
Abstract
Hereditary breast and ovarian cancer (HBOC) can be identified by genetic testing of cancer-causing genes. In this study, we identified a spectrum of genetic variations among 76 individuals of Armenian descent either with a family history of cancer or breast cancer before the age of 40. We screened 76 suspected HBOC patients and family members as well as four healthy controls using a targeted and hereditary comprehensive cancer panel (127 genes). We found 26 pathogenic (path) and 6 likely pathogenic (LPath)variants in 6 genes in 44 patients (58%); these variants were found in BRCA1 (17), BRCA2 (19), CHEK2 (4), PALB2 (2), and NBN (1). A few different variants were found in unrelated individuals; most notably, variant p.Trp1815Ter in the BRCA1 gene occurred in four unrelated patients. We did not find any known significant variants in five patients. Comprehensive cancer panel testing revealed pathogenic variants in cancer genes other than BRCA1 and BRCA2, suggesting that testing only BRCA1 and BRCA2 would have missed 8 out of 44 suspected HBOC patients (18%). These data also confirm that a comprehensive cancer panel testing approach could be an appropriate way to identify most of the variants associated with hereditary breast cancer.
Similar content being viewed by others


Introduction
Hereditary breast and ovarian cancer (HBOC) and their causative variants have become one of the most studied hereditary cancers. Although the BRCA1 (NM_007294.3) and BRCA2 (NM_000059.3) gene variants were the primary focus of such studies in the past, hereditary cancer panel testing has recently replaced the approach involving only the BRCA1 and BRCA2 genes. In fact, it has been shown that close to 10% of hereditary breast cancer can be caused by variants in genes such as CHEK2 (NM_007194.3), PALB2 (NM_024675.3), ATM (NM_000051.3), and MUTYH (NM_001128425.1)1. There are hundreds of genes that could be involved in tumorigenesis and cancer, yet not all are involved in every cancer. Thus, there are specific hereditary cancer panels for breast, ovarian, colorectal, endometrial, prostate, and other cancers. These panels contain some of the most prevalently mutated genes in each specific cancer, yet there could be genes that may cause hereditary cancer in a specific population that may not be included in general cancer gene panels. Therefore, ethnic-specific population studies could be important in the identification and inclusion of genes that may cause hereditary cancers in a specific population.
Additionally, there are several cancer-causing variants that could be more prevalent in an ethnic or specific population. The most famous of these variants are the three BRCA1 and BRCA2 variants (i.e., 5382insC and 185delAG in BRCA1 and 6174delT in BRCA2) that are prevalent in the Ashkenazi Jewish population2. Ethnic-specific studies have shown that there may be variants classified as founder mutations, as well as unique variants in suspected HBOC patients. In fact, the number of founder mutations compared to unique variants is quite small, indicating that some populations will not benefit from targeted gene testing. Recently, gene panels have been shown to be the better approach, expanding HBOC variant discovery to several genes from different pathways (Breast Cancer Information Core, BIC). This approach has resulted in the identification of many pathogenic (Path) and likely pathogenic (LPath) variants, and variants of unknown significance (VUSs) from genes other than BRCA1 and BRCA2. Path and LPath variants are important in drug discovery and treatment options, while VUSs could be significant in the initiation of more studies to identify their cancer-causing roles. Some deleterious variants could introduce premature termination codons through frameshift deletions or insertions, nonsense or splice junction mutations, or large deletions or duplications. Some splice site mutations and large rearrangements do not change the reading frame but result in a loss or gain of one to several exons, which could potentially have an impact on gene function. Deleterious missense mutations are typically confined within specific residues of functional motifs. However, the risk contribution of numerous other sequence variants remains unclear. Some VUSs include missense changes and small in-frame deletions and insertions that mostly lead to one amino acid change without a frameshift as well as alterations in noncoding intervening sequences or in untranslated exonic regions3.
There have been two small-scale studies on breast cancer genetics in the Armenian population. One study attempted to find known BRCA1 and BRCA2 founder mutations in 46 suspected HBOC patients, but no founder mutations were found4. Another study attempted to identify BRCA1 and BRCA2 mutations in six suspected HBOC patients of Armenian descent using targeted panel testing. No pathogenic variants were found in these patients5.
In this study, we analyzed the mutation spectrum of 76 patients of Armenian descent with suspicion of hereditary breast cancer (family history of cancer) or breast cancer under the age of 40, selected according to the National Comprehensive Cancer Network (NCCN) guidelines, using a comprehensive 127-gene hereditary cancer panel. The purpose of this study was not only to identify the mutational spectrum of HBOC in the Armenian population but also to identify the genes that could have breast cancer-causing variants in the Armenian population. We intended to identify these genes to provide insights for an Armenian-specific HBOC-testing gene panel and a comparison with genes in panels for other ethnic groups. We also anticipated finding novel variants that could potentially cause breast cancer due to potential different distributions of disease-causing variants in the Armenian population. This is the first large-scale study of breast cancer-causing variants in the Armenian population.
Materials and methods
Patient selection and BRCAPRO score
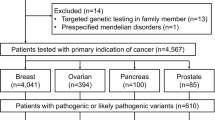
We tested 76 suspected HBOC patients or family members and 4 healthy controls using a targeted test (24 patients) and a hereditary comprehensive cancer panel (127 genes). Overall, 34 patients were <40 years of age (22 with a family history of cancer, 63%), 16 patients were between 40 and 50 years old (10 with a family history of cancer, 63%), and 26 patients were >50 years of age (25 with a family history of cancer, 96%). All patients were selected according to the criteria provided in the NCCN Clinical Practice Guidelines in Oncology (Version 2.2019-July 30.2018). Only patients with a confirmed histopathologic diagnosis of invasive breast cancer (BC) were included in the study. Following genetic counseling, the probability for each female patient to be a mutation carrier in one or both of the BRCA genes was estimated using the BRCAPRO model of the University of Texas Southwestern Medical Center at Dallas CancerGene software, version 5.1 (http://www4.utsouthwestern.edu/breasthealth/cagene/). The BRCAPRO risk is derived through the Bayesian probability model and takes into account the first and second degree relatives of a patient, age at the time of diagnosis of BC and/or ovarian cancer (OC), and ages of unaffected family members6. The male patients with a diagnosis of BC were not subject to BRCAPRO risk calculation given that the model values for male BC patients were significantly higher than those for female BC patients. Patients were all ethnically Armenian. This study was approved by the Institutional Review Board of the Center of Medical Genetics and Primary Health Care. Informed consent was obtained from all human subjects participating in this research.
Gene panel sequencing and bioinformatics
Variant analysis was performed using an advanced bioinformatics pipeline and manual curation.
A 127-gene comprehensive cancer panel (AIP, ALK, APC, ATM, ATR, AXIN2, BAP1, BARD1, BLM, BMPR1A, BRCA1, BRCA1, BRIP1, BUB1B, CASR, CDC73, CDH1, CDK4, CDKN1B, CDKN1C, CDKN2A, CEBPA, CHEK2, CTC1, CTNNA1, CYLD, DDB2, DICER1, DIS3L2, DKC1, EGLN1, EPCAM, ERCC1, ERCC2, ERCC3, ERCC4, ERCC5, EXT1, EXT2, EZH2, FAN1, FANCA, FANCB, FANCC, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCL, FANCM, FH, FLCN, GALNT12, GATA2, GPC3, GREM1, HOXB13, HRAS, KIF1B, KIT, LZTR1, MAX, MC1R, MEN1, MET, MITF, MLH1, MLH3, MRE11, MSH2, MSH6, MUTYH, NBN, NF1, NF2, NHP2, NOP10, NTHL1, PALB2, PDGFRA, PHOX2B, PMS2, POLD1, POLE, POLH, POT1, PRKAR1A, PRSS1, PTCH1, PTCH2, PTEN, RAD50, RAD51C, RAD51D, RB1, RECQL4, RET, RUNX1, SDHA, SDHAF2, SDHB, SDHC, SDHD, SLC45A2, SLX4, SMAD4, SMARCA4, SMARCB1, SMARCE1, STK11, SUFU, TERC, TERT, TINF2, TMEM127, TP53, TSC1, TSC2, TYR, VHL, WRAP53, WRN, WT1, XPA, XPC, and XRCC2) offered by Fulgent Diagnostics (Temple City, CA) was used to sequence 76 patients and 4 healthy controls. Next-generation sequencing (NGS) was performed on a HiSeq instrument from Illumina, La Jolla, CA. The average depth of sequencing depth/mean coverage was between 1100x and 1300x. The assay mainly covers the exonic coding regions 10–20 bp into the flanking introns. The promoter region of the PTEN gene was sequenced, and the promoter regions of the BRCA1 and BRCA2 genes were not included in the panel. PMS2 pseudogene detection was performed by Fulgent’s proprietary “Misalignment Tool”, which analyzed both reads from the real gene and the pseudogene. Some patients were also confirmed by long-range PCR for mutations in exon 13 to exon 15 of the PMS2 gene. After sequencing, the Fulgent bioinformatics pipeline was used to obtain a list of variants with data from standards and guidelines for the interpretation of sequence variants recommended by the American College of Medical Genetics and Genomics (ACMG)7,8. Each variant for every sample was analyzed in detail before any classification was made.
FATHMM, SIFT, PolyPhen-2, and CADD
FATHMM
The program Functional Analysis Through Hidden Markov Models (FATHMM) predicts whether single nucleotide variants (SNVs) in the human genome are likely to be functional or nonfunctional in inherited diseases. FATHMM uses distinct models for coding and noncoding regions to improve overall accuracy. The coding predictor is based on six groups of features representing sequence conservation, nucleotide sequence characteristics, genomic features (codons, splice sites, etc.), amino acid features and expression levels in different tissues. We used a threshold of 0, which generated a sensitivity of 0.949.
Sorting intolerant from tolerant (SIFT)
The program sorting intolerant from tolerant (SIFT) predicts whether an amino acid substitution is likely to affect protein function based on sequence homology and the physicochemical similarity between the alternate amino acids10. The data we provide for each amino acid substitution are a score and a qualitative prediction (either “tolerated” or “deleterious”). The score is the normalized probability that the amino acid change is tolerated, so scores closer to zero are more likely to be deleterious. The qualitative prediction is derived from this score such that substitutions with a score <0.05 are called “deleterious”, and all others are called “tolerated”. We used the criteria of <0.05 as damaging and between 0.05–0.07 as probably damaging.
PolyPhen-2
The program PolyPhen-2 predicts the effects of an amino acid substitution on the structure and function of a protein using sequence homology, Pfam annotations, 3D structures from PDB where available, and a number of other databases and tools (including DSSP and ncoils). The PolyPhen score represents the probability that a substitution is damaging, so values closer to one are more confidently predicted to be deleterious11. The qualitative prediction is based on the false- positive rate of the classifier model used to make the predictions. We used the following criteria: scores of 0.446–0.908 as probably damaging and scores of 0.908 or more as damaging.
Combined annotation-dependent depletion (CADD)
The combined annotation-dependent depletion (CADD) tool scores the predicted deleteriousness of single nucleotide variants and insertion/deletion variants in the human genome by integrating multiple annotations, including conservation and functional information, into one metric. Phred-style CADD raw scores are displayed, and variants with higher scores are more likely to be deleterious. CADD provides genome wide scores12. CADD provides a ranking rather than a prediction or default cutoff, with higher scores more likely to be damaging. We used the following criteria: scores of 10–20 as probably damaging and scores of >20 as damaging.
Variants
All variants reported in this study were described according to current HGVS mutation nomenclature guidelines and were verified using Variant Validator13. All variants identified in this study were submitted to the ClinVar database.
Results
Pathogenic or likely pathogenic variants
We found 44 patients with pathogenic or likely pathogenic variants (32 unique variants) in 76 tested patients or family members (58%). The number of patients with BRCA1 and BRCA2 variants was 36, ~82% of this group. These variants were closely split, with 19 patients with BRCA2 (14 unique) and 17 patients with BRCA1 (10 unique) variants. The remaining variants (i.e., 9) were identified in the following genes: BRIP1 (1), CHEK2 (4), NBN (1), and PALB2 (2). The majority of the BRCA1 variants were either frameshift or nonsense mutations with 6 frameshift, 1 nonsense, 1 missense, and 2 intronic mutations. The BRCA2 gene had a slightly different variant comprising 8 frameshift, 4 nonsense, and 2 missense mutations. Variants in the rest of the genes were a mix of frameshift, nonsense, and missense mutations (Table 1). Variant p.Trp1815Ter in the BRCA1 gene was seen in four unrelated patients, all with a family history of cancer, indicating a possible founder mutation in the Armenian population. Variant p.Ile1159Metfs in BRCA1 was identified in the same family members from two different families, and p.Leu1669Ter in BRCA2 was identified in two sisters and one unrelated individual. Additionally, each one of the variants p.Cys1146Leufs in BRCA1 and p.Ala938Profs in BRCA2 were present in two unrelated patients. These data suggest that these variants may also be founder mutations. BRCA1 gene variants were found in three different exons: exon 5 (1), exon 11 (6), and exon 23 (1). The BRCA2 gene variants were found in seven different exons: exon 7 (1), exon 10 (2), exon 11 (7), exon 16 (1), exon 22 (1), exon 23 (1), and exon 25 (1). Two of the pathogenic variants in the BRCA1 gene were splice acceptor site variants (NM_007294.3: c.302-1G>A) and (NM_007294.3: c.4358-2A>G). These splice site mutations could result in a truncated or altered protein, potentially interfering with its function in DNA repair. Other mutations have been reported in the splice site at positions BRCA1 c.302-1 or c.302-2 (source, ClinVar).
We found four patients in this patient cohort that did not report any family history of cancer yet had pathogenic or likely pathogenic variants identified. Three of them had bilateral breast cancer, and one had unilateral breast cancer before the age of 35. Additionally, to the best of our knowledge, one of the identified variants, p.Val875Leu, in BRCA2 has not been reported in any patient suspected of HBOC. A detailed explanation of every pathogenic and likely pathogenic variant annotation is presented in the supplementary material.
Variants of unknown significance (VUSs)
We found 37 variants of unknown significance (VUSs) in 32 patients, and based on our bioinformatics pipeline designation, these variants had indications of being significant but are not well studied or reported (Table 2). We reported a 45% VUS rate, which is higher than the industry-reported 30–40% VUS rate. A detailed explanation of every VUS annotation is presented in the supplementary material. We did not analyze the following VUSs: (MUTYH p.Leu420Met; CHEK2 p.Asp438Tyr; SLX4 p.Cys1805Arg; and MLH1 p.His318Gln) in patients who also carried likely pathogenic variants; these variants are only reported in the Supplemental Table.
Patients with two variants
We also found ten patients with two variants; five patients had one likely pathogenic variant and one VUS, and another five had two VUSs. Four of the patients reported cancer in both maternal and paternal relatives, and two did not report a family history. The other four patients reported cancer only in their maternal relatives, but two of these patients were sisters (Supplemental Table 1).
Patients and genes with no variant and healthy controls
We did not find any VUSs, pathogenic variants, or likely pathogenic variants in six breast cancer patients, and they only had benign/likely benign variants (Supplemental Table 1). Breast cancer in these patients could have been due to environmental factors or possibly genomic structural changes such as large deletions, duplications, and inversions, which were not investigated in this study. Additionally, none of the 76 patients had a VUS, pathogenic, or likely pathogenic variant in any of the following 37 genes: AIP, BAP1, CDC73, CDK4, CDKN1B, CDKN2A, CTC1, CYLD, EPCAM, ERCC1, ERCC3, FAN1, FANCG, FLCN, GALNT12, GATA2, HOXB13, HRAS, MAX, NHP2, PDGFRA, POT1, PRKAR1A, RAD51C, RAD51D, RUNX1, SDHA, SDHAF2, SMAD4, SMARCB1, SMARCE1, SUFU, TERC, TMEM127, TP53, VHL, and XPA.
Three of the four healthy controls were a 92-year-old grandmother and her 72-year-old daughter with no family history of cancer and her 50-year-old grandson with two paternal aunts with breast cancer at ages 75 and 76. The other healthy control was an unrelated 50-year-old female had no family history of cancer. The testing of the 92-year-old grandmother and her 72-year-old daughter showed no pathogenic variants or VUSs, yet the tested 50-year-old grandson had CHEK2 (p.Arg145Trp) likely pathogenic variant, evidently from his paternal side as two of his paternal aunts had breast cancer. The other healthy female control, who was unrelated to the rest of the healthy controls, had no pathogenic variants or VUSs.
Age distribution of variants
We analyzed the age distribution of patients with path/LPath variants. The average age for the Path/LPath group was almost 46 years old. This group had more BRCA1 variants in <40-year-old patients than BRCA2 variants, and these variants were present more often in older populations (up to 70 years old). Interestingly, the non-BRCA variants present in this group were found mostly in the 40- and 50-year-old patients. The average age for BRCA1 variants was 41 years (range: 26–65 years), and the average age for BRCA2 variants was 48 years (range: 29–81 years). The average age for other variants was 50 years (range: 34–68 years) (Fig. 1).

Age distribution of pathogenic and likely pathogenic variants identified in BRCA1, BRCA2, and the other genes.
FATHMM, SIFT, PolyPhen2, and CADD
We used FATHMM, SIFT, PolyPhen2, and CADD prediction programs to assess the functional effects of Path/LPath classifications (Supplementary Table 2) and the VUS classifications (Table 3). One variant (FANCD2 p.Pro593Ser) had a tolerated classification from all four prediction programs, and two other variants (ATR p.Asp331Gly and AXIN2 p.Ala695Ser) were classified as tolerated by the 3 out of 4 programs. Nine of the VUSs had a FATHMM prediction as tolerant, while they had damaging predictions from the other three programs, with the exception of three, had another tolerant prediction (Table 3). These programs use different algorithms; thus, we consider all of the designations significant. All predictions for the Path/LPath variants, except variant p.Val875Leu in BRCA2, were damaging (D) or probably damaging (PD) with significant scores (Supplementary Table 2). Notably, one of the VUS mutations was SLX4 c.5413T>C (p.Cys1805Arg) variant, which has been shown to disrupt SLX4-SLX1 complex formation in functional studies10. The FATHMM classification of this variant was tolerant, yet it had a significant score from the other three programs and was consequently predicted to be damaging. We observed the same pattern in three variants classified as a VUS (ATR c.1602G>C, p.Trp534Cys; PALB2, c.2821A>G, p.Ile941Val; and PALB2 c.3428T>A, p.Leu1143His) (Table 3). Such a comparison could be used for further assessments of variants classified as a VUS. All the variants with a FATHMM classification of a negative score had a risk estimate of being damaging, which indicates that these substitutions are likely to interfere with the function of the protein. Almost all the variants that we classified as a VUS had scores as low as the known pathogenic variants for the SIFT, PolyPhen2, and CADD programs. The variant c.838G>A (p.Ala280Thr) in the WRAP53 gene, which we classified as a VUS, had a damaging or probably damaging score from all the programs. This variant has not been reported in breast cancer, but according to this analysis, it could interfere with the function of the protein.
Discussion
In this study, we reported 44 variants that are responsible for hereditary breast cancer and 37 VUSs that may be involved in hereditary and/or early onset of breast cancer in the Armenian population. The pathogenic/likely pathogenic group had 45 variants, 33 of which were unique, indicating that focusing on so-called founder mutations will miss the majority of the pathogenic/likely pathogenic variants causing breast cancer. We also reported 36 VUSs (31 unique), which were found in functional domains and mostly in mutational hotspots with the potential to interfere with protein function. These variants were found in known cancer-causing genes, such as ATR, BRCA1, CHEK2, MLH1, and MUTYH, and in two less prominent genes, SLX4 and WRAP53. The SLX4 complex is required for the efficient repair of DNA interstrand crosslinks (ICLs)14. The importance of SLX4 for ICL repair was underscored by the findings that biallelic mutations in SLX4 in humans cause Fanconi anemia (FA)15. The SLX1-SLX4 complex has a preference for 5’-flap structures and promotes symmetrical cleavage of static and migrating Holliday junctions (HJs). Finally, Wilson et al. reported that SLX1 foci could not be detected when they overexpressed a mutant form of SLX4 (p.Cys1805Arg) that is incapable of interacting with SLX116. The Variant SLX4 p.Cys1805Arg allele frequency in GnomAD exomes is 0.000008, which does not exceed the estimated maximal expected allele frequency for a pathogenic SLX4 variant of 0.0001, and the variant was not found in GnomAD genomes (PM2 Pathogenic Moderate). Pathogenic predictions from several programs support its deleterious effect (PP3 Pathogenic Supporting). Variant CHEK2 p.Asp438Tyr is in a hotspot region of 7 pathogenic nonsense and frameshift variants (PM1 Pathogenic Moderate). It has been shown to exhibit a 70% reduction in kinase activity in a cell-based assay when compared to that of wild-type CHEK217, suggesting that the variant may have a functional impact. However, it is unclear whether such a reduction in kinase activity would be sufficient to contribute to cancer risk. The allele frequencies of this variant in European and Finnish subpopulations are slightly higher than the estimated maximal expected allele frequency of a pathogenic CHEK2 variant (0.0000284), suggesting that this variant is likely a benign polymorphism. However, population data in this region of CHEK2 are not considered reliable due to high pseudogene homology18. The variant has been reported in breast, ovarian, and prostate cancer patients in the literature, without strong evidence for causality. However, one case–control study reported a significant association with prostate cancer19. It was reported as a VUS in male breast cancer20. In our study, this variant was found in two sisters with breast cancer at ages 44 and 51, and both carry the BRCA2 p.Val875Leu likely pathogenic variant. Variant MLH1 p.His318Gln is in a mutation hotspot of 22 pathogenic nonsense and frameshift variants (PM1 Pathogenic Moderate). This variant is not reported in either GnomAD exomes or in GnomAD genomes (PM2 Pathogenic Moderate). A total of 207 out of 273 non-VUS missense mutations in the MLH1 gene are pathogenic (PP2 Pathogenic Supporting). Pathogenicity prediction software does not agree on the potential impact of this missense change. The variant is reported in ClinVar as a VUS (Variation ID: 187486). This variant has not been reported in the literature in individuals with MLH1-related disease. The MLH1 p.His318Gln variant was identified in a patient (i.e., P-34) who also had the LPath variant p.Ala2951Ser in the BRCA2 gene. Since the evidence on the contribution of pathogenic variants in mismatch repair genes for breast cancer is not very strong, it is more likely that the LPath variant in BRCA2 plays a crucial role in the development of familial breast cancer, and the involvement of the variant p.His318Gln in the MLH1 gene in the patient’s breast cancer may be marginal or limited.
Variant MUTYH p.Leu420Met is in a mutation hotspot along with 12 pathogenic variants (PM1 Pathogenic Moderate). Additionally, 61 of 104 non-VUS missense mutations in the MUTYH gene were pathogenic (PP2 Pathogenic Supporting). The variant was observed in ExAC with an allele frequency of 71/120676 (1/1699), which does not exceed the estimated maximal expected allele frequency for a pathogenic MUTYH variant of 1/219. Pathogenic predictions from multiple programs support its deleterious effect (PP3 Pathogenic Supporting). This variant has been reported in a family with HNPCC and breast, pancreatic, and colorectal cancers. A functional study indicated that the variant acted comparable to wild-type21. Meanwhile, MUTYH heterozygous or homozygous mutations among breast cancer patients with or without a history of the disease evidenced an association of MUTYH with an increased risk of BC22. The MUTYH p.Leu420Met variant was identified in two patients (i.e., P-13 and P-70). Patient 13 also carried the LPath variant c.4358-2A>G in the BRCA1 gene, which could play a more crucial role in the development of this patient’s breast cancer. Therefore, the role of the MUTYH p.Leu420Met variant may also be marginal.
Variant p.Trp1815Ter in the BRCA1 gene was found in three unrelated female patients who were 26, 29, and 38 years old. In contrast, the male patient who also had this variant was 65 years old, a late onset for this variant in male patients. Variant p.Ile1159Metfs was found in two sets of siblings, two sisters at ages 31 and 32, and a brother/sister pair at ages 54 and 57. This difference in the age of onset for these two sibling pairs suggests a variable penetrance for this mutation. Perhaps the twin sisters had another genetic or environmental contributing factor for their early onset of breast cancer. Variant p.Leu1669Ter in BRCA2 was also seen in two sisters with breast cancer at ages 26 and 56; it was also seen in an 81-year-old female. This large age gap between these three patients is another example of deleterious mutations in a BRCA gene that manifest variable penetrance.
During analysis of the age distribution for variants, we realized that more BRCA1 and BRCA2 variants were classified in the pathogenic group. This was expected since these two genes are studied incomparably more than others and were the first genes to be tested for HBOC. This study and similar studies emphasize the significance of comprehensive gene panels to identify non-BRCA variants that could be involved in hereditary breast cancer. The percentage of found VUSs using a comprehensive cancer panel with 127 genes did increase the number of VUSs, and it allowed us to find several variants implicated in hereditary cancers, 8 of which have not been reported in suspected HBOC patients. VUS rates have been recently reported as 34.8% in a combined study conducted in Greece, Turkey, and Romania using a 36-gene panel23. Testing of 127 genes resulted in the identification of a 45% VUS rate, which is higher than the reported rate in the recent study where a 36-gene panel was used. However, this rate is quite reasonable, considering a threefold increase in the number of genes tested compared to the currently used panels.
In conclusion, our study identified variants involved in breast cancer in the Armenian population. We also reported nine novel variants (Tables 1 and 2) that, to the best of our knowledge, had not been reported previously in patients with breast cancer. We realized that variants with a higher frequency or possible founder mutations represented only 10% of the variants, thereby missing the rest. Thus, we concluded that testing with comprehensive cancer panels increases the chances of finding cancer-causing variants in genes that are not routinely tested for in breast cancer patients. These patients and perhaps their family members would need genetic counseling before and after testing to help them understand their treatment and prevention measures, such as surgical intervention, targeted therapy, and surveillance strategies.
References
LaDuca, H. et al. Utilization of multigene panels in hereditary cancer predisposition testing: analysis of more than 2,000 patients. Genet. Med. 43, 295–305 (2014).
Dillenburg, C. V. et al. Prevalence of 185delAG and 5382insC mutations in BRCA1, and 6174delT in BRCA2 in women of Ashkenazi Jewish origin in southern Brazil. Genet. Mol. Biol. 35, 599–602 (2012).
Borg, A. et al. Characterization of BRCA1 and BRCA2 deleterious mutations and variants of unknown clinical significance in unilateral and bilateral breast cancer.: the WECARE study. Hum. Mutat. 31, E1200–E1240 (2010).
Babikyan, D. T. & Sarkisian, T. F. Preliminary genetic investigation of high-risk breast cancer patients in Armenia. Eur. J. Hum. Genet. 17, 191 (2009).
Atshemyan, S., Chavushyan, A., Berberian, N., Sahakyan, A. & Zakharyan, R. Characterization of BRCA1/2 mutations in patients with family history of breast cancer in Armenia. F1000Res 10, 29 (2017).
Ready, K. J. et al. Accuracy of the BRCAPRO model among women with bilateral breast cancer. Cancer 115, 725–730 (2009).
Maxwell, K. N. et al. Evaluation of ACMG-guideline-based variant classification of cancer susceptibility and non-cancer-associated genes in families affected by breast cancer. Am. J. Hum. Genet. 98, 801–817 (2016).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Shihab, H. A. et al. An integrative approach to predicting the functional consequences of non-coding and coding sequence variation. Bioinformatics 31, 1536–43 (2015).
Vaser, R., Adusumalli, S., Leng, S., Sikic, M. & Ng, P. SIFT missense predictions for genomes. Nat. Protoc. 11, 1–9 (2016).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Rentzsch P., Witten D., Cooper G. M., Shendure J., Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 29. https://doi.org/10.1093/nar/gky1016. (2018).
Freeman, P. J., Hart, R. K., Gretton, L. J., Brookes, A. J. & Dalgleish, R. Variant validator: accurate validation, mapping and formatting of sequence variation descriptions. Hum. Mutat. 39, 61–68 (2018).
Wilson, J. S. et al. Localization-dependent and independent roles of SLX4 in regulating telomeres. Cell Rep. 4, 853–60 (2013).
Fekairi, S. et al. Human SLX4 is a Holliday junction resolvase subunit that binds multiple DNA repair/recombination endonucleases. Cell 138, 78–79 (2009).
Kim, Y. et al. Mutations of the SLX4 gene in fanconi anemia. Nat. Genet. 43, 142–146 (2011).
Bell, D. W. et al. Genetic and functional analysis of CHEK2 (CHK2) variants in multiethnic cohorts. Int. J. Cancer 121, 2661–2667 (2007).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–91, https://doi.org/10.1038/nature19057 (2016).
Southey, M. C. et al. PALB2, CHEK2 and ATM rare variants and cancer risk: data from COGS. J. Med. Genet. 53, 800–811 (2016).
Pritzlaff, M. et al. Male breast cancer in a multi-gene panel testing cohort: insights and unexpected results. Breast Cancer Res. Treat. 161, 575–586 (2017).
Komine, K. et al. Functional Complementation assay for 47 MUTYH variants in a MutY-disrupted Escherichia coli strain. Hum. Mutat. 36, 704–711 (2015).
Dutil, J. et al. Germline variants in cancer genes in high-risk non-BRCA patients from Puerto Rico. Sci. Rep. 9, 17769 (2019).
Tsaousis, G. N. et al. Analysis of hereditary cancer syndromes by using a panel of genes: novel and multiple pathogenic mutations. BMC Cancer 19, 535, https://doi.org/10.1186/s12885-019-5756-4 (2019).
Acknowledgements
This work was made possible by contributions from the Armenian American Medical Society (AAMS), Center for Medical Genetics and Primary Health Care, Morava Scientific and Technology Services, Analysis Research & Planning for Armenia (ARPA), Armenian Relief Society (ARS Glendale, CA), and Urmia Armenian Society. We would like to acknowledge Dr. Anatole Ghazalpour for his constructive comments.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Moradian, M.M., Babikyan, D.T., Markarian, S. et al. Germline mutational spectrum in Armenian breast cancer patients suspected of hereditary breast and ovarian cancer. Hum Genome Var 8, 9 (2021). https://doi.org/10.1038/s41439-021-00140-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-021-00140-2
This article is cited by
-
Reclassifying BRCA1 c.4358-2A > G and BRCA2 c.475 + 5G > C variants from “Uncertain Significance” to “Pathogenic” based on minigene assays and clinical evidence
Journal of Cancer Research and Clinical Oncology (2024)
-
Ethnicity-specific BRCA1, BRCA2, PALB2, and ATM pathogenic alleles in breast and ovarian cancer patients from the North Caucasus
Breast Cancer Research and Treatment (2024)
-
Rare germline variants in POLE and POLD1 encoding the catalytic subunits of DNA polymerases ε and δ in glioma families
Acta Neuropathologica Communications (2023)
