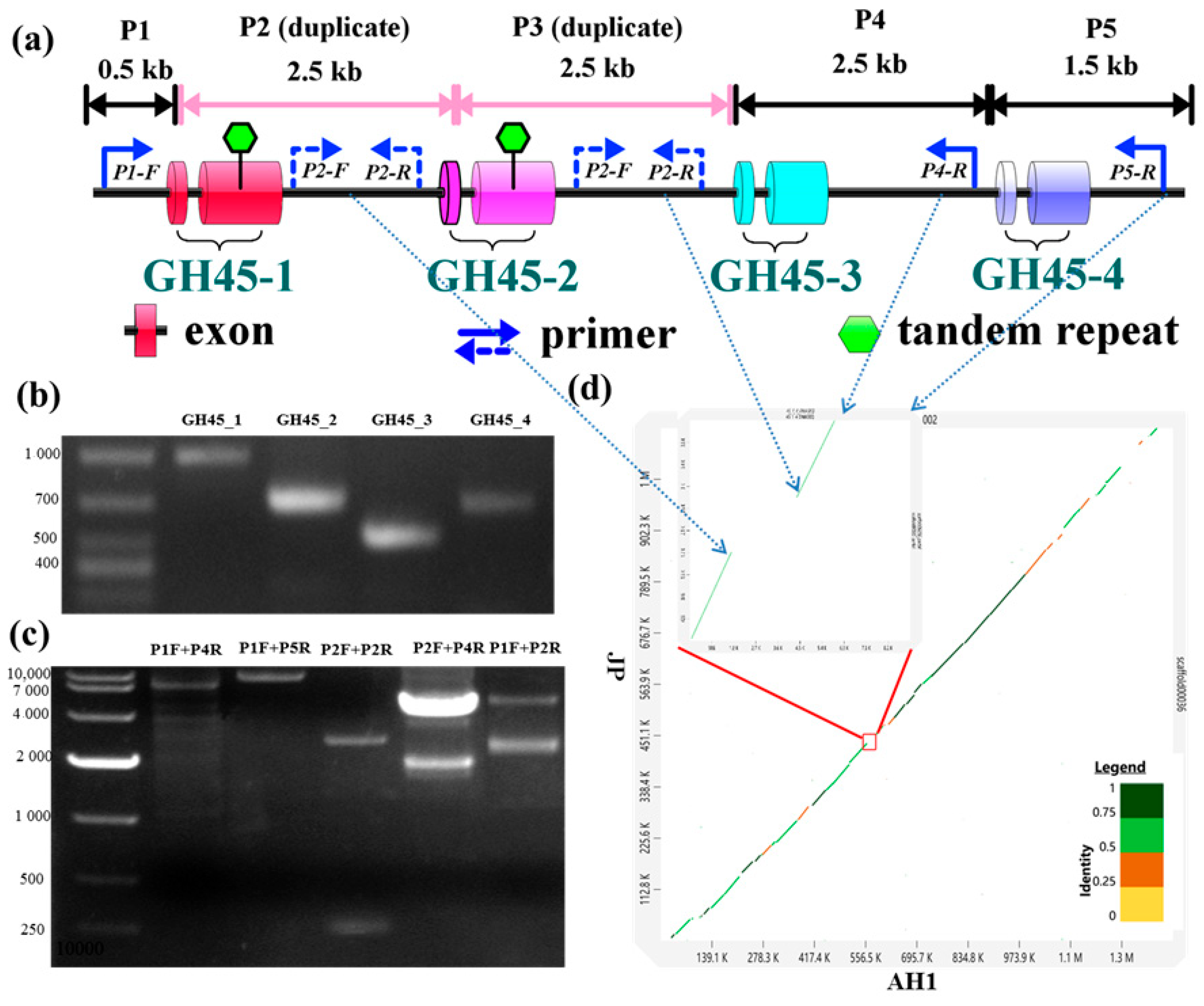
3.1. Identification of Novel HGT Gene Families, Glycoside Hydrolase 45
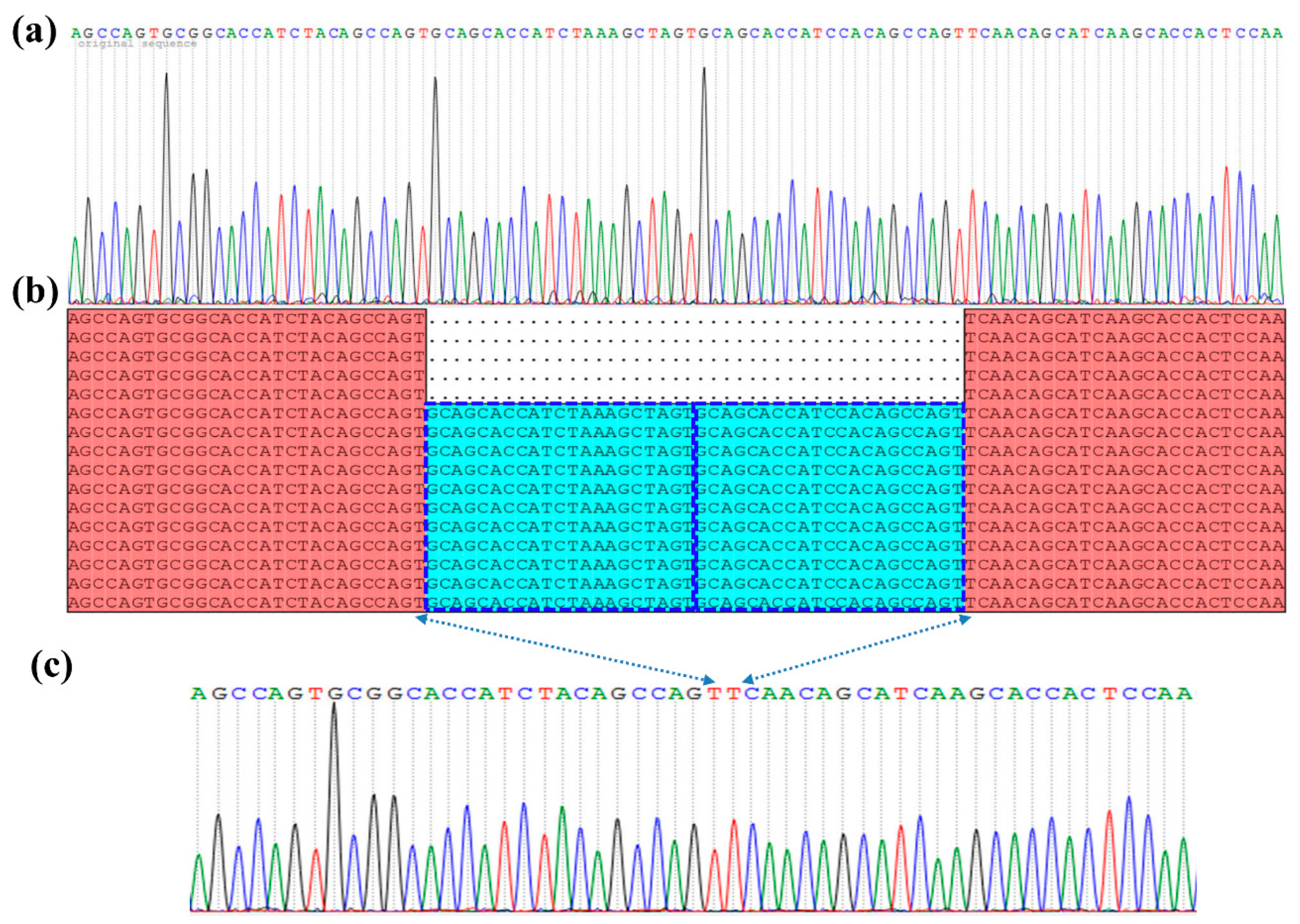
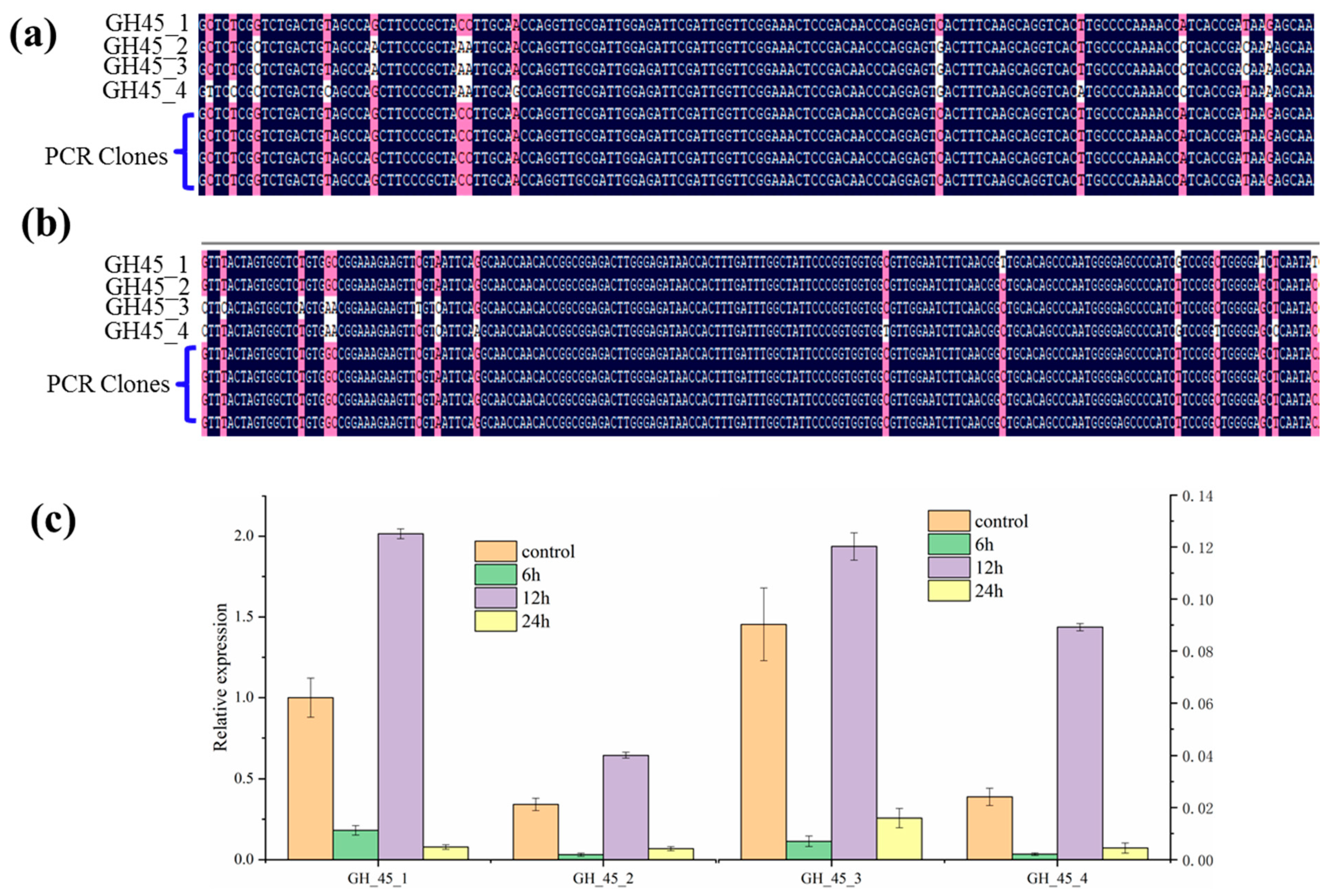
3.2. Sequence Characterizations of Different GH45 Copies
3.3. Expression Profiling of Novel Multi-Copy GH45 Genes
3.4. Copy Number Variations of GH45 Genes Among Different B. xylophilus Strains
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mamiya, Y. Pathology of the pine wilt disease caused by Bursaphelenchus xylophilus. Annu. Rev. Phytopathol. 1983, 21, 201–220. [Google Scholar] [CrossRef] [PubMed]
- Soliman, T.; Mourits, M.C.M.; van der Werf, W.; Hengeveld, G.M.; Robinet, C.; Lansink, A.G.J.M.O. Framework for modelling economic impacts of invasive species, applied to pine wood nematode in Europe. PLoS ONE 2012, 7, e45505. [Google Scholar] [CrossRef]
- Espada, M.; Eves-van den Akker, S.; Maier, T.; Vijayapalani, P.; Baum, T.; Mota, M.; Jones, J.T. STATAWAARS: A promoter motif associated with spatial expression in the major effector-producing tissues of the plant-parasitic nematode Bursaphelenchus xylophilus. BMC Genom. 2018, 19, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Espada, M.; Silva, A.C.; Eves van den Akker, S.; Cock, P.J.; Mota, M.; Jones, J.T. Identification and characterization of parasitism genes from the pinewood nematode Bursaphelenchus xylophilus reveals a multilayered detoxification strategy. Mol. Plant Pathol. 2016, 17, 286–295. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, S.E.; Dayi, M.; Maeda, Y.; Tsai, I.J.; Tanaka, R.; Bligh, M.; Takeuchi-Kaneko, Y.; Fukuda, K.; Kanzaki, N.; Kikuchi, T. Stage-specific transcriptome of Bursaphelenchus xylophilus reveals temporal regulation of effector genes and roles of the dauer-like stages in the lifecycle. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Tsai, I.J.; Tanaka, R.; Kanzaki, N.; Akiba, M.; Yokoi, T.; Espada, M.; Jones, J.T.; Kikuchi, T. Transcriptional and morphological changes in the transition from mycetophagous to phytophagous phase in the plant-parasitic nematode Bursaphelenchus xylophilus. Mol. Plant Pathol. 2016, 17, 77–83. [Google Scholar] [CrossRef]
- Kikuchi, T.; Cotton, J.A.; Dalzell, J.J.; Hasegawa, K.; Kanzaki, N.; McVeigh, P.; Takanashi, T.; Tsai, I.J.; Assefa, S.A.; Cock, P.J.A.; et al. Genomic insights into the origin of parasitism in the emerging plant pathogen Bursaphelenchus xylophilus. PLoS Pathog 2011, 7, e1002219. [Google Scholar] [CrossRef] [Green Version]
- Figueiredo, J.; Simoes, M.J.; Gomes, P.; Barroso, C.; Pinho, D.; Conceicao, L.; Fonseca, L.; Abrantes, I.; Pinheiro, M.; Egas, C. Assessment of the geographic origins of pinewood nematode isolates via single nucleotide polymorphism in effector genes. PLoS ONE 2013, 8, e83542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palomares-Rius, J.E.; Tsai, I.J.; Karim, N.; Akiba, M.; Kato, T.; Maruyama, H.; Takeuchi, Y.; Kikuchi, T. Genome-wide variation in the pinewood nematode Bursaphelenchus xylophilus and its relationship with pathogenic traits. BMC Genom. 2015, 16, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.X.; Meng, F.L.; Deng, X.; Wang, X.; Feng, Y.Q.; Zhang, W.; Pan, L.; Zhang, X.Y. Comparative transcriptome analysis of the pinewood nematode Bursaphelenchus xylophilus reveals the molecular mechanism underlying its defense response to host-derived -pinene. Int. J. Mol. Sci. 2019, 20, 911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, X.L.; Ye, J.R.; Wu, X.Q.; Huang, L.; Zhu, L.H.; Lin, S.X. Deep sequencing analyses of pine wood nematode Bursaphelenchus xylophilus microRNAs reveal distinct miRNA expression patterns during the pathological process of pine wilt disease. Gene 2015, 555, 346–356. [Google Scholar] [CrossRef]
- Ding, X.; Ye, J.; Lin, S.; Wu, X.; Li, D.; Nian, B. Deciphering the molecular variations of pine wood nematode Bursaphelenchus xylophilus with different virulence. PLoS ONE 2016, 11, e0156040. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, T.; Jones, J.T.; Aikawa, T.; Kosaka, H.; Ogura, N. A family of glycosyl hydrolase family 45 cellulases from the pine wood nematode Bursaphelenchus xylophilus. FEBS Lett. 2004, 572, 201–205. [Google Scholar] [CrossRef] [Green Version]
- Palomares-Rius, J.E.; Hirooka, Y.; Tsai, I.J.; Masuya, H.; Hino, A.; Kanzaki, N.; Jones, J.T.; Kikuchi, T. Distribution and evolution of glycoside hydrolase family 45 cellulases in nematodes and fungi. BMC Evol. Biol. 2014, 14, 69. [Google Scholar] [CrossRef] [Green Version]
- O’Dushlaine, C.T.; Edwards, R.J.; Park, S.D.; Shields, D.C. Tandem repeat copy-number variation in protein-coding regions of human genes. Genome Biol. 2005, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Brahmachary, M.; Guilmatre, A.; Quilez, J.; Hasson, D.; Borel, C.; Warburton, P.; Sharp, A.J. Digital genotyping of macrosatellites and multicopy genes reveals novel biological functions associated with copy number variation of large tandem repeats. PLoS Genet 2014, 10, e1004418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orgel, L.E.; Crick, F.H. Selfish DNA: The ultimate parasite. Nature 1980, 284, 604–607. [Google Scholar] [CrossRef]
- Duda, T.F., Jr.; Palumbi, S.R. Molecular genetics of ecological diversification: Duplication and rapid evolution of toxin genes of the venomous gastropod Conus. Proc. Natl. Acad. Sci. USA 1999, 96, 6820–6823. [Google Scholar] [CrossRef] [Green Version]
- Verstrepen, K.J.; Jansen, A.; Lewitter, F.; Fink, G.R. Intragenic tandem repeats generate functional variability. Nat. Genet. 2005, 37, 986–990. [Google Scholar] [CrossRef] [Green Version]
- Gymrek, M.; Willems, T.; Guilmatre, A.; Zeng, H.Y.; Markus, B.; Georgiev, S.; Daly, M.J.; Price, A.L.; Pritchard, J.K.; Sharp, A.J.; et al. Abundant contribution of short tandem repeats to gene expression variation in humans. Nat. Genet. 2016, 48, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Quilez, J.; Guilmatre, A.; Garg, P.; Highnam, G.; Gymrek, M.; Erlich, Y.; Joshi, R.S.; Mittelman, D.; Sharp, A.J. Polymorphic tandem repeats within gene promoters act as modifiers of gene expression and DNA methylation in humans. Nucleic Acids Res. 2016, 44, 3750–3762. [Google Scholar] [CrossRef] [Green Version]
- Cantsilieris, S.; Baird, P.N.; White, S.J. Molecular methods for genotyping complex copy number polymorphisms. Genomics 2013, 101, 86–93. [Google Scholar] [CrossRef] [Green Version]
- Dayi, M.; Sun, S.; Maeda, Y.; Tanaka, R.; Yoshida, A.; Tsai, I.J.; Kikuchi, T. Nearly Complete Genome Assembly of the Pinewood Nematode Bursaphelenchus xylophilus Strain Ka4C1. Microbiol. Res. Announc. 2020, 9, e01002–e01020. [Google Scholar] [CrossRef]
- Fan, H.; Chu, J.Y. A brief review of short tandem repeat mutation. Genom. Proteom. Bioinform. 2007, 5, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Paques, F.; Leung, W.Y.; Haber, J.E. Expansions and contractions in a tandem repeat induced by double-strand break repair. Mol. Cell. Biol. 1998, 18, 2045–2054. [Google Scholar] [CrossRef] [Green Version]
- Fiston-Lavier, A.S.; Anxolabehere, D.; Quesneville, H. A model of segmental duplication formation in Drosophila melanogaster. Genome Res. 2007, 17, 1458–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fondon, J.W., 3rd; Garner, H.R. Molecular origins of rapid and continuous morphological evolution. Proc. Natl. Acad. Sci. USA 2004, 101, 18058–18063. [Google Scholar] [CrossRef] [Green Version]
- Nasrallah, M.P.; Cho, G.; Simonet, J.C.; Putt, M.E.; Kitamura, K.; Golden, J.A. Differential effects of a polyalanine tract expansion in Arx on neural development and gene expression. Hum. Mol. Genet. 2012, 21, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, N.; Yoshida, H.; Kumaki, Y.; Kamiya, M.; Tanaka, T.; Izumi, Y.; Kretsinger, R.H. Flexible structures and ligand interactions of tandem repeats consisting of proline, glycine, asparagine, serine, and/or threonine rich oligopeptides in proteins. Curr. Protein Pept. Sci. 2008, 9, 591–610. [Google Scholar] [CrossRef] [PubMed]
- Young, E.T.; Sloan, J.S.; Van Riper, K. Trinucleotide repeats are clustered in regulatory genes in Saccharomyces cerevisiae. Genetics 2000, 154, 1053–1068. [Google Scholar]
- Yu, L.-Z.; Wu, X.-Q.; Ye, J.-R.; Zhang, S.-N.; Wang, C. NOS-like-mediated nitric oxide is involved in Pinus thunbergii response to the invasion of Bursaphelenchus xylophilus. Plant Cell Rep. 2012, 31, 1813–1821. [Google Scholar] [CrossRef]
- Mallez, S.; Castagnone, C.; Espada, M.; Vieira, P.; Eisenback, J.D.; Harrell, M.; Mota, M.; Aikawa, T.; Akiba, M.; Kosaka, H.; et al. Worldwide invasion routes of the pinewood nematode: What can we infer from population genetics analyses? Biol. Invasions 2015, 17, 1199–1213. [Google Scholar] [CrossRef] [Green Version]
- Takemoto, S.; Kanzaki, N.; Futai, K. PCR-RFLP image analysis: A practical method for estimating isolate-specific allele frequency in a population consisting of two different strains of the pinewood nematode, Bursaphelenchus xylophilus (Aphelenchida: Aphelencoididae). Appl. Entomol. Zool. 2005, 40, 529–535. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.Y.; Cheng, F.X.; Xu, R.M.; Xie, B.Y. Genetic variation in the invasive process of Bursaphelenchus xylophilus (Aphelenchida: Aphelenchoididae) and its possible spread routes in China. Heredity 2008, 100, 356–365. [Google Scholar] [CrossRef]
- Zhou, L.F.; Chen, F.M.; Xie, L.Y.; Pan, H.Y.; Ye, J.R. Genetic diversity of pine-parasitic nematodes Bursaphelenchus xylophilus and Bursaphelenchus mucronatus in China. Forest Pathol. 2017, 47, e12334. [Google Scholar] [CrossRef]
- Pereira, F.; Moreira, C.; Fonseca, L.; van Asch, B.; Mota, M.; Abrantes, I.; Amorim, A. New insights into the phylogeny and worldwide dispersion of two closely related nematode species, Bursaphelenchus xylophilus and Bursaphelenchus mucronatus. PLoS ONE 2013, 8, e56288. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.; Han, H.; Ryu, S.H.; Kim, W. Microsatellite variation in the pinewood nematode, Bursaphelenchus xylophilus (Steiner and Buhrer) Nickle in South Korea. Genes Genom. 2010, 32, 151–158. [Google Scholar] [CrossRef]
- Vieira, P.; Castagnone, C.; Mallez, S.; Espada, M.; Navas, A.; Mota, M.; Castagnone-Sereno, P. Sequence variability of the Mspi satellite DNA family of the pinewood nematode Bursaphelenchus xylophilus at different geographic scales. J. Nematol. 2013, 45, 324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, M.W. Distinguishing Among Evolutionary Models for the Maintenance of Gene Duplicates. J. Hered. 2009, 100, 605–617. [Google Scholar] [CrossRef] [Green Version]
- Redon, R.; Ishikawa, S.; Fitch, K.R.; Feuk, L.; Perry, G.H.; Andrews, T.D.; Fiegler, H.; Shapero, M.H.; Carson, A.R.; Chen, W.; et al. Global variation in copy number in the human genome. Nature 2006, 444, 444–454. [Google Scholar] [CrossRef] [Green Version]
- Sackton, T.B.; Lazzaro, B.P.; Schlenke, T.A.; Evans, J.D.; Hultmark, D.; Clark, A.G. Dynamic evolution of the innate immune system in Drosophila. Nat. Genet. 2007, 39, 1461–1468. [Google Scholar] [CrossRef]
- Kontogiannatos, D.; Swevers, L.; Kourti, A. Recent gene multiplication and evolution of a juvenile hormone esterase-related gene in a lepidopteran pest. Gene Rep. 2016, 4, 139–152. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GH45_1 | GH45_2 | GH45_3 | GH45_4 | |
|---|---|---|---|---|
| Length (bp) | 1177 | 1091 | 912 | 1101 |
| Exon number | 2 | 2 | 2 | 2 |
| SP site * | Yes | Yes | NA | NA |
| Copies of TR | 6 | 4 | NA | NA |
| ORF Length | 1056 | 1014 | 162 | 288 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, X.; Wang, Q.; Guo, Y.; Li, Y.; Lin, S.; Zeng, Q.; Sun, F.; Li, D.-W.; Ye, J. Copy Number Variations of Glycoside Hydrolase 45 Genes in Bursaphelenchus xylophilus and Their Impact on the Pathogenesis of Pine Wilt Disease. Forests 2021, 12, 275. https://doi.org/10.3390/f12030275
Ding X, Wang Q, Guo Y, Li Y, Lin S, Zeng Q, Sun F, Li D-W, Ye J. Copy Number Variations of Glycoside Hydrolase 45 Genes in Bursaphelenchus xylophilus and Their Impact on the Pathogenesis of Pine Wilt Disease. Forests. 2021; 12(3):275. https://doi.org/10.3390/f12030275
Chicago/Turabian StyleDing, Xiaolei, Qingtong Wang, Yunfei Guo, Yulong Li, Sixi Lin, Qingwei Zeng, Feijian Sun, De-Wei Li, and Jianren Ye. 2021. "Copy Number Variations of Glycoside Hydrolase 45 Genes in Bursaphelenchus xylophilus and Their Impact on the Pathogenesis of Pine Wilt Disease" Forests 12, no. 3: 275. https://doi.org/10.3390/f12030275