
Abstract
Worldwide, black shales and shale waste are known to be a potential source of metals to the environment. This project demonstrates ongoing weathering and evaluates leaching processes at a 100-m-high shale waste deposit closed in the 1960s. Some deep parts of the deposit are still burning with temperatures exceeding 500 °C. To demonstrate ongoing weathering and leaching, analyses of groundwater and solid samples of shale and shale waste have been undertaken. Largest impact on groundwater quality was observed downstream the deposit, where elevated temperatures also indicate a direct impact from the burning waste deposit. Groundwater quality is largely controlled by pH and redox conditions (e.g., for arsenic, nickel, molybdenum, uranium and vanadium), and the mixture of different waste materials, including pyrite (acidic leachates) and carbonates (neutralizing and buffering pH). Analyses of shale waste from the deposit confirm the expected pyrite weathering with high concentrations of iron, nickel and uranium in the leachates. No general time trends could be distinguished for the groundwater quality from the monitoring in 2004–2019. This study has shown that black shale waste deposits can have a complex long-term impact on the surrounding environment.
Similar content being viewed by others

Introduction
Black shale is the denomination of a heterogeneous group of dark-coloured sedimentary rocks containing organic matter (Vine and Tourtelot 1969) which to various extents are enriched in sulfides and metals. Ketris and Yudovich (2009) have made estimates of median concentrations based on reported data in black shales globally, e.g., 30 mg/kg for arsenic, 20 mg/kg for molybdenum, 70 mg/kg for nickel, 8.5 mg/kg for uranium and 205 mg/kg for vanadium. Weathering and leaching of shale residues on mining sites and from shale waste dumps lead to release of elements resulting in local and regional environmental impacts. This is reported from numerous sites worldwide, e.g., in Denmark, Norway, Finland, Estonia and Germany in Europe and also from the USA and China (e.g., Loukola-Ruskeeniemi et al. 1998; Puura 1998; Puura et al. 1999; Woo et al. 2002; Peng et al. 2004; Burkhardt et al. 2009; Grawunder et al. 2009; Jüriado et al. 2012; Paikaray 2012; Peng et al. 2014; Schovsbo et al. 2014; Phan et al. 2015; Liu et al. 2017; Lerat et al. 2018; Parviainen and Loukola-Ruskeeniemi 2019; Stuckman et al. 2020; Waersted et al. 2020).
The black shales in Sweden (denoted alum shales) are mainly composed of muscovite-illite, quartz and feldspar with up to 15% pyrite and up to 20% organic material (kerogen). The shales are enriched in trace elements such as molybdenum, nickel, uranium and vanadium (Armands 1972; Andersson et al. 1985). Mining of alum shale from the 18th to the 20th centuries has led to dispersion of elements from weathering of shale residues, still in progress, notably at Degerhamn, Ranstad and Kvarntorp (e.g., Allard et al. 1991, 2011; Falk et al. 2006; Kalinowski et al. 2006; Lavergren 2008; Lavergren et al. 2009; Åhlgren et al. 2020).
In Kvarntorp, 200 km south-west of Stockholm, Sweden, alum shale was mined in open pits and processed for oil production during 1942–1966. Most of the shale residues from the mining period (unprocessed fine-grained shale denoted fines, semi-combusted shale denoted semi-coke and fully burned shale denoted shale ash, as well as minor amounts of limestone) were deposited on site, either backfilling of open pits, or on a single deposit, denoted Kvarntorpshögen (the Kvarntorp waste deposit). The deposit is presently 100 m high and covers an area of 450,000 m2. It is estimated that the deposit contains 3 Mtonnes of fines, 2 Mtonnes of semi-coke and 23 Mtonnes of shale ash, as well as limestone in some parts (Bäckström 2010). The deposit was closed in 1966, but temperatures are still elevated, caused by pyrite oxidation in the fines (temperatures up to 100 °C), as well as kerogen oxidation (temperatures above 500 °C).
It is of general interest to monitor the release of leachates from shale residues, particularly from well-defined deposits, for several reasons: (1) assessment of present and future environmental impact and health risks associated with spreading of elements from the deposit and (2) enhanced understanding of relevant processes—weathering and leaching in situ, element mobilization and transport, element accumulation in soil and sediments.
The Kvarntorp site is of interest for extended studies of metal releases from alum shale in situ for several reasons: (1) the alum shale in Kvarntorp generally contains higher trace element concentrations than the global median levels except for arsenic which is quite similar and nickel with large variations in concentration (cf. Allard et al. 2011). (2) There is a continuously progressing weathering of shale in the deposit. Oxidation of pyrite and other metal sulfides leads to releases of metals by run-off and into the local groundwater aquifers. This has been going on for more than 50 years. (3) The deposit is still burning which means that large parts of the deposit are dry. Increased leaching of metals from the area is expected after cooling of the deposit and water exposure to parts that presently are dry. (4) It may be possible to assess the reactivity and leachability in situ of different shale fractions—fines, semi-coke and shale ash. (5) No remediation activities have been undertaken to control and reduce metal releases from the deposit since the end of mining 54 years ago. Element immobilization due to sorption and secondary mineral formations are important factors that can be demonstrated, and internal drainage mixing processes can be assessed (cf. Vriens et al. 2019).
The objective of the present project is to demonstrate the ongoing weathering and leaching processes in the Kvarntorp alum shale waste deposit and the resulting impact on the local and regional groundwater quality. Present and long-term impacts on groundwater chemistry are assessed largely based on groundwater quality monitoring in the vicinity of the deposit starting in 2004.
Description of the area
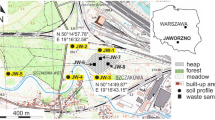
The location of Kvarntorp in Sweden is shown in Fig. 1a, as well as a map showing the mining area with the shale waste deposit (Kvarntorpshögen), pit lakes and watercourses leading in to and out from the area (Fig. 1b).

a Map showing the location of Kvarntorp. b Map showing the Kvarntorp area with the waste deposit Kvarntorpshögen (K) and the pit lake Norrtorpssjön (N), location for sampling of shale and limestone. c Extent of the waste deposit from 1947 to 1966, when operations ended. Final size: 100 m high, covers an area of 450,000 m2, contains some 3 Mtonnes of fines, 2 Mtonnes of semi-coke and 23 Mtonnes of shale ash. d Location of groundwater wells G1–G11 around the deposit and locations for sampling of solid samples (A, B = shale ash; C, D = fines (D used in humidity cells, Sartz et al. in prep), E = shale ash (red ash used in humidity cells) and F = shale ash (black ash used in humidity cells)). Average levels for groundwater depths in the wells (metres above sea level) are given. (Maps are processed with the software QGIS Desktop 2.14.13, with data from Lantmäteriet (Swedish Land Survey) as a basis)
The crystalline basement in the area is superimposed by lower Cambrian Mickwitzia sandstone followed by Lingulid sandstone beneath layers of phosphorite and glauconite sandstone. Higher up in the strata, green shale (mudstone) is followed by late Cambrian alum shale and Ordovician limestone covered by till on top (Bengtson 1971; Nielsen and Schovsbo 2007, 2015). The uppermost and youngest layers have been eroded and are not represented all the way to the northern limit of the sedimentary outlier since the bedrock is tilted towards the south (Hessland and Armands 1978).
A marshy meadow in a small valley in the northern part of the area contains no alum shale. The top layer of the bedrock is a 5- to 6-m-thick layer of the green shale above the sandstone layers, covered by 4–5 m of till. This area became the location for the shale waste deposit (Kvarntorpshögen) in 1942. The green shale constitutes an almost impermeable layer under the deposit that largely prevents water exchange between the deposit and the sandstone aquifer (Holm et al. 2005). Fig. 1c shows the evolution of the deposit during 1947–1966. The exact locations in the deposit and the quantities of the various shale residues (fines, semi-coke and shale ash) are not known since the records about the waste are sparse or non-existing.
The locations of groundwater wells G1–G11 around the deposit are given in Fig. 1d. These wells were installed in 1997 (G11), 2003 (G4 and G5) and 2004 (G1–G3 and G6–G10).
Material and methods
Shale sampling
Solid samples, two shale ashes (A and B) and fines (C), were collected from the waste deposit (Åhlgren et al. 2018) (see Fig. 1d for locations) and from an outcrop at an open pit (five samples of alum shale and two samples of limestone; location N in Fig. 1b). An additional six samples of fines from the deposit were also analysed (see Fig. 1d for location) as well as three alum shale samples from old exploration drill cores kept by the Swedish Geological Survey.
Groundwater sampling
Two aquifers make up the groundwater in the area—a shallow aquifer in the till underneath and around the deposit with a dominant flow direction from south to north, as indicated by the water levels (Fig. 1d), and a lower aquifer in the sandstone layer (Bengtson 1971). Most of the groundwater wells (G1, G3–G6 and G8–G11) are shallow and installed in the upper aquifer, in the till layer and in the shale waste, while two wells (G2 and G7) are installed in the lower sandstone aquifer (Holm et al. 2005). G2 and G7 were installed using rotational drilling, and the contact between the alum shale aquifer and the sandstone aquifer was sealed with cement. Description of the wells is given in Table 1. Filter sections were generally 1 m long, and washed sand (grain size 0.6–1 mm) was added around the sections. Bentonite was added around the top section in order to prevent infiltration of surface water.
Bailer samplers were used for sampling of water, regularly since December 2015 in all of the wells, G1–G11 (six times annually from 2015 until October 2017 and then three times annually until 2019), totally 176 groundwater samples. Groundwater levels were measured, and the wells were emptied the day prior to sampling. Sterile polypropylene tubes were used for storing the samples at 5–6 °C until analysis. Groundwater temperature was measured in 2017. Additionally, temperature data for G1–G10 from previous studies in 2004 and 2005 were also available (Holm et al. 2005).
Two surface watercourses pass through the area, of which the westerly one is close to the groundwater wells north and north-east of the deposit and may receive run-off water from the deposit (Fig. 1b). A serpentine pond system north of the deposit was constructed in the 1940s, mainly in order to remove organic compounds such as phenols from the industrial process water outlets in the area but is also receiving surface run-off from the deposit. Water quality in the surface waters close to the deposit and in the pit lakes (Fig. 1b) is monitored regularly, as described in Åhlgren et al. (2020) and in Åhlgren and Bäckström (in preparation). The present field program reported here was, however, largely focusing on assessing the direct impact of leachates from the deposit on the local groundwater aquifers, and the impact on surface water quality is outside the present scope.
Analysis of solid samples
All solid samples were sent to MS Analytical (Vancouver) for total element quantification. Samples were fused with borate flux in a muffle furnace before the resulting beads were dissolved in dilute mineral acid. Inductively Coupled Plasma Optical Emission Spectrometry (ICP-OES) was then used for the analysis of major elements (Al, Ca, Fe, K, Mg, Mn and Na; for detection limits see Table 2). Samples were also digested in aqua regia or in a four-acid mixture and analysed for trace elements (As, Cd, Co, Cu, Mo, Ni, Pb, U and V) by ICP-MS. Total sulfur and carbon were quantified by a Leco carbon and sulfur analyser (for detection limits see Table 2).
Quantitative phase analysis by X-ray diffractometry (XRD) was made on the shale samples A, B and C from the waste deposit (Åhlgren et al. 2018). Grinding under ethanol in a vibratory McCrone Micronizing Mill during 10 minutes reduced the sample size to < 10 μm. Step-scan X-ray powder-diffraction data were collected over a range 3–80°2θ with CoKα radiation on a Bruker D8 Advance Bragg–Brentano diffractometer equipped with an Fe monochromator foil, 0.6-mm (0.3°) divergence slit, incident- and diffracted-beam Soller slits and a LynxEye-XE detector. The long fine-focus Co X-ray tube was operated at 35 kV and 40 mA, with a take-off angle of 6°. The X-ray diffractograms were analysed using the International Centre for Diffraction Database PDF-4 and Search-Match software by Bruker. The diffraction data were refined with Rietveld program Topas 4.2 (Bruker AXS).
Analysis of water samples
Electrical conductivity (Radiometer CDC836T-6, with temperature compensation), pH (Metrohm 6.0257.000 with temperature compensation), redox potential (during a few sampling occasions in 2017; Thermo Scientific REDOX/ORP 9678BNWP) and alkalinity were measured in the lab within 12 hours after sampling. Elements were quantified after filtration (0.20-μm polypropylene) and acidification (1 % nitric acid), by Inductively Coupled Plasma Mass Spectrometry (ICP-MS, Agilent 7500cx. Elements and detection limits are given in Table 3. Inorganic anions (chloride and sulfate) were quantified by capillary electrophoresis (2015–2017) using sodium chromate buffer (50 mM) containing TTAB (5 mM) and a 40 cm ∗ 50 μm silica capillary (from 2015 to 2017) or by ion chromatography, SS-EN ISO 10304-1:2009 (2018, 2019).
Ferrous iron (Fe(II)) was analysed in 2017 using photometry and the blue complex of Fe(II) and TPTZ (2,4,6-tri(2′-pyridyl)-1,3,5-triazin). A Hewlett Packard 8453 was used, and the absorbance at 595 nm was recorded. To 4 mL of sample was added 0.25-mL HCl (6 M), followed by the addition of 0.5-mL NH4F (2 M), 0.5-mL TPTZ (2.4 mM) and finally 1-mL buffer solution (NH4Ac (2 M)/HAc (2 M)). The sample was shaken vigorously between each addition. The mix was left for 10 minutes prior to measurement. Calibration was performed by analysis of standards containing Fe(II)(NH4)2-sulfate, prepared to cover a ferrous iron concentration range of 0.1–11 mg/L.
Chemical speciation calculations were performed on the measured concentrations in all groundwaters, using the geochemical code PHREEQC Interactive (version 3.4.0.12927; Parkhurst and Appelo 1999) with the MINTEQ.v4 database. Saturation indices (SI is the logarithm of the ratio between the ion activity product and the solubility constant) were also obtained from the geochemical calculations.
Principal component analysis (PCA) was performed on all groundwater data, in order to determine the relationship between the different waters, using The Unscrambler X (version 10.5) (Camo Software AS, Oslo, Norway). Data from humidity cells (day 7) performed on fines, red ash and black ash from the area (Sartz et al. in prep; sampling locations shown in Fig. 1d) were also included in the PCA in order to determine the impact from different waste materials on the groundwater composition. All data (except pH) was log transformed and normalized prior to calculations. PCA has previously been used to determine flow and relationship between different waters from mining sites (Bäckström and Sartz 2016).
Results
Composition of the shale
The mineralogic composition of the shale ash samples A and B and of the fine sample C, representing unprocessed shale, is given in Table 4 (crystalline phases, neglecting the organic fraction). Major mineral components in the fines are the silicates (quartz, feldspars and clay minerals), totally some 73% of the inorganic mass. The transformation of iron in the pyrite and marcasite in the fines into goethite, hematite and jarosite in the shale ash as a result of the pyrolysis is evident.
Element concentrations in fines and shale ashes from the deposit and in shale and limestone from the exposed outcrop (N, Fig. 1b) and from the exploration drill core are given in Table 5. The differences between the two shale ashes are significant (S, Fe and Ca among the major components) as well as the differences between the four shale samples (two fines and two alum shales; the same elements). However, concentrations in the shale vary in general between different layers (Andersson et al. 1985). Also, weathering and leaching of shale that is exposed in outcrops would lead to redistribution of elements and changes in composition (Chi Fru et al. 2016). Thus, the observed variations between the shale samples may reflect different locations, as well as differences in weathering state.
It is clear from the total concentrations in drill cores and shale ashes that processing of the shale varied and was incomplete in some cases. Concentrations of sulfur are for instance still quite high in shale ash A compared to shale ash B (Table 5). Sulfur is expected to be lost as SO2 during the pyrolysis process. Some trace elements could also be expected to be lost during the process, for instance, zinc that has a low boiling point (907 °C) and therefore could be volatilized during pyrolysis (Saqib and Bäckström 2014). Concentrations of zinc are lower in the ashes compared both to fines and drill core shale but on the same level as in the weathered shale from the pit site (Table 5).
Groundwater chemistry
Shale residues have been deposited on the same site since the early 1940s (see Fig. 1c). Surface waters, as well as local shallow groundwaters, must have been contaminated by shale leachates already since that time. There was, however, no regular monitoring of surface water or groundwater quality in the area until 1993, when monitoring started in the small stream north of the deposit, and not until 2003–2004 in groundwaters in the vicinity of the deposit (from wells G1–G10).
Major components
Concentrations of major components (cations: Na, K, Mg, Ca, Al, Mn and Fe; also Li and Sr; anions: HCO3− and SO42−; pH) from 2004 (Holm et al. 2005) and from 2015–2019, are given for selected groundwaters of different types (G1, G6, G8, G9 and G 10) in Fig. 2, which illustrates the variation of concentrations with time during the period 2004–2019.


Temporal changes for pH, concentrations of hydrolysable elements (Al, Mn and Fe) and major anions (HCO3 and SO4) in selected groundwater samples for 2004 (according to Holm et al. 2005) and for 2015–2019. Alkali metals (Li, Na and K) and alkaline earth metals (Mg, Ca and Sr)
All measurements during the period for all 11 wells G1–G11 are summarized in Table 6: median and min–max values for the sampling period 2015–2019, as well as median values from 2004 (Holm et al. 2005).
Variations in water chemistry between the groundwater wells are substantial, e.g., with pH ranging from 3.7 to 12 (from 3.2 to 12.2 in 2004), total carbonate from 0 to 900 mg/L and sulfate from 36 to 4400 mg/L during the sampling period. G1 has the lowest pH and is evidently highly affected by shale leachates with high concentrations of sulfate and iron, as well as trace elements (see below) throughout the entire time period.
Redox conditions for the groundwaters indicate two groups: G2, G6 and G8 (average ferrous iron concentration 0.2 mg/L and average redox potential 320 mV) and G1, G3, G4, G5, G7, G9, G10 and G11 (average ferrous iron concentration 8.6 mg/L and average redox potential 190 mV). The larger group is somewhat more reducing as indicated by both the redox potential and the dissolved ferrous iron concentrations.
From Table 6, and considering the depth and positions of the wells and the various layers around and below the wells (Table 1), it can be assumed that G2 and G7 may represent deep groundwater from the sandstone layer while G9 and G10 may represent shallow groundwater from the till surrounding the deposit. No alum shale waste was deposited nearby the G9 site, and the ground does not seem to be reworked since it is covered by undisturbed quaternary deposits (till). It is therefore assumed that G9 may represent local background conditions. Water from G10, however, also from the till horizon, appears to be affected by shale leachates as indicated by higher levels of major components (sulfate, potassium and magnesium in particular) and trace metals (Sr, Li, Co and Ni) and with slightly lower pH.
Trace elements
Concentrations of some of the trace elements (Co, Ni, Cu, Zn, V, As, Mo and U; also Li and Sr in Fig. 2) from 2004 and from 2015–2019 are given for the selected groundwaters (G1, G6, G8, G9 and G10) in Fig. 3.


Temporal changes for hydrolysable trace metals (Co, Cu, Ni and Zn) in selected groundwater samples for 2004 (according to Holm et al. 2005) and for 2015–2019. Redox sensitive trace elements (V, As, Mo and U)
Concentrations of the trace elements are summarized in Table 7 (as in Table 6): median and min–max values for the sampling period 2015–2019, as well as median values from 2004.
The large variation of concentrations between wells G1 and G11 for some of the trace elements and sulfate is further illustrated in Fig. 4, with concentration levels from October 2018. Highest levels (median values, 2015–2019) in groundwaters G1–G11 are obtained in G1 (with the lowest pH in the series) for Cd, Co, Ni, Zn and V, and the highest levels for Cu, Li, As and Mo are obtained in G3. Levels from the measurements in 2018 largely confirm this general trend (Fig. 3). The highest uranium levels are obtained in G8, in 2004, 2015–2019 and 2018. The differences in temperatures indicate that the deposit is hot, primarily in the north-eastern part (G1 and G3) and to some extent also in the north-western part (G5, G6 and G8).

Spatial distribution of concentrations for As, Fe, Mo, Ni, U, V and SO42− in groundwaters (except deep groundwater) around the waste deposit (cf. Fig. 1d). Concentration levels from October 2018 and water temperatures at 1-m depth from November 2017 (no temperature data available for G4 and G11)
Groundwater classification
Chemistry profiles
Analysis of the chemistry of groundwaters G1–G11 (pH, major components and trace elements, Figs. 2 and 3 and Tables 6 and 7) and combining with data on the location and positions of the wells (Fig. 1d and Table 1), as well as data on the composition of the shale waste categories (Table 5) may be the basis for classification of the groundwaters (cf. discussion under Results).
Groundwaters G2 and G7 come from the lower aquifer, in the sandstone layer. G7 seems to be unaffected by shale leachates from the deposit. G2 has higher levels of sulfate, lithium and strontium as well as higher pH, up to 10 or above in 2018 and in 2004, giving lower levels of manganese and iron. Alkaline pH may, however, indicate contact with cement used to seal the contact between the two aquifers, not observed in G7.
Groundwaters G6, G9 and G10 come from the upper aquifer, in the till layer. G9 seems to be largely unaffected by leachates from the deposit. The concentrations show a good correspondence with national values for unaffected groundwater where the span for the 5th to 95th percentile for, e.g., arsenic is 0.03–0.92, cobalt 0.01–1.2 and nickel 0.07–4.0 μg/L. Sulfate (median 200 mg/L in G9 compared to 45 mg/L) and iron (median 1.28 mg/L in G9 compared to 1.1 mg/L) surpass the 95th percentile for unaffected groundwater (SGU 2013). Median pH for G9 is 7.09. There is a possibility of natural presence of shale and pyrite in the till, which could explain the elevated sulfate concentrations that reflect the general background conditions at the Kvarntorp site.
Groundwaters G1 and G3 from the upper aquifer are clearly distinguished from the other groundwaters. Both have high sulfate, nickel and lithium concentrations and low pH indicating a progressing weathering of the shale waste. G1 has the water with the highest concentrations of iron (median 326 mg/L) and sulfate (median 3785 mg/L) and with the lowest pH during 2015–2019 (below 4). In 2004, also G3 was acidic (pH 3.6–4.3), but during 2015–2019, pH was higher, in the range 5.82 to 6.42. Pyrite containing shale and fines are expected to generate acidic leachates and subsequent metal releases, as documented in numerous laboratory tests and measurements in the field at shale mining sites (e.g., Bäckström 2010; Allard et al. 2011; Karlsson et al. 2012; Wilke et al. 2015; Chi Fru et al. 2016; Åhlgren et al. 2020; Sartz et al. in preparation). The data from G3 might indicate that the pyrite weathering has decreased, but increased electrical conductivity and higher sulfate and iron concentrations contradict this. Also, potassium and magnesium concentrations are higher in 2015–2019 compared to 2004. The pH increase may possibly be due to illite buffering, as has been reported elsewhere (Puura 1998) or carbonate buffering.
All other groundwaters, G4, G5, G8 and G11, can be considered till water (G9) from the upper aquifer contaminated with leachates from the shale wastes as indicated by the levels of trace elements from the shale. However, pH is generally above 6.5. This indicates and confirms that weathering of shale ashes, and possibly semi-coke, generates leachates that are not acidic. Weathering is progressing even in the absence of acid generating sulfides, in agreement with observations in laboratory leaching tests in progress (Sartz et al. in preparation). Progressing shale weathering has previously been observed in buffered aquatic systems in the field at the Kvarntorp site, with pH above 7–8 (Allard et al. 2011; Karlsson et al. 2013; Åhlgren et al. 2020).
Groundwater G6 belongs to the sequence G4, G5 and G8 but with the difference that median pH is 9.13 during 2015–2019 (maximum 12.0) and maximum 12.2 in 2004. There is no cement seal around the well that could be the source of alkalinity. Estonian oil shale ash is rich in free lime and anhydrite and produces alkaline leachates with pH 12–13 (Velts et al. 2011) which is generally not the case for the Kvarntorp shale ash which generally produces a circumneutral or slightly acidic leachate (Karlsson et al. 2012), and G6 is the only shallow groundwater well in Kvarntorp where alkaline conditions have been found. For the Estonian oil shale, it is the high content of limestone in the shale (calcite 44%) which when heated (800 or 1400 °C depending on method) generates calcium oxide and pH above 12 in contact with water (Pihu et al. 2019). The shale used for oil production in Kvarntorp is not as rich in carbonates as the Estonian oil shale. Calcite is not a major mineral component in the shale, but limestone waste was disposed at the Kvarntorp deposit together with the shale residues. The high pH found in G6 may reflect the presence of calcium hydroxide from the decomposition of calcium oxide originating from the degradation of calcium carbonate in the deposit, still burning with temperatures exceeding 500 °C. The pH variation in G2 from the lower aquifer (sandstone; between 6.89 and 10.0), however, could be caused by cement leaching from the sealing material (Table 1). Also G7 might have been influenced by cement resulting in much higher alkalinity and pH in 2004 compared to the period 2015–2019.
Groundwater G8 has the highest uranium levels, median values above 1 mg/L during 2015–2019. The uranium comes from sludges of possibly processed shale residues that are deposited close to the filter section of the pipe in the well.
PCA
A PCA based on data for all groundwater samples, as well as data from humidity cell leaching tests (Sartz et al., work in progress) is given in Fig. 5 (score plot) together with the loading plot showing element relationships.

a Principal component analysis (score plot) showing all groundwater samples and data from day 7 from the humidity cells (Sartz et al. in prep), where D = fines, E = red shale ash and F = black shale ash. The first two principal components explain 65% of all the variation in the data set. b Loading plot showing the relationship between the different elements
The PCA score plot confirms the assessments based on the measured concentrations in the groundwaters combined with information on the location and depths of the wells (Fig. 1d and Table 1). Almost all the groundwaters are unique, as illustrated by the score plot, although the G4, G5 and G8 groundwaters are fairly similar in composition. Principal component 1 (PC1, 54%) explains significantly more of the data variation compared to the second principal component (11%). Groundwaters G2 (average PC1 score − 3.43) and G7 (average PC1 score − 4.75) are located in the sandstone aquifer and G9 (average PC1 score − 3.12) is located in the till upstream the waste deposit. These unaffected groundwaters (compared to the other groundwaters in the area) have highly negative PC1 score values. Groundwaters G6 (average PC1 score − 0.93) and G11 (average PC1 score − 0.55) are slightly affected by contaminants and found at some distance from the deposit. Groundwater G10 (average PC1 score − 0.76) is affected by surface run-off from parts of the deposit with fines. Groundwaters G4 (average PC1 score 1.32), G5 (average PC1 score 0.27) and G8 (average PC1 score 1.65) are more closely located towards the deposit and are very tightly grouped together in the PCA. Groundwaters G1 (average PC1 score 5.35) and G3 (average PC1 score 3.89) are located towards the right (high PC1 scores) in the PCA plot and close to fines and black ash from the humidity cell experiments (D and F). This indicates a significant impact from the waste materials on these groundwaters. Groundwater G1 is closest to fines while G3 is found closer to shale ashes (black and red) in the deposit. Groundwater G1 is thus the most affected groundwater positioned straight north of the fines located along the eastern side of the waste deposit (Fig. 1d). This confirms the fact that G1 is affected mainly by leachates from fines, even if it is not possible, solely from these data, to determine the exact proportion between fines leachates and black/red shale ash leachates. Score values from PC1 are thus tentatively interpreted as describing the input of contaminated leachates from the shale waste in the deposit. This is confirmed by the loading plot (Fig. 5b) having high values for nickel and uranium in PC1. PC2 is probably governed by redox reactions as dissolved iron and manganese are found with strongly negative loading values in PC2. This is also strengthened by the fact that groundwaters G2, G6 and G8 with more oxidizing conditions have positive scores in PC2 (Fig. 5a). There is a clear relationship between the average score values for PC1 and logarithmic nickel concentrations (r2 0.77) and logarithmic uranium concentrations (r2 0.64) indicating that PC1 is a good proxy for the level of contamination in the groundwaters. This clearly shows that the combination of PCA and humidity cell experiments can provide information about what materials affect the different groundwaters. PCA has been used with success in other groundwater systems to identify contamination sources (Sappa et al. 2014; Rao 2014; Bäckström and Sartz 2016). In total, the PCA confirms the groundwater classifications assessed solely from the monitoring data (water chemistry) combined with data on the wells (location, depths etc.).
Discussion
Element distribution and speciation
Figs. 2 and 3 (concentrations of major elements and trace elements in selected groundwaters, 2015–2019), Tables 6 and 7 (concentrations of all of the elements in all of G1–G11, median and max–min values, 2015–2019) and Table 5 (composition of the solid materials, shale ash and fines from the deposit, shale from the pit lake area) illustrate and define the overall distribution and exchange of elements between solid materials and groundwater in the shale waste/groundwater system that constitutes the Kvarntorp deposit. Most important chemical parameter in this system that largely governs the leaching/weathering of the solid materials and concentrations and element speciation in the groundwater is pH (Table 6 and Figs. 2 and 3).
The groundwater G9 from the upper aquifer in the till is dominated by Ca–HCO3–SO4 with pH around 7.07 and total concentrations of dissolved solids around 530 mg/L (median values, Table 6). The groundwater G7 from the lower aquifer in the sandstone is also dominated by Ca–HCO3–SO4 with pH around 6.81 and concentrations of dissolved solids around 160 mg/L (median values, Table 7). The location of the G9 well is upstream the deposit. There is no significant change in chemistry in the water during the period 2004–2019 and no evident impact of contamination from shale leachates. Thus, G9 may serve as a background and reference for the assessment of the impact of leachates on the other groundwaters except G7 and G2. There is no apparent contamination of elements from the shale in the G7 water. This indicates that the green shale layer between the deposit and the sandstone horizon 31 m below the surface prevents exchange of water between the upper aquifer in the till/shale waste and the lower aquifer in the sandstone.
Major components—Na, K, Mg, Ca, HCO3 −, SO4 2−, (Cl) and pH
All shallow groundwaters, with the exception of G9, exhibit significant contributions of shale leachates, affecting to various extent all of the measured parameters. The highest contributions of leachates and largest effects on the concentrations of major elements and sulfate, as well as on pH, are observed in G1 (lowest pH), followed by G3 (Fig. 2 and Table 6).
The high pH in G6, most likely from calcium hydroxide originating from degraded calcium carbonate at high temperature (see discussion under Chemistry profiles), is decreasing with time from 12 in 2015 to 8 in 2019, indicating a depletion of calcium hydroxide but possibly equilibrium with calcium carbonate.
G1 and G3 differ from the other groundwaters when calcium concentrations are plotted against sulfate (Fig. 6a). Calcium concentrations are roughly only half of what would be expected compared to the trend for the other groundwaters. This pattern might indicate that there are no carbonates enough in the vicinity of G1 and G3 to buffer the acid being generated, which is consistent with leaching tests from Degerhamn where calcium was less present in acidic samples than in non-acidic lime-burned shale ash with dissolution of gypsum (Lavergren 2008). Calcium removal from the samples is likely due to precipitation of gypsum (saturation index − 0.14 and − 0.04 for G1 and G3, respectively) at high sulfate concentrations.

a Calcium concentrations (meq/L) versus sulfate concentrations (meq/L) in groundwaters G1–G11. b Magnesium/calcium ratios versus potassium concentrations (meq/L) in groundwaters G1–G11
Mg/Ca ratios plotted against potassium concentrations (Fig. 6b) provide information about ongoing redox activities in the deposit. A low Mg/Ca ratio suggests (1) that no magnesium bearing lime reacts, (2) that magnesium has been washed out or (3) that the calcium concentrations are limited by gypsum equilibrium. There are two possible reasons for lime not to react. Either there is no formation of sulfuric acid or all lime is consumed (Puura 1998). G1, G3 and G4 (located immediately downstream the waste deposit) display Mg/Ca ratios close to 1 or greater which is an indication of redox activities. These groundwaters are slightly reducing, though, with ferrous iron concentrations between 6 and 10 mg/L indicating that oxidizing reactions take place in the deposit upstream the groundwater wells. More reducing conditions close to the wells will, however, reduce the possibility for secondary immobilization when iron oxyhydroxides are being dissolved. Weathering of illite generates potassium in a slow process. It has been suggested that monitoring of potassium concentrations in combination with magnesium and calcium could provide information about the development in the deposit (Holm et al. 2005). A decreasing Mg/Ca ratio combined with an increasing potassium concentration would indicate a decrease in lime buffering and ongoing sulfuric acid production being buffered by illite instead. No such trends can be distinguished in the time span 2004–2019. There is a correlation (r2 0.72) between the Mg/Ca ratio and the potassium concentration when all 11 locations are included and an even stronger correlation (r2 0.94) when G6 and G10 are excluded. For G6, the Mg/Ca ratio is lower than for other wells with similar potassium concentrations. For G6, this could be due to equilibrium with brucite (saturation index − 0.02) since G6 is the only groundwater in equilibrium with brucite (see below). G10 displays the opposite relationship between Mg/Ca and potassium compared to G6, but since 2004, G10 has approached the linearity between Mg/Ca and potassium shown by the other groundwaters. This is an indication of decreased weathering at this site upstream the deposit, since also the electrical conductivity has decreased. For G3 and G4, both the Mg/Ca ratio and the potassium concentrations have increased since 2004 suggesting that weathering have increased in these waters. This is further strengthened by an increase in electrical conductivity in 2015–2019 compared to 2004. From Mg/Ca ratios, no general conclusion can be drawn for the entire area about changing weathering. Instead, a suggestion is that weathering rates are decreasing at some parts and increasing at other.
Solubility control
SI from the geochemical calculations for the groundwaters are given in Table 8. Positive SI (> 0.5) indicates oversaturation, negative SI (< 0.5) indicates undersaturation and SI in the range − 0.5 to 0.5 is considered to be in equilibrium with the considered solid phase. Solid phases likely found at equilibrium with the groundwaters are calcite, celestite, dolomite, gibbsite, gypsum, jarosite and siderite. The presence of jarosite and gypsum has been confirmed to exist in the different waste materials within the waste deposit (Table 4).
Other high concentration elements—Al, Fe, Mn, Li and Sr
Highest concentrations of iron are found in the acidic G1 (reaching 379 mg/L in 2015–2019), and G3 (167 mg/L) and G10 (202 mg/L) have high concentrations (Table 6) indicating the presence of ferrous iron. In the case of G10, the pipe is made of steel, but presumably, the high concentrations are explained to a large part by water affected by alum shale. The ditch north of the waste deposit shows spots with both elevated temperatures and electrical conductivity, which indicates that leachates from warm parts of the deposit are reaching the ditch through the groundwater. Iron concentrations in the ditch are much lower though, than in the groundwater, supposedly due to precipitation of iron oxyhydroxides when reaching neutral and oxidizing surface water (cf. Armstrong et al. 2019).
A comparison between all sampling occasions in all groundwaters indicates a negative correlation with pH for manganese (r2 0.78, logged data) and a weaker negative correlation (r2 0.53) for iron (logged data). Other elements show even weaker correlations with pH. Perkins and Mason (2015) found that manganese is highly mobile in weathering shale and suggested that manganese loss from carbonate poor black shales can be an indicator of weathering even if no other signs of weathering are clearly noted. In Kvarntorp, G2, G7 (the deep wells) and G6 (highest pH) have manganese concentrations in the range 0.0002–0.4 mg/L; G9 (local background reference) generally has below 1 mg/L and G8 normally around 1 mg/L. Groundwaters G5, G10 and G11 have concentrations between 1 and 6 mg/L while G1 and G3 have concentrations between 10 and 22 mg/L. Groundwaters with elevated manganese concentrations in Kvarntorp could be due to weathering and/or reducing conditions in the groundwater.
Lithium concentrations of about 5 μg/L (median) in the reference background well G9 are below the levels in the sandstone aquifer (10–60 μg/L in G2 and G7). Groundwaters G5, G6, G8, G10 and G11 have concentrations between 10 and 600 μg/L, while G1, G3 and G4, that are believed to be highly affected by the shale leachate, have concentrations between 1000 and 2000 μg/L. Lithium concentrations in alum shale has previously been reported to be in the range 4- to 24-mg/kg dw (Allard et al. 2011), which means that alum shale is not enriched in lithium compared to other black shales (world median 31-mg/kg dw; Ketris and Yudovich 2009). Leaching tests have shown that lithium is quite easily leached from alum shale, but not from shale ash (Åhlgren et al. in preparation), which strengthens the suggestion of contact with partly pyrolysed shale or fines for G1 and G3 but possibly also for G4. Lithium has been reported to be found in high concentrations in flowback water after fracking in the Marcellus shale (USA) with median concentrations of 95 mg/L after 14 days (Haluszczak et al. 2013) and is considered for recovery (Lee and Chung 2020). These concentrations largely surpass the groundwater in Kvarntorp.
Regional strontium background level in surface water upstream the Kvarntorp area is below 0.1 mg/L (Åhlgren et al. 2020). Only the upstream well G9 has concentrations that low. Groundwaters G1, G3, G4, G7, G10 and G11 have concentrations between 0.2 and 0.7 mg/L, while G5, G6 and G8 exceed 1 mg/L. Highest concentrations are found in the deep groundwater G2 (1.5–2.2 mg/L). Even though the strontium concentrations are higher in the deep groundwater, the levels are not high when comparing with Danish drinking water works (median 1.45 mg/L, range 0.02 to 30 mg/L) and Danish groundwater wells (median 1.52 mg/L) (Frei et al. 2020).
Hydrolyzable divalent trace elements—Co, Ni, Cu and Zn
Concentrations of nickel (as well as manganese, iron, vanadium, molybdenum and uranium) as a function of pH are given in Fig. 7. A pH influence is evident for nickel, as expected, with high concentrations at low pH (G1) decreasing with increasing pH (G3, G4 and G10). The higher levels in G10 in comparison with G3 and G4 may indicate reducing conditions.

Diagrams showing concentrations vs. pH for hydrolysable major elements and trace elements (Mn, Fe and Ni) as well as redox sensitive trace elements (V, Mo and U) in groundwaters G1–G11
Total concentrations of the hydrolysable elements cobalt, nickel, copper and zinc are high in fines and shale ash (Table 5). Concentrations of cobalt, nickel and zinc in the shale waters from G1 to G3, with low pH, are higher than in the other groundwaters (Table 7), even though for nickel, the correlation with pH is negative but fairly weak (r2 0.32) when all groundwater wells are included. High levels of cobalt and nickel are observed in G10. High concentration of particularly nickel, but also cobalt and zinc, in the groundwaters are evidently indications of shale weathering and contribution of contaminated leachates. Soils underlain by black shales elsewhere have shown to be enriched in easily mobilized metals with cadmium and copper being of particular concern (Yu et al. 2012), and cadmium has been found in elevated concentrations in rice in black shale areas (Duan et al. 2020). Both cadmium (median level 12 μg/L) and copper (median level 460 μg/L) have been found at high concentrations in Degerhamn in acidic groundwater (Lavergren 2008), whereas in Kvarntorp, the median for cadmium is lower (6 μg/L for G1) and copper much lower (median 2.4 μg/L for G1 and 5.5 μg/L for G3). Part of the explanation could be in concentration differences in the shale material, since both elements display higher concentrations in the Degerhamn shale bedrock (3-mg/kg dw cadmium and 170-mg/kg dw copper; Lavergren 2008) compared to Kvarntorp (median 1.14-mg/kg dw cadmium and 137-mg/kg dw copper in drill cores).
Leaching tests (Åhlgren et al. 2018) with fines and shale ash (the same material as given in Table 5) showed that nickel was not released from the shale ash to any greater extent, whereas there was a significant release of nickel from the fines at low pH.
Redox and pH sensitive trace elements—V, As, Mo and U
Kvarntorp alum shale is enriched in the redox sensitive elements vanadium, arsenic, molybdenum and uranium (Table 5). Elevated concentrations of these elements in the groundwaters (Table 7) are indicators of shale weathering and further mobilization into the groundwater (see Fig. 4 for the spatial distribution).
Vanadium and arsenic can be incorporated in pyrite which is the probable origin of these elements in alum shale. In addition, vanadium is also trapped in clays (Lerat et al. 2018). Shale residues from the waste deposit show a range of 420- to 750-mg/kg dw vanadium and 80- to 150-mg/kg dw arsenic, and these elements have a correlation of 0.49 (r2, logged data) in the groundwaters. It is, however, not the same groundwater well that has the highest concentrations of both vanadium and arsenic. Neither of these two elements seems to be leached to any greater extent, except for vanadium in G1 with up to 130 μg/L and arsenic in G3 with up to 170 μg/L. This is in line with results from studies of alum shale, shale residues and groundwater in Degerhamn, Öland, which also showed low leachability of arsenic and vanadium (Lavergren 2008). Leaching tests at pH 12, however, have shown more important leaching of vanadium from shale ash where 28% of the vanadium was leached (Karlsson et al. 2013) confirming that vanadium is most mobile under oxidized and alkaline conditions (Wright and Belitz 2010). Arsenic mobility in fractured rock with pyritic black shales has earlier shown to be determined by concentrations of sulfide and available oxidant where microbial sulfide production is suggested to enhance arsenic mobilization (Zhu et al. 2008).
Yu et al. (2014) suggest that arsenic released from oxidation of arsenian pyrite is adsorbed onto the surface of, or coprecipitated with, secondary iron minerals. A possible explanation for the differences in arsenic concentrations in G1 and G3 could be precipitation of schwertmannite in G1 favoured by the lower pH. Arsenic is effectively adsorbed to schwertmannite (Acero et al. 2006), and arsenic is thus removed from the groundwater through either coprecipitation or sorption to schwertmannite in G1. In 2004, arsenic concentrations and pH were lower in G3, indicating possible precipitation of schwertmannite and removal of arsenic also in G3 at that time.
Molybdenum has reduced mobility in sulfidic environments due to the formation of MoS2 simultaneously with FeS and FeS2 (Smedley et al. 2014). Molybdenum, acting as the molybdate anion with concentrations of 130- to 210-mg/kg dw in fines and ashes (Table 5) shows restricted solubility at low pH and has increased from below 5 to 170–630 μg/L in G3 since the pH increased from 3.6–4.3 in 2004 to 5.8–6.4 in 2015–2019. Molybdenum concentrations are below 36 μg/L, despite the presumed high weathering rate in the well, which could be due to the low pH. G6, with median pH above 9, has fairly high concentration of molybdenum (maximum level above 100 μg/L), which would be due to the high pH, while G8 shows even higher concentrations (maximum level almost 600 μg/L for the period 2015–2019).
Molybdenum concentrations in G6 show enhanced leaching at high pH, whereas in G3 and G8, redox processes are the likely reason for elevated concentrations. Uranium displays leaching due to low pH in G1 and governed by high carbonate levels in G8 where it covaries with molybdenum but not iron. Also vanadium is leached due to low pH in G1.
It is interesting to note that molybdate might be solubility controlled by for instance CaMoO4 and FeMoO4. (Table 8). There is no apparent solubility control of strontium by celestite (SrSO4), nickel by NiMoO4, vanadium by Fe(VO3)2 or uranium by uraninite (UO2).
Uranium occurs in four manners in alum shale according to Armands (1972): (1) in authigenic K-feldspar, (2) in detrital zircon and titanite, (3) with sulfur and pyrite and (4) organically bound to carbon, whereas Lecomte et al. (2017) propose that uranium was accumulated in organic matter and biogenic phosphate nodules. Uranium concentrations in shale ash and fines are in the range 160- to 240-mg/kg dw. Elevated concentrations of uranium are found in G8 (up to 1760 μg/L) which has a circumneutral pH but the highest carbonate concentrations of all waters, indicating formation of soluble uranium carbonate complexes (Bernhard et al. 1996). High uranium concentrations have previously been observed in one of the pit lakes in the Kvarntorp area, with circumneutral pH and high carbonate level (Allard et al. 2011; Åhlgren et al. 2020).
Leaching tests (Åhlgren et al. 2018) with fines and shale ash (the same material as given in Table 5) showed that uranium leached from both fines and ashes at low pH.
Temperatures
Temperatures were elevated in groundwaters at the north-eastern part of the deposit, both in 2005 and in 2017. Fig. 4, bottom right, shows the temperatures at a depth of 1 m below the water surface in November 2017. Measurements in the ditch just north of the deposit show higher temperatures (18 °C in June 2019) close to well G3 than further to the west (12 °C), indicating that warm water from the deposit is reaching the ditch at this site. Temperatures in the groundwater have not changed notably since 2005. Either the heat generating reactions in the waste deposit have not decreased, or the heat storage capacity in the waste deposit is so significant that the cooling process is very slow. Modelling of the temperature decrease performed in 2005 (Holm et al. 2005) indicated an expected cooling time period of up to several hundred years when treating the waste deposit as a passive heat storage. This estimate most likely underestimated the time period as the presence of reactive shale with chemical energy has the capacity to produce energy as heat was not included in this modelling.
Time trends
Generally, there is no evident time trend indicating significant changes in releases of metals from the shale to groundwaters G1–G6, G8, G10 and G11. The greatest disparity between different years is found when data from 2004 are compared with those from 2015–2019, as, for example, much higher cobalt and nickel concentrations in G10 in 2004 compared to 2015–2019. When comparing results only from 2015–2019 vanadium, nickel and uranium display a decreasing trend in G1. When results for 2004 are included, the trend is not evident since some of the concentrations from that year are lower than in 2015. G8 displays varying concentrations for molybdenum. A possible increased weathering during 2015–2019 compared to 2004 is suggested for G3 and G4 (both downstream the deposit) since both electrical conductivity and Mg/Ca ratios agree with such a conclusion.
G10 shows a decrease in sulfate (from 3000 to around 2000 mg/L), magnesium (from 400 to around 150 mg/L), cobalt (from > 300 to 1–67 μg/L) and nickel (from > 2400 to 4–300 μg/L) after 2004. This may be an indication that this well is presently less affected by shale weathering. The well is situated upstream the deposit and is not expected to receive leachates from the deposit itself but rather from the surrounding dumped fines through surface run-off.
The deposit was still unvegetated until the beginning of the 1980s, but vegetation has gradually colonized the deposit since then, and today, primarily birches (Betula) are common. Other sites have shown dissolution of goethite and release of trace elements due to reductive conditions caused by organic cover (Paktunc 2013) and leaching tests of alum shale fines from Kvarntorp have shown enhanced leaching of vanadium and molybdenum when covered by organic material (Sjöberg and Karlsson 2015).
A general conclusion is that changes in groundwater chemistry with time in terms of increasing or decreasing levels of contaminants originating from the deposit cannot be quantitatively assessed. There are at least three uncertainties that prevent predictions of the development with time: (1) lack of data from the initial period from 1942 to 2003. (2) Uncertainties in the prediction of how the temperature in the deposit will develop with time. Wet or even water saturated parts of the deposit will be depleted with respect to leachable elements, but significant parts of the deposit are still hot and dry. An open question remains whether releases of elements will increase or possibly decrease when the deposit cools down and more water starts to infiltrate. (3) The heterogeneity of the deposit with three categories of shale residues with different weathering potential. The exact locations and distribution of these different categories within the deposit are not known in detail. However, it is reasonable to expect effects on the releases of element-enriched leachates from the deposit area once it has cooled down and is infiltrated with water in all parts.
Conclusions
The shallow groundwaters around the Kvarntorp waste deposit (upper aquifer, in the till horizon) show clear impacts from shale waste in terms of elevated levels of elements which obviously are released from weathering shale and shale ashes. PCA of the groundwater data in combination with leaching data from humidity cells of specific waste materials from the area provides information about potential sources for trace elements to the groundwater. This analysis confirms assessments solely based on water monitoring data combined with data regarding the water sampling wells (locations, depths, lining etc). For eight of the sampled groundwaters, the concentrations of elements characteristic of alum shale, such as molybdenum, nickel, uranium and to some extent also arsenic, are well above background levels by up to three orders of magnitude (vanadium in one well with pH below 4). Groundwater in the lower aquifer (depth 31 m, in the sandstone layer) is generally not affected.
Presence of groundwater with low pH (below 4) in particularly one groundwater indicates high loadings of leachates from fines containing pyrite that is being oxidized, which produces acid. Presence of highly alkaline pH (up to 12) in particularly one groundwater is suggested to be due to presence of calcium hydroxide from the degradation of calcium carbonate at high temperatures (above 500 °C) in the deposit. Most of the waste is composed of shale ash which also left signatures in the water like arsenic, molybdenum, uranium and to some extent vanadium, however, at near neutral pH (above 6.5).
Concentrations in the shallow groundwaters downstream the deposit reached high levels, e.g., up to 172-μg/L As, 633-μg/L Mo, 1900-μg/L Ni, 1760-μg/L U, 135-μg/L V and 4400-mg/L SO42−. Substantial quantities of these elements are released from the waste deposit. If distributed into the regional groundwater system, such concentrations could constitute a potential health hazard, but the groundwater distribution seems to be rather limited since the lower aquifer is not affected.
Evidently, there is a continued and progressing impact of shale leachates on the groundwater around the deposit since at least 2004 and probably during the whole lifetime of the deposit since the 1940s. The time span between 2015 and 2019 is too short to distinguish a significant trend. There are indications of increased activity and weathering in some of the wells, supported by both increased electrical conductivity and increased Mg/Ca ratios in combination with increased potassium concentrations. Most groundwaters, though, do not show any clear changes in levels of elements from shale waste weathering which suggests that no general leaching trend for the area can be discerned based on data from the short time span 2004–2019. A reasonable conclusion is, nevertheless, that leaching will progress at about the same level for a long time and that the current release of elements to downstream water will continue. The waste deposit has affected the surroundings for more than half a century and will probably continue to do so, possibly for centuries to come.
Presence of different waste types makes the picture in Kvarntorp complex, but it has been demonstrated that processed black shale has long-term impact on the surrounding environment with high levels of several elements, notably arsenic, molybdenum, uranium and to some extent nickel.
Data availability
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Acero P, Ayora C, Torrentó C, Nieto J-M (2006) The behavior of trace elements during schwertmannite precipitation and subsequent transformation into goethite and jarosite. Geochim Cosmochim Acta 70:4130–4139
Åhlgren K, Sjöberg V, Bäckström M (2018) Leaching of U, V, Ni and Mo from alum shale waste as a function of redox and pH – suggestion for a leaching method. In: Wolkersdorfer C, Sartz L, Weber A, Burgess J, Tremblay G (eds) Mine water: risk to opportunity. Proc, 11th ICARD/IMWA 2018, Pretoria, South Africa, pp 782–787
Åhlgren K, Sjöberg V, Allard B, Bäckström M (2020) Chemistry of acidic and neutralized alum shale pit lakes 50 years after mine closure, Kvarntorp Sweden. Mine Water Environ 39:481–497
Allard B, Arsenie I, Håkansson K, Karlsson S, Ahlberg A-C, Lundgren T, Collin M, Rasmuson A, Strandell E (1991) Effects of weathering on metal releases from an engineered deposit for alum shale leaching residues. Water Air Soil Pollut 57-58:431–440
Allard B, Bäckström M, Häller S, Karlsson S, Panova E, Grawunder A (2011) Water chemistry and trace metal concentrations in an acidic alum shale pit lake – effects of liming. In: Rüde TR, Freund A, Wolkersdorfer C (eds) Mine Water: managing the challenges. Proc, 11th IMWA, Aachen, pp 503–508
Andersson A, Dahlman B, Gee DG, Snäll S (1985) The Scandinavian alum shales. Swedish Geol Survey SER Ca Nr 56, Uppsala
Armands G (1972) Geochemical studies of uranium, molybdenum and vanadium in a Swedish alum shale. Stockh Contrib Geol 27:1–148
Armstrong JGT Parnell J, Bullock LA, Boyce AJ, Perez M, Feldmann J (2019) Mobilisation of arsenic, selenium and uranium from Carboniferous black shales in west Ireland. Appl Geochem 109: 13 pages, 104401
Bäckström M (2010) Environmental impact from an alum shale deposit, Kvarntorp Sweden – present and future scenarios. In: Wolkersdorfer C, Freund A (eds) Mine water & innovative thinking. Proc, IMWA Symp. Cape Breton Univ Press, Sydney, pp 551–554
Bäckström M, Sartz L (2016) Use of multivariate statistics in order to understand the flow of acid rock drainage from an abandoned mining site. J Environ Prot 7:358–371
Bengtson P (1971) The hydrogeology of the Kvarntorp area. Swedish Geol Survey, SER C, Nr 667, Uppsala [Kvarntorpsområdets hydrogeology, In Swedish with English summary]
Bernhard G, Geipel G, Brendler V, Nitsche H (1996) Speciation of uranium in seepage waters of a mine tailing pile studied by time-resolved laser-induced fluoresence spectroscopy (TRLFS). Radiochim Acta 74:87–91
Blomquist T, Ekholm D (1997) Additional sampling in the Kvarntorp area. VBB Viak, Örebro, project number 15203497 [Kompletterande provtagningar i Kvarntorpsområdet, In Swedish]
Burkhardt E-M, Meissner S, Merten D, Büchel G, Küsel K (2009) Heavy metal retention and microbial activities in geochemical barriers formed in glacial sediments subjacent to a former uranium mining leaching heap. Chem Erde 69:21–34
Chi Fru E, Hemmingsson C, Callac N, Perez N, Panova EG, Broman C, El Albani A (2016) Atmospheric weathering of Scandinavian alum shales and the fractionation of C, N and S isotopes. Appl Geochem 74:94–108
Duan Y, Yang Z, Yu T, Yang Q, Liu X, Ji W, Jiang H, Zhuo X, Wu T, Qin J, Wang L (2020) Geogenic cadmium pollution in multi-medians caused by black shales in Luzhai, Guangxi. Environ Pollut 260. https://doi.org/10.1016/j.envpol.2019.113905
Falk H, Lavergren U, Bergbäck B (2006) Metal mobility in alum shale from Öland, Sweden. J Geochem Explor 90:157–165
Frei R, Frei KM, Munch Kristiansen S, Jessen S, Schullehner J, Hansen B (2020) The link between surface water and groundwater-based drinking water – strontium isotope spatial distribution patterns and their relationships to Danish sediments. Appl Geochem 121. https://doi.org/10.1016/j.apgeochem.2020.104698
Grawunder A, Lonschinski M, Merten D, Büchel G (2009) Distribution and bonding of residual contamination in glacial sediments at the former uranium mining leaching heap of Gessen/Thuringia, Germany. Chem Erde 69:5–19
Haluszczak LO, Rose AW, Kump LR (2013) Geochemical evaluation of flowback brine from Marcellus gas wells in Pennsylvania, USA. Appl Geochem 28:55–61
Hessland I, Armands G (1978) Alum shale – geology. Statens industriverk, Stockholm: SIND PM 1978:3 [Alunskiffer: Underlagsmaterial geologi, in Swedish]
Holm T, Ekholm D, Johansson L, Bäckström M, Rehn I, Forsmark T, Axenhamn L, Sundblad B (2005) Kvarntorp area – study of the waste deposit. SWECO VIAK AB and SGU, Göteborg, pp 56 [Kvarntorpsområdet – Studie av Kvarntorpshögen, In Swedish]
Jüriado K, Raukas A, Petersell V (2012) Alum shales causing radon risks on the examples of Maardu area, North-Estonia. Oil Shale 29:76–84
Kalinowski BE, Johnsson A, Arlingr J, Pedersen K, Ödegaard-Jensen A, Edberg F (2006) Microbial mobilization of uranium from shale mine waste. Geomicrobiol J 23:3–4
Karlsson L, Bäckström M, Allard B (2012) Leaching of sulfidic alum shale waste at different temperatures. In: Price WA, Hogan C, Tremblay G (eds) Proc, 9th ICARD 2012. Curran Associates, Inc., Ottawa, pp 812–821
Karlsson L, Karlsson S, Allard B, Sjöberg V, Bäckström M (2013) Release of metals from unprocessed and processed black shale due to natural weathering. In: Brown A, Figuero L, Wolkersdorfer C (eds) Reliable mine water technology, vol I. Publication Printers, Denver, Colorado, USA, pp 391–397
Ketris MP, Yudovich YE (2009) Estimations of Clarkes for Carbonaceous biolithes: world average for trace element contents in black shales and coals. Int J Coal Geol 78:135–148
Lavergren U (2008) Metal dispersion from natural and processed black shale. Dissertation, University of Kalmar, Sweden
Lavergren U, Åström ME, Falk H, Bergbäck B (2009) Metal dispersion in groundwater in an area with natural and processed black shale – nationwide perspective and comparison with acid sulfate soils. Appl Geochem 24:359–369
Lecomte A, Cathelineau M, Michels R, Peiffert C, Brouand M (2017) Uranium mineralization in the Alum Shale Formation (Sweden): evolution of a U-rich marine black shale from sedimentation to metamorphism. Ore Geol Rev 88:71–98
Lee J, Chung E (2020) Lithium recovery by solvent extraction from simulated shale gas produces water – impact of organic compounds. Appl Geochem 116. https://doi.org/10.1016/j.apgeochem.2020.104571
Lerat JG, Sterpenich J, Mosser-Ruck R, Lorgeoux C, Bihannic I, Fialips CI, Schovsbo NH, Pironon J, Gaucher EC (2018) Metals and radionuclides (MaR) in the alum shale of Denmark: identification of MaR-bearing phases for the better management of hydraulic fracturing waters. J Nat Gas Sci Eng 53:139–152
Liu Y, Xiao T, Perkins RB, Zhu J, Zhu Z, Xiong Y, Ning Z (2017) Geogenic cadmium pollution and potential health risks, with emphasis on black shale. J Geochem Explor 176:42–49
Loukola-Ruskeeniemi K, Uutela A, Tenhola M, Paukola T (1998) Environmental impact of metalliferous black shales at Talvivaara in Finland, with indication of lake acidification 9000 years ago. J Geochem Explor 64:395–407
Nielsen AT, Schovsbo NH (2007) Cambrian to basal Ordovician lithostratigraphy in southern Scandinavia. Bull Geol Soc Den 53:47–92
Nielsen AT, Schovsbo NH (2015) The regressive Early-Mid Cambrian ‘Hawke Bay Event’ in Baltoscandia: epeirogenic uplift in concert with eustasy. Earth Sci Rev 151:288–350
Paikaray S (2012) Environmental hazards of arsenic associated with black shales: a review on geochemistry, enrichment and leaching mechanism. Rev Environ Sci Biotechnol 11:289–303
Paktunc D (2013) Mobilization of arsenic from mine tailings through reductive dissolution of goethite influenced by organic cover. Appl Geochem 36:49–56
Parkhurst DL, Appelo CAJ (1999) User’s guide to PHREEQC (Version 2) – a computer program for speciation, batch-reaction, one-dimensional transport, and inverse geochemical calculations. U.S. Geological Survey Water-Resources Investigations, Denver Colorado. Report. Vol. 99-4259
Parviainen A, Loukola-Ruskeeniemi K (2019) Environmental impact of mineralised black shales. Earth Sci Rev 192:65–90
Peng B, Song Z, Tu X, Xiao M, Wu F, Lv H (2004) Release of heavy metals during weathering of the Lower Cambrian black shales in western Hunan, China. Environ Geol 45:1137–1147
Peng B, Rate A, Song Z, Yu C, Tang X, Xie S, Tu X, Tan C (2014) Geochemistry of major and trace elements and Pb-Sr isotopes of a weathering profile developed on the Lower Cambrian black shales in central Hunan, China. Appl Geochem 51:191–203
Perkins RB, Mason CE (2015) The relative mobility of trace elements from short-term weathering of a black shale. Appl Geochem 56:67–79
Phan TT, Capo RC, Stewart BW, Graney JR, Johnson JD, Sharma S, Toro J (2015) Trace metal distribution and mobility in drill cuttings and produced waters from Marcellus Shale gas extraction: uranium, arsenic, barium. Appl Geochem 60:89–103
Pihu T, Konist A, Puura E, Liira M, Kirismäe K (2019) Properties and environmental impact of oil shale ash landfills. Oil Shale 36:257–270
Puura E (1998) Weathering of mining waste rock containing alum shale and limestone: a case-study of the Maardu dumps, Estonia. Ph.D. Diss, Royal Inst of Technology, Stockholm
Puura E, Neretnieks I, Kirsimäe K (1999) Atmospheric oxidation of the pyritic waste rock in Maardu, Estonia. 1 field study and modelling. Environ Geol 39:1–19
Rao NS (2014) Spatial control of groundwater contamination, using principal component analysis. J Earth Syst Sci 123:715–728
Sappa G, Ergul S, Ferranti F (2014) Geochemical modelling and multivariate statistical evaluation of trace elements in arsenic contaminated groundwater systems of Viterbo Area (Central Italy). SpringerPlus 3:237. https://doi.org/10.1186/2193-1801-3-237
Saqib N, Bäckström M (2014) Trace element partitioning of solid waste incineration ashes with respect to fuel composition, chlorine content and boiler temperature. Waste Manag 34:2505–2519
Schovsbo NH, Nielsen AT, Gautier DL (2014) The Lower Palaeozoic shale gas play in Denmark. Geol Surv Denmark and Greenland Bulletin 31:19–22
SGU (Swedish Geological Survey) (2013) Assessement criteria for groundwater. SGU report 2013:01, 238 pages. [In Swedish, Bedömningsgrunder för grundvatten]
Sjöberg V, Karlsson S (2015) Impact of organic carbon on the leachability of vanadium, manganese, iron and molybdenum from shale residues. Miner Eng 75:100–109
Smedley PL, Cooper DM, Ander EL, Milne CJ, Lapworth DJ (2014) Occurrence of molybdenum in British surface water and groundwater: distributions, controls and implications for water supply. Appl Geochem 40:144–154
Stuckman MY, Lopano CL, Berry SM, Hakala JA (2020) Geochemical solid characterization of drill cuttings, core and drilling mud from Marcellus Shale Energy development. J Nat Gas Sci Eng 68:1–14
Velts O, Uibu M, Kallas J, Kuusik R (2011) Waste oil shale ash as a novel source of calcium for precipitated calcium carbonate: carbonation mechanism, modeling, and product characterization. J Hazard Mater 195:139–146
Vine JD, Tourtelot EB (1969) Geochemical investigations of some black shales and associated rocks: the content and association of minor elements in some black shales deposited in various types of geologic environments. Contributions to Geochemistry. United States government printing office, Washngton, Geological Survey Bulletin 1314-A
Vriens B, Peterson H, Laurenzi L, Smith L, Aranda C, Mayer KU, Beckie RD (2019) Long-term monitoring of waste-rock weathering at the Antamina mine, Peru. Chemosphere 215:858–869
Waersted FM, Riss PJ, Skipperud L (2020) The effect of water exchange on the leaching of alum shale. Appl Geochem 119:104610
Wilke FDH, Vieth-Hillebrand A, Naumann R, Erzinger J, Horsfield B (2015) Induced mobility of inorganic and organic solutes from black shales using water extraction: implications for shale gas exploitation. Appl Geochem 63:158–168
Woo NC, Choi MJ, Lee KS (2002) Assessement of groundwater quality and contamination from uranium-bearing black shale in Gosean-Boeun areas, Korea. Environ Geochem Health 24:261–273
Wright MT, Belitz K (2010) Factors controlling the regional distribution of vanadium in groundwater. Groundwater 48:515–525
Yu C, Peng B, Peltola P, Tang X, Xie S (2012) Effect of weathering on abundance and release of potentially toxic elements in soils developed on Lower Cambrian black shales, P. R. China. Environ Geochem Health 34:375–390
Yu C, Lavergren U, Peltola P, Drake H, Bergbäck B, Åström ME (2014) Retention and transport of arsenic, uranium and nickel in a black shale setting revealed by a long-term humidity cell test and sequential chemical extractions. Chem Geol 363:134–144
Zhu W, Young LY, Yee N, Serfes M, Rhine ED, Reinfelder JR (2008) Sulfide driven arsenic mobilization from arsenopyrite and black shale pyrite. Geochim Cosmochim Acta 72:5243–5250
Acknowledgements
Kumla municipality is gratefully acknowledged for access to the area and permission to take samples, and the Swedish Geological Survey for providing access to the old exploration drill cores from the area.
Funding
Open access funding provided by Örebro University.
Author information
Authors and Affiliations
Contributions
KÅ and MB performed the study conception and design, and KÅ performed the water sampling, sample preparations and analyses. VS performed the metal analysis. KÅ and MB performed the sampling of solid materials. The first draft was written by KÅ with contributions from BA and MB; all authors commented on previous versions of the manuscript and all authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Responsible Editor: Xianliang Yi
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Åhlgren, K., Sjöberg, V., Allard, B. et al. Groundwater chemistry affected by trace elements (As, Mo, Ni, U and V) from a burning alum shale waste deposit, Kvarntorp, Sweden. Environ Sci Pollut Res 28, 30219–30241 (2021). https://doi.org/10.1007/s11356-021-12784-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-021-12784-2