
Abstract
Complementary to hydrophobic five membered ring β-amino acids (e.g. ACPC), β-sugar amino acids (β-SAAs) have found increasing application as hydrophilic building blocks of foldamers and α/β chimeric peptides. Fmoc-protected β-SAAs [e.g. Fmoc-RibAFU(ip)-OH] are indeed useful Lego elements, ready to use for SPPS. The removal of 1,2-OH isopropylidene protecting group increasing the hydrophilicity of such SAA is presented here. We first used N3-RibAFU(ip)-OH model compound to optimize mild deprotection conditions. The formation of the 1,2-OH free product N3-RibAFU-OH and its methyl glycoside methyl ester, N3-RibAFU(Me)-OMe were monitored by RP-HPLC and found that either 50% TFA or 8 eqv. Amberlite IR-120 H+ resin in MeOH are optimal reagents for the effective deprotection. These conditions were then successfully applied for the synthesis of chimeric oligopeptide: -GG-X-GG- [X=RibAFU(ip)]. We found the established conditions to be effective and—at the same time—sufficiently mild to remove 1,2-O-isopropylidene protection and thus, it is proposed to be used in the synthesis of oligo- and polypeptides of complex sequence combination.
Similar content being viewed by others

Introduction
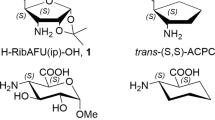
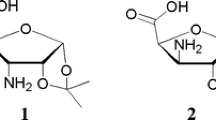
Oligopeptides containing β-amino acids (β-peptides) have favorable backbone folding properties as foldamers (Gellman 1998; Hill et al. 2001; Seebach et al. 1996). Numerous oligomers made from diastereomers of 2-aminocyclopentanecarboxylic acid (ACPC) (Abraham et al. 2010; Appella et al. 1999a; Martinek et al. 2002) and 2-aminocyclohexanecarboxylic acid (ACHC) (Appella et al. 1996; Appella et al. 1999b; Hetényi et al. 2005) are regarded as benchmark nanosystems incorporating cyclic β-amino acids. However, the hydrophobic character enhanced by ACPC and ACHC residues is a serious drawback during their potential physiological application. In fact, their homooligomers are insoluble in water (Hetényi et al. 2009). This can be amended by introducing sugar amino acids (SAAs) (Risseeuw et al. 2009, 2013) which are more hydrophilic by nature. It has been shown particularly, both for five- and six-membered cyclic SAAs (H-SAA-OHs) to behave as appropriate building blocks (Nagy et al. 2017; Csordás et al. 2016; Goldschmidt Gőz et al. 2018; Suhara et al. 2006; Chandrasekhar et al. 2004; Gruner et al. 2002a, b; Pandey et al. 2011). Furanoid β-SAAs, e.g., 3-amino-3-deoxy-d-furanuronic acids (AFUs), primarily, d-xylo (1, 2) and d-ribo (3, 4) epimeric pairs (Nagy et al. 2017) are hydrophilic analogs of cis- and trans-ACPC (Fig. 1). The cost-effective synthesis of oligopeptides incorporating both furanoid and pyranoid β-SAAs were recently done in our group (Csordás et al. 2016; Nagy et al. 2019).

Some hydrophobic β-AAs, the cis- and the trans-ACPC and their hydrophilic furanoid analogues (β-SAAs): H-XylAFU(ip)-OH (1), H-XylAFU-OH (2), H-RibAFU(ip)-OH (3) and H-RibAFU-OH (4)
The stable protection of the hydroxyl groups of β-SAA is essential during the synthesis of oligo- and polypeptides. Afterwards, the removal of their protecting groups is of a special interest to enhance the hydrophilicity of the parent chimera peptides (Suhara et al. 2006; Schweizer 2002; Gruner et al. 2002a, b; Chakraborty et al. 2002). In principle, the hydrophilic moieties ensure the better compatibility with the living organism of aqueous media. In addition, the introduction of more hydrophilic residues makes these types of β-SAA building blocks tunable and more versatile and thus, enhanced biocompatibility could be achieved, otherwise crucial for drug delivery and biomedical applications.
Both the N3-RibAFU(ip)-OH (7) and Fmoc-RibAFU(ip)-OH (8) β-SAA have their 1,2-O-isopropylidene protecting groups originating from 1,2;5,6-di-O-isopropylidene-d-glucofuranose (5) (Scheme 1) (Nagy et al. 2017). Note that, the original 5,6-O-isopropylidene group was removed in one of the intermediary steps. The selective deprotection of 5,6-O-isopropylidene acetals of d-Glc is described using protic acids: e.g., H2SO4 (Rbaa et al. 2020; Miljkovic 2009; Sukumar et al. 1986), HClO4 on silica gel (Agarwal and Vankar 2005), polyphosphoric acid on silica gel (Nikam and Gore 2020) or AcOH (Ma et al. 2020; Pikas et al. 2020; Ferreira et al. 2020; Ravn et al. 2019; Gruner et al. 2002a, b; Yadav, Chander and Reddy 1992). The application of some Lewis acids was also reported, such as FeCl3·6H2O/SiO2 (Kim et al. 1986), CuCl2·2H2O (Iwata and Ohrui 1981), Zn(NO3)2·6H2O (Vijayasaradhi, Singh and Aidhen 2000), Yb(OTf)3·H2O (Yadav et al. 2001) just as BiCl3 (Swamy and Venkateswarlu 2002). Alternative methods for the selective removal of the isopropylidene protection under relatively mild conditions were studied more recently as aqueous tert-butyl hydroperoxide (Maddani and Prabhu 2011), CBr4-photoirradiation (Chen et al. 2004) and acid zeolites (Rauter et al. 2010). The removal of 2,3-O-isopropylidene acetals is also presented in acidic conditions of TFA (Bornaghi et al. 2004; Ganapati and Arvind 2020; Ferreira et al. 2020; Gelin et al. 2020; Pogula et al. 2020; Ahmed-Belkacem et al. 2020), AcOH (Decultot et al. 2020), HCl (Ko et al. 2017) or BCl3 (Yamamoto et al. 2019; Yoo et al. 2018). For deprotection of 1,2-O-isopropylidene moieties, fewer approaches were mentioned, including H2SO4 (Masamune et al. 2001; Yanaisaka et al. 1970), HCl (Sorensen et al. 2001; Yanaisaka et al. 1970), TsOH (Yuan et al. 2020; Sukumar et al. 1986; Rosenthal and Cliff 1980), aqueous TFA (Piccini et al. 2020; Fernandez-Bolanos and Lopez 2007) or cationic exchange resins (e.g. Dowex-50 H+) (Weber et al. 1986; Fleet and Smith 1985). The deprotection conditions for various di-O-isopropylidene of sugar scaffolds are summarized in Table 1.

The synthesis of the furanoid β-SAA, Fmoc-RibAFU(ip)-OH (8) starts from d-Glc, turned subsequently into the azido derivative (7). Reagents and conditions: a (CH3)2CO, H2SO4; b NaH, Im2SO2, DMF; c NaN3, Bu4NBr, toluene; d 75% AcOH; e 1. NaIO4, MeOH-H2O, 2. KMnO4, 50% AcOH; f 10% Pd/C, MeOH (H-Cube®); g Fmoc-OSu, THF, MeOH-H2O; h CH3NH2.HCl, EDC.HCl, DIEA, HOBt, DCM
Although several papers described various conditions for the deprotection of isopropylidene acetals, no case of such 1,2-O-isopropylidene removal from SAAs oligopeptides is known. Only 2,3-O-isopropylidene removal was reported, when cyclic dimer of protected 5-aminomethyl-3,4-dihydroxy-tetrahydrofuran-2-yl-acetic acid (H-SAA-OH) without 2,3-O-isopropylidene acetal was formed in aqueous TFA (Bornaghi et al. 2004). In a patent of antibody–drug conjugates, a linker as MC-SAA-Phe-Cit-APEA containing 5-azidomethyl-3,4-dihydroxy-tetrahydrofuran-2-yl-acetic acid (N3-SAA-OH) was synthesized and the 2,3-O-isopropylidene protection was removed with aqueous HCl, following the peptide synthesis (Ko et al. 2017). Linkers having high hydrophilic SAAs were designed to reduce significantly or even avoid the use of organic solvents during the conjugation process. This can enhance high conjugation efficiency and the stability of conjugate products, which is important in drug development.
Therefore, it seems essential to develop applicable approaches for peptides having 1,2-O-isopropylidene protected SAA building blocks. The main goals we set out to: (1) free the furanoid 1,2-OHs to increase the hydrophilicity of the molecule and (2) form methyl glycoside to prevent furanoid ring opening. Consequently, we synthesized our model oligopeptide Ac-GG-X-GG-R (Nagy et al. 2019) this time with X=RibAFU(ip) and R=OH or NH2 and the removal conditions of the 1,2-O-isopropylidene protecting group were successfully probed.
Results and discussion
Realizing the two main goals, we took into account the essential criteria in both cases: only the planned reactions might happen and unwished transformations in the polypeptide chain (chain scission, rearrangement, elimination, etc.) cannot occur.
To work out the conditions suitable for removal of 1,2-O-isopropylidene protection, the easily accessible intermediate N3-RibAFU(ip)-OH (7) (Scheme 1) was selected and probed. In addition, N3-RibAFU(ip)-NHMe (11) (Nagy et al. 2017), the amide derivative of 7, was also used as proper model to provide the possibility of the comparison with the related oligopeptides.
An efficient multigram synthesis of the sugar amino acid Fmoc-RibAFU(ip)-OH (8) was completed previously from d-Glc (Nagy et al. 2017). The deprotection of the 5,6-O-isopropylidene moiety was achieved with diluted acetic acid (Scheme 1, step d). Then, N3-RibAFU(ip)-OH (7) was obtained from 6 and in the final steps Fmoc-RibAFU(ip)-OH (8) product was achieved. We have shown that Fmoc-RibAFU(ip)-OH (8) is a suitable monomer for solid-phase peptide synthesis (SPPS), as both the 1,2-OHs and the amino function are protected (Nagy et al. 2019).
The deprotection was carried out under strong acidic conditions (Scheme 2) and the reactions were followed by RP-HPLC. Chromatograms were well resolved and clearly showed the steadily decrease in concentration of the starting 7 (retention time 11.9 min).

Derivatives 7 and 11 used for optimizing conditions of the 1,2-O-isopropylidene removal. Route A (see SFig. 22, SFig. 23, SFig. 24, SFig. 26); Route B (SFig. 27); Route C (Fig. 2, Fig. 3, Table 2, SFig. 20, SFig. 21, SFig. 25, STable 1, STable 2)
In Route A: Compound 7 was dissolved in the mixture of TFA in DCM of 50%, 70% and 90% concentrations in parallels together with a small amount of H2O (2.5%) and TIS (2.5%) as scavengers. All reactions were carried out at room temperature. Analysis of RP-HPLC diagrams resulted in evaluable data of the reaction with varied concentration of TFA. Since component 9 has hydrophilic character during the deprotection reactions, instead of a conventional C-18 column, Hydro-RP LC column was used for RP-HPLC system. The hydrolysis of 7 (retention time 21.6 min) was almost completed and no intermediate was detected (SFig. 22-24). The new signal at 4.3 min was indicated and the reaction mixture was processed to give white oil, however, attempts to isolate a pure product failed.
Due to the free OH-1 in 9 such mixture of the anomers could be expected. This fact was indicated by ESI–MS exhibiting a peak at m/z 188.03029 (SFig. 7) of the unprotected carboxylic acid (9). Further studies with HILIC LC–MS (Hydrophilic Interaction Chromatography) corroborated the presence of 9 in the mixture. In these chromatograms, the signal at 2.13 min was assigned to the starting 7 based on the ESI–MS m/z 228.06094 [M−H]− (SFig. 14). Furthermore, the signal at 10.46 min was attributed to the product 9 in accordance with ESI–MS m/z 188.03057 [M−H]− (SFig. 15). This expected product (9) might be also highly unstable, as solely the related O-protected derivatives were described in the literature (Nagy et al. 2017; Csordás et al. 2016; Gruner et al. 2002a, b; Masamune et al. 2001; Dauban et al. 1996). Both the starting 7 and the product 9 could be detected with their exact masses, however, the retention time of 7 was short and the peak shape of 9 was not sufficient sharp. Indeed, the separation has to be further optimized to obtain better quality chromatographic peaks, thus, the integration and comparison can be made for following the reactions.
Analogue reaction of N3-RibAFU(ip)-NHMe (11) in TFA/DCM (50%) with TIS (2.5%) and H2O (2.5%) was stirred at room temperature for 1 h to be completed according to TLC and RP-HPLC (SFig. 26). The signal of the expected product 12 appeared at about 2 min and indicated the mixture of products also in this case. Working up the reaction mixture gave a solid which could not be separated to give pure anomers. Formation of more products was attributed to the free OH-1 in 12—as in the case of 9. Additional reasons for the dispersity of the peak at 2 min was revealed by ESI–MS exhibiting the isotopic peak of an isobaric contaminant at m/z 203.822 beside of the peak of the mixture of 12 at m/z 203.07803 (SFig. 10).
In Route B: Compound 7 was dissolved in the solution of TFA/dried MeOH in parallel concentrations of 30%, 50% and 70%. The mixtures were kept for 18 h at room temperature then were processed to give oily products from each sample. Analysis with RP-HPLC revealed complex mixtures in all cases (SFig. 27). Only the decreasing signal of the starting 7 was unambiguously assigned between 11.7 and 12 min, respectively, depending on the TFA concentration. Further components—intermediates and side products—were detected in all cases. The formation of the intermediate 14 in the reaction mixture of concentration of 30% TFA was identified (SFig. 5). Attempted separation of the components failed from the oily mixtures, thus, Route B was not continued.
In Route C: These reactions of the starting 7 were carried out under heterogeneous conditions: Amberlite IR-120 H+ resin was used as the acid component in dried MeOH. The mol ratio was in parallel reactions: 2, 4, 8 and 12 eqv. The mixtures were kept at 60 °C, 40 °C and at room temperature till to the reaction was complete. Analysis of RP-HPLC chromatograms revealed that 1,2-O-isopropylidene deprotection on 7 (11.9 min) occurred, parallel, with the formation of the methyl ester methyl furanoside (10) as final main product (9.0 min). Besides, two further peaks were detected at 6.8 min and at 15.3 min, respectively. These were reasonably assigned to the temporary intermediate methyl esters (14 and 15). The best results were obtained from the reaction with 8 eqv. Amberlite IR-120 H+ at 60 °C when almost complete transformation occurred in 360 min (Fig. 2). Parallel reactions at 40 °C and at room temperature terminated after 1 day and 4 days, respectively.

The 1,2-O-isopropylidene removal from N3-RibAFU(ip)-OH as function of the time resolved by RP-HPLC: compound 7 treated with 8 eqv. of Amberlite IR-120 H+ resin in MeOH at 60 °C. Decreasing concentration of the starting 7 and the increasing one of the main product (10) were assigned with 1H-NMR and MS data
The conversion to product 10 was near to 90% in each case (Table 2, STable 1, STable 2). Quantitative analysis of the RP-HPLC diagram of the best experiment revealed the disappearance of the starting 7 after 60 min, and that of the intermediate 14 after 240 min (Fig. 3). The mixture of products contained mainly the final product 10 (89%) and also intermediate 15 (~ 10%; not isolated) (Table 2).

a The gradual concentration changes of the starting 7, intermediates (14 and 15) and the final product (10) as function of time (0 → t(min) → 360) at 60 °C, with 8 eqv. Amberlite IR-120 H+ in MeOH. b The concentration decrease of the starting 7 as function of time (0 → t(min) → 120) at 60 °C, with different equivalents of Amberlite IR-120 H+ in MeOH
Working up the mixture gave inseparable oil. The 1H NMR and ESI–MS were measured with the best mixture. The 1H NMR signals of the 1,2-O-isopropylidene group of the starting 7 (at δ 1.28 and 1.44 ppm) disappeared in the spectrum of the methyl glycoside product (10). At the same time, the spectrum of the latter exhibited the characteristic methyl signals of the methyl ester and the methyl O-glycoside at 3.84 and 3.42 ppm, respectively (Fig. 4 and SFig. 4). The ratio of the anomers calculated from the H-1 signals shows that α-anomer (3J coupling of H1-H2: < 1.0 Hz) is the main component and the β-anomer (3J couplings of H1-H2: 11.5 Hz) is the minor one. ESI–MS exhibited m/z [M+Na]+ 240.1 for C7H11N3O5 supporting the molecular composition of 10. The small quantity of the minor component did not allow the detailed analysis, however, the evident presence of 15 in the reaction mixture was supported by the mechanism of the complex reaction (Scheme 1).

a 1H NMR spectra of the starting (7) showing 1,2-O-isopropylidene signals at 1.28 and 1.44 ppm, respectively. b The characteristic methyl group resonances of the methyl ester and the methyl O-glycoside of (10) at 3.42 and 3.84 ppm, respectively
Analogue reaction of N3-RibAFU(ip)-NHMe (11) was carried out under the best conditions as in the case of 7: with Amberlite IR-120 H+ (8 eqv.)/dried MeOH at 60 °C and completed within 300 min. Analysis of RP-HPLC diagrams revealed that 1,2-O-isopropylidene deprotection on 11 (9.9 min) occurred, parallel, with the formation of the expected methyl furanoside (13) as final main product (at 4.5 min, SFig. 25). Besides, a second product was observed at 3 min, probably, the 1,2-(OH)2 derivative (12). Working up the reaction mixture gave an inseparable oily product. The main component of the mixture was identified by ESI–MS which exhibited two strong peaks (m/z 217.09303 [M+H]+ and m/z 239.07487 [M+Na]+) corresponding to the structure of 13. Other two peaks were also measured (m/z 203.07725 [M′+H]+ and m/z 225.19593 [M′+ Na]+) which were attributed to the side product 12 in the mixture (SFig. 11).
Synthesis and deprotection of a model chimera peptide
Our ultimate goal was to apply the currently fine-tuned deprotection method for making various peptides and chimera sequences. The previously successfully applied -GG-X-GG- model system was synthesized here with X=RibAFU(ip) and conditions to remove its 1,2-O-isopropylidene protecting group were probed. The synthesis of -GG-RibAFU(ip)-GG- was carried out with Fmoc-strategy introducing Fmoc-RibAFU(ip)-OH (8) on RAM-Tentagel® or 2-Cl-Trt-Cl resin (Scheme 3) by PyBOP/DIEA which was found as one of the most effective coupling reagents for SAAs (Nagy et al. 2019; Goldschmidt Gőz et al. 2019). The N-terminus of this model peptide were protected to avoid unwanted side-reactions (e.g. esterification). Peptide 18 was used for evaluation of peptide synthesis method and an intermediate of methyl glycoside formation. The deprotection of 1,2-O-isopropylidene moiety was carried out with the mixture of TFA/DCM/TIS/H2O which is in fact a common cleavage cocktail in SPPS. The 50% TFA condition was used to remove peptides from the resin and in parallel to complete the 1,2-O-isopropylidene deprotection (Scheme 3, step e). The removal of the 1,2-O-isopropylidene group gave successfully both pentapeptides with free 1,2-OHs (16, 17). In the mixture of the crude product, 4:1/α:β anomeric ratio was observed at 11.29 min and 12.04 min, respectively (Fig. 5). The formation of methyl glycoside was accomplished after the final cleavage (Scheme 3, step g and h) using two routes. The Amberlite IR-120 H+ (8 eqv.)/MeOH condition was executed on compound 18 with 1,2-O-isopropylidene protection (Route D) and on compound 17 with fully unprotected SAA (Route E). In both cases, the Ac-GG-RibAFU(Me)-GG-OMe (19) pentapeptide was observed. However, Route E was faster for methyl glycoside formation, with retention times of 4.75 min and 5.54 min presenting a 4:1/α:β anomeric ratio, respectively (SFig. 19).

Solid-phase chimera oligopeptide synthesis of -GGXGG- on RAM-Tentagel® or 2-Cl-Trt-Cl resin. The removal of the 1,2-O-isopropylidene protection from Ac-GG-RibAFU(ip)-GG- was successful using the following reagents and conditions: a Fmoc-RibAFU(ip)-OH (8) with PyBOP/DIEA; b Fmoc-GG-OH with PyBOP/DIEA; c Piperidine (2%)/DBU (2%) in DMF; d Ac2O:DIEA:DMF (1:1.2:3); e TFA (50%)/DCM (45%)/TIS (2.5%)/H2O (2.5%); f AcOH:MeOH:DCM (1:1:8); g IR-120 H+ (8 eqv.)/MeOH, 60 °C, 6 h (see SFig. 19); h IR-120 H+ (8 eqv.)/MeOH, 60 °C, 3 h from compound 17 (see SFig. 18). Ratios of α/β anomers were determined by HILIC LC–UV–MS

HILIC LC–UV–MS chromatogram of Ac-GG-RibAFU-GG-NH2 (16) and HCD MS/MS spectrum of its singly protonated compound acquired at 20 eV collision energy
Although RP-HPLC was used to follow the deprotection reaction, there were components (9, 12, 13) and peptides (16, 17, 19) having a short retention time due to their increased hydrophilicity. The analysis of these polar compounds was a challenge, as their interaction with C-18 stationary phases was insufficient, leading to a very difficult product isolation. Products (9, 16, 17, 19) were characterized via HILIC LC–UV–MS (Jablonski, Hudalla and Fountain 2012; Yoshida 2004) measurements with BEH Amide column capable of analyzing hydrophilic SAAs or sequences (Fig. 5). The optimized condition was found to enhance the retention of hydrophilic polypeptides, making analysis and isolation more adequate.
Conclusions
A rather general 1,2-O-isopropylidene deprotection method was worked out for α/β-chimera peptides using a suitable d-ribo furanoid β-SAA model system, namely the N3-RibAFU(ip)-OH synthetic intermediate. To obtain the unprotected derivatives, two different conditions were studied successfully, namely (1) various TFA concentrations to form the free 1,2-OH product (9) and (2) various equivalents of Amberlite IR-120 H+ resin or TFA in MeOH forming methyl glycoside (10) to prevent the furanoid ring opening. In the first case, 50% TFA in DCM with TIS and H2O as scavengers was found to give the fully unprotected compound (N3-RibAFU-OH, 9). Furthermore, 8 eqv. Amberlite IR-120 H+ resin in MeOH at 60 °C turned out to be optimum to furnish methyl glycoside methyl ester N3-RibAFU(Me)-OMe (10). These optimized methods, 50% TFA and H+ resin/MeOH, were applied successfully during the synthesis of α/β-chimera oligopeptide [-GG-RibAFU(ip)-GG-] to form the Ac-GG-RibAFU-GG-NH2 (16) without 1,2-O-isopropylidene protection and the methyl glycoside variant, namely the Ac-GG-RibAFU(Me)-GG-OMe (19). The described, fine-tuned conditions of deprotection were shown to be appropriate and sufficiently mild to remove 1,2-O-isopropylidene protection in oligo- and polypeptides of more complex amino acid sequences.
Experimental section
Reagents and instrumentations
Reagents, materials and solvents were obtained from Sigma-Aldrich, Merck, Reanal and VWR. Moisture-sensitive solvents were dried on molecular sieve (3 Å). The capacity of Amberlite IR-120 H+ resin was 1.80 eq/L. Reactions were followed by RP-HPLC on a Phenomenex Jupiter C-18 column or Synergy™ 4 μm Hydro-RP 80 Å LC column with eluents 0.1% TFA in H2O (A) and 0.08% TFA, 95% acetonitrile/5% H2O (B), flow rate 1.0 ml/min and UV detection at 220 and 280 nm. Gradient were as follows: 0 min: 0% B, 30 min: 60% B, 32 min: 95% B, 33 min: 0% B, 45 min: 0% B and 45.1 min: 0% B for Phenomenex Jupiter C-18 column, and 0 min: 0% B, 30 min: 60% B, 33 min: 95% B, 39 min: 95% B, 40 min: 0% B, 45 min: 0% B and 45.2 min: 0% B for Synergy™ 4 μm Hydro-RP 80 Å LC column. MS spectra were performed with Bruker Esquire 3000 + tandem quadrupole mass spectrometer equipped with an electrospray ion source. FTIR spectra were recorded on a Bruker IFS 28 spectrometer by ATR technique. 1H NMR measurements were implemented with Bruker Avance 250 spectrometer in CDCl3 or DMSO-d6 at room temperature. Deuterated solvents were purchased from Eurisotop. Hydrophilic compounds were analyzed by HILIC LC–UV–MS approach. The measurements were executed on a column Waters Acquity BEH Amide UPLC (2.1 × 150 mm, 1.7 µm) using 20 mM ammonium acetate (A) and 100% acetonitrile (B) with flow rate 250 µl/min, UV detection at 210 and 280 nm and 40 °C column temperature. Dionex 3000 UHPLC was coupled to a Q Exactive Focus orbitrap mass spectrometer (Thermo Scientific, Bremen, Germany). ESI–MS spectra were acquired in m/z 200–800 (spray voltage: 3.5 kV; sheath gas: 46 au; aux. gas: 11 au; capillary temp: 360 °C; probe heater: 406 °C). MS/MS spectra were acquired using higher energy collision induced dissociation (HCD) at 20 eV. Gradients were as follows: 0 min: 90% B, 2 min: 90% B, 22 min: 40% B, 23 min: 40% B, 24 min: 90% B and 30 min: 90% B for peptides, and 0 min: 95% B, 2 min: 95% B, 22 min: 60% B, 23 min: 60% B, 24 min: 95% B and 30 min: 95% B for SAAs.
1,2-O-Isopropylidene-3-azido-3-deoxy-α-d-ribofuranuronic acid (7)
Compound 7 was prepared based on previous results of our group (Nagy et al. 2017). RP-HPLC: 11.9 min or 21.6 min; FTIR-ATR: cm−1 νmax: 3500–2400 (OH), 2112 (N3), 1724 (C=O); ESI–MS: m/z calculated for C8H11N3O5 [M−H]− 228.06205, found 228.06207; 1H NMR (DMSO-d6, 250 MHz) δ ppm 5.84 (d, J = 3.3 Hz, 1H), 4.78 (m, 1H), 4.35 (d, J = 9.7 Hz, 1H), 3.85 (dd, J = 9.6 and 4.5 Hz, 1H), 1.44 (s, 3H), 1.28 (s, 3H); HILIC LC–UV–MS: 2.13 min, m/z calculated for C8H11N3O5 [M−H]− 228.06204, found 228.06094.
1,2-O-Isopropylidene-N-(9-fluorenylmethoxy-carbonyl)-3-amino-3-deoxy-α-d-ribofuranuronic acid (8)
Compound 8 was prepared based on previous results of our group (Nagy et al. 2017). RP-HPLC: 21.0 min; FTIR-ATR: cm−1 νmax: 3500–2400 (OH), 3367 (NH), 1720 (C=O), 1692 (aromatic); ESI–MS: m/z calculated for C23H23NO7 [M+H]+ 426.2, found 426.2; 1H NMR (DMSO-d6, 250 MHz) δ ppm 7.89 (d, 2H), 7.69 (m, 3H), 7.36 (m, 5H), 5.85 (s, 1H), 4.61 (s, 1H), 4.29 (m, 3H), 4.01 (m, 1H), 1.46 (s, 3H), 1.26 (s, 3H).
N-Methyl-1,2-O-isopropylidene-3-azido-3-deoxy-α-d-ribofuranuronamide (11)
1,2-O-isopropylidene-3-azido-3-deoxy-α-d-ribofuranuronic acid (7, 200 mg; 0.87 mmol) was dissolved in DCM (49 ml), then CH3NH2·HCl (120 mg), EDC·HCl (671 mg), DIEA (0.6 ml) and HOBt (235 mg) were added. The mixture was stirred at room temperature for 3 h (EtOAc-Hex 2:1). After the reaction was complete, DCM (25 ml) was added to the mixture and extracted with 2 N HCl (2 × 50 ml), then washed with H2O (1 × 50 ml), saturated NaHCO3 solution (1 × 50 ml) and H2O (1 × 50 ml). The organic phase was dried (Na2SO4), filtered and concentrated in vacuo. The residue was treated by hexane to afford the product as white solid (160 mg, 76%). RP-HPLC: 9.9 min; ESI–MS: m/z calculated for C9H14N4O4 [M+H]+ 243.10933 and [M+Na]+ 265.09128, found 243.10859 and 265.09035, respectively; 1H NMR (CDCl3, 250 MHz) δ ppm 6.43 (s, 1H), 5.84 (d, J = 3.3 Hz, 1H), 4.71 (t, J = 3.6 Hz, 1H), 4.48 (d, J = 9.5 Hz, 1H), 3.65 (dd, J = 9.3 and 4.3 Hz, 1H), 2.86 (d, J = 4.9 Hz, 3H), 1.57 (s, 3H), 1.37 (s, 3H).
The removal of 1,2-O-isopropylidene protection from the azido derivative (7)
3-Azido-3-deoxy-d-ribofuranuronic acid (9)
(A) 1,2-O-Isopropylidene-3-azido-3-deoxy-α-d-ribofuranuronic acid (7, 100 mg; 0.44 mmol) was dissolved in 5 ml of different concentrations of TFA (50%, 70%) in DCM (45% and 25%, respectively) with TIS (2.5%) and H2O (2.5%). The mixtures were stirred at room temperature for 3 h and evaporated in vacuo. The residues were dissolved in 1,2-dimethoxyethane (5 ml) and concentrated to obtain white oils in both cases. RP-HPLC: 4.3 min; ESI–MS: m/z calculated for C5H7N3O5 [M−H]− 188.03075, found 188.03029; HILIC LC–UV–MS: 10.46 min, m/z calculated for C5H7N3O5 [M−H]− 188.03074, found 188.03057.
(B) 1,2-O-Isopropylidene-3-azido-3-deoxy-α-d-ribofuranuronic acid (7, 50 mg; 0.22 mmol) was added into 2.5 ml mixtures of TFA (50%, 70%, 90%) in DCM (45%, 25%, 5%, respectively), TIS (2.5%) and H2O (2.5%). Reactions were stirred at room temperature. Fractions of reaction mixtures were concentrated by air blowing, diluted in water then measured by RP-HPLC (SFig. 22, SFig. 23, SFig. 24).
Methyl 3-azido-3-deoxy-d-ribofuranuronate methyl ester (10)
(A) 1,2-O-Isopropylidene-3-azido-3-deoxy-α-d-ribofuranuronic acid (7, 50 mg; 0.22 mmol) was dissolved in dried MeOH (2 ml) and Amberlite IR-120 H+ resin (2, 4, 8 and 12 eqv. corresponding to 100, 200, 400 or 600 mg) were added. The mixtures were heated to 60 °C for 6 h. The reactions were followed by RP-HPLC (SFig. 20, SFig. 21). The mixtures were filtered and washed with MeOH. Filtrates were evaporated in vacuo to obtain oil products. The mixture of products containing mainly the final product 10 (~ 90%) was characterized. RP-HPLC: 9.0 min; ESI–MS: m/z calculated for C7H11N3O5 [M+Na]+ 240.1, found 240.1; 1H NMR (CDCl3, 250 MHz) δ ppm 4.93 (s, 1H), 4.58 (d, J = 7.2 Hz, 1H), 4.36 (m, 1H), 4.16 (d, J = 4.4 Hz, 1H), 3.83 (s, 3H), 3.42 (s, 3H).
(B) 1,2-O-Isopropylidene-3-azido-3-deoxy-α-d-ribofuranuronic acid (7, 50 mg; 0.22 mmol) was dissolved in dried MeOH (2 ml) and Amberlite IR-120 H+ resin (400 mg, 8 eqv.) was added to the solution. The reaction mixture was stirred at different temperatures (60 °C, 40 °C and RT). Fractions of reaction mixtures were neutralized (NaHCO3), centrifuged, diluted in MeOH then measured by RP-HPLC (STable 1, STable 2).
(C) 1,2-O-Isopropylidene-3-azido-3-deoxy-α-d-ribofuranuronic acid (7, 50 mg; 0.22 mmol) was added into 2 ml mixtures of TFA (30%, 50%, 70%) and dried MeOH (70%, 50%, 30%, respectively). Reactions were stirred at room temperature overnight and evaporated in vacuo to obtain oil product. Mixtures were measured by RP-HPLC (SFig. 27).
The removal of 1,2-O-isopropylidene protection from methylamide derivative (11)
N-Methyl-3-azido-3-deoxy-d-ribofuranuronamide (12)
Compound 11 (40 mg; 0.17 mmol) was dissolved in the mixture (2 ml) of TFA (50%), DCM (45%), TIS (2.5%) and H2O (2.5%) and stirred at room temperature for 1 h. The reaction was monitored by TLC (EtOAc-Hex 2:1) and RP-HPLC (SFig. 26). The solvent was removed in vacuo and the residue was treated with cold Et2O to precipitate the solid product. ESI–MS: m/z calculated for C6H10N4O4 [M+H]+ 203.07803, found 203.07722.
Methyl N-methyl-3-azido-3-deoxy-d-ribofuranosiduronamide (13)
Compound 11 (40 mg; 0.17 mmol) was dissolved in dried MeOH (1.5 ml) and Amberlite IR-120 H+ resin (300 mg, 8 eqv.) was added to the solution. The reaction was stirred at 60 °C for 6 h, monitored by TLC (EtOAc-Hex 2:1) and RP-HPLC. The mixture was filtered and washed with MeOH. The filtrate was concentrated in vacuo to achieve the oil product. ESI–MS: m/z calculated for C7H12N4O4 [M+Na]+ 239.07563 and [M+H]+ 217.09368, found [M+Na]+ 239.07487 and [M+H]+ 217.09303, respectively (SFig. 25).
Model peptide
SPPS of the model peptide was executed manually on RAM-Tentagel® or 2-Cl-Trt-Cl resin with the standard methodology using Fmoc-strategy. Resins were swollen in DCM. Coupling of Fmoc-Gly-Gly-OH to resins was implemented in two different methods. For RAM-Tentagel® resin, the first step was the removal of Fmoc-group conducted with 2% piperidine and 2% DBU in DMF (10 + 20 min). The coupling was accomplished using Fmoc-Gly-Gly-OH (3 eqv. to the nominal capacity of the resin ~ 0.24 mmol/g) dissolved in DMF and PyBOP (3 eqv.)/DIEA (6 eqv.) added to the solution. In the case of 2-Cl-Trt-Cl resin, the coupling was made with Fmoc-Gly-Gly-OH (1.5 eqv. to the nominal capacity of the resin ~ 1.60 mmol/g to tune down to 0.28 mmol/g) which was dissolved in DMF and DIEA (3.75 eqv.) was added to the solution. Afterwards, amino acids were coupled to resins using reagent pairs PyBOP/DIEA in DMF. Coupling of amino acids lasted for 1 h, whereas that of Fmoc-RibAFU(ip)-OH finished in 3 h. After coupling, resins were washed with 3 × DMF, 3 × DCM, 2 × MeOH, 1 × Et2O and dried in vacuo. The capacity of the resin was determined by spectrometric measurement of the amount of Fmoc chromophore (Fmoc-piperidine adduct) released upon treatment of the resin with 50% piperidine in DMF (Chan and White 2000). Fmoc-deprotection was done by 2% piperidine and 2% DBU in DMF (3 + 17 min). The successful removal was analyzed by Kaiser test. The acetylation was performed with Ac2O:DIEA:DMF (1:1.2:3) for 30 min.
Ac-Gly-Gly-RibAFU-Gly-Gly-NH2 (16)
The peptide was cleaved from RAM-Tentagel® resin (50 mg) with TFA (50%), DCM (45%), TIS (2.5%) and H2O (2.5%) for 3 h. The resin was washed with 3 × DCM and 3 × MeOH, then solvent was removed in vacuo. The residue was treated with cold Et2O to precipitate white solid product (4.9 mg). HILIC LC–UV–MS: 11.29 min and 12.04 min, m/z calculated for C15H24N6O9 [M+H]+ 433.1683 and [M+Na]+ 455.1502, found 433.1676 and 455.1493, respectively.
Ac-Gly-Gly-RibAFU-Gly-Gly-OH (17)
The peptide was cleaved from 2-Cl-Trt-Cl resin (150 mg) by a mixture of TFA (50%), DCM (45%), TIS (2.5%) and H2O (2.5%) for 3 h. The resin was washed with 3 × DCM and 3 × MeOH, then solvent was removed in vacuo. By treating the residue with cold Et2O, the white solid product was precipitated (14.1 mg). HILIC LC–UV–MS: 12.68 min and 12.94 min, m/z calculated for C15H23N5O10 [M+H]+ 434.15232, [M+Na]+ 456.13426 and [M+H–H2O]+ 416.14176, found 434.15109, 456.413304 and 416.14060, respectively.
Ac-Gly-Gly-RibAFU(ip)-Gly-Gly-OH (18)
The peptide was cleaved from 2-Cl-Trt-Cl resin (150 mg) with AcOH:MeOH:DCM (1:1:8) for 3 h. The resin was washed with 3 × DCM, 3 × iPrOH and 1 × Et2O. The solvent was removed in vacuo. The residue was precipitated in cold Et2O to obtain white solid product (11.4 mg, 58%). HILIC LC–UV–MS: 9.91 min, m/z calculated for C18H27N5O10 [M+H]+ 474.18362, found 474.18276.
Ac-Gly-Gly-RibAFU(Me)-Gly-Gly-OMe (19)
(A) Peptide 17 (13 mg) was dissolved in dried MeOH with Amberlite IR-120 H+ (8 eqv.). The mixture was stirred at 60 °C for 3 h, filtered and washed with MeOH. The filtrate was concentrated in vacuo. The product as white solid was precipitated with cold Et2O (7.8 mg). HILIC LC–UV–MS: 4.75 min and 5.54 min, m/z calculated for C17H27N5O10 [M+H]+ 462.18362 and [M+H–CH3OH]+ 430.15741, found 462.18214 and 430.15641, respectively.
(B) Peptide 18 (10 mg) was dissolved in dried MeOH with Amberlite IR-120 H+ (8 eqv.). The mixture was stirred at 60 °C for 6 h, filtered and washed with MeOH. The filtrate was concentrated in vacuo and the residue was treated in cold Et2O to obtain white solid product (6.7 mg).
Abbreviations
- ACPC:
-
2-Aminocyclopentanecarboxylic acid
- ACHC:
-
2-Aminocyclohexanecarboxylic acid
- β-SAA:
-
β-Sugar amino acid
- SPPS:
-
Solid-phase peptide synthesis
- H-XylAFU(ip)-OH:
-
1,2-O-Isopropylidene-3-amino-3-deoxy-α-d-xylofuranuronic acid
- H-XylAFU-OH:
-
3-Amino-3-deoxy-α-d-xylofuranuronic acid
- H-RibAFU(ip)-OH:
-
1,2-O-Isopropylidene-3-amino-3-deoxy-α-d-ribofuranuronic acid
- H-RibAFU-OH:
-
3-Amino-3-deoxy-α-d-ribofuranuronic acid
- N3-RibAFU(ip)-OH:
-
1,2-O-Isopropylidene-3-azido-3-deoxy-α-d-ribofuranuronic acid
- Fmoc-RibAFU(ip)-OH:
-
1,2-O-Isopropylidene-N-(9-fluorenylmethoxy-carbonyl)-3-amino-3-deoxy-α-d-ribofuranuronic acid
- N3-RibAFU(ip)-NHMe:
-
N-Methyl-1,2-O-isopropylidene-3-azido-3-deoxy-d-ribofuranuronamide
- N3-RibAFU-OH:
-
3-Azido-3-deoxy-α-d-ribofuranuronic acid
- N3-RibAFU-NHMe:
-
N-Methyl-3-azido-3-deoxy-d-ribofuranuronamide
- N3-RibAFU(Me)-OMe:
-
Methyl 3-azido-3-deoxy-d-ribofuranuronate methyl ester
- N3-RibAFU(Me)-NHMe:
-
Methyl N-methyl-3-azido-3-deoxy-d-ribofuranosiduronamide
References
Abraham E et al (2010) A systematic study of the solid state and solution phase conformational preferences of β-peptides derived from transpentacin. Tetrahedron Asymmetry 21:1797–1815. https://doi.org/10.1016/j.tetasy.2010.12.007
Agarwal A, Vankar YD (2005) Selective deprotection of terminal isopropylidene acetals and trityl ethers using HClO4 supported on silica gel. Carbohydr Res 340:1661–1667. https://doi.org/10.1016/j.carres.2005.04.005
Ahmed AMA, Mohammed AI, Read RW (2020) Towards functional fluorous surfactants. Synthesis and spectroscopic features of systematically modified sugar-substituted fluorous 1,2,3-triazoles. J Fluor Chem. https://doi.org/10.1016/j.jfluchem.2020.109519
Ahmed-Belkacem R, Sutto-Ortiz P, Guiraud M, Canard B, Vasseur J-J, Decroly E, Debart F (2020) Synthesis of adenine dinucleosides SAM analogs as specific inhibitors of SARS-CoV nsp14 RNA cap guanine-N7-methyltransferase. Eur J Med Chem. https://doi.org/10.1016/j.ejmech.2020.112557
Appella DH, Christianson LA, Karle IL, Powel DR, Gellman SH (1996) β-peptide foldamers: robust helix formation in a new family of β-amino acid oligomers. J Am Chem Soc 118:13071–13072. https://doi.org/10.1021/ja963290l
Appella DH, Christianson LA, Karle IL, Powell DR, Gellman SH (1999b) Synthesis and characterization of trans-2-aminocyclohexanecarboxylic acid oligomers: an unnatural helical secondary structure and implications for β-peptide tertiary structure. J Am Chem Soc 121:6206–6212. https://doi.org/10.1021/ja990748l
Appella DH, Christianson LA, Klein DA, Richards MR, Powell DR, Gellman SH (1999a) Synthesis and structural characterization of helix-forming β-peptides: trans-2-aminocyclopentanecarboxylic acid oligomers. J Am Chem Soc 121:7574–7581. https://doi.org/10.1021/ja991185g
Bornaghi LF, Wilkinson BL, Kiefel MJ, Poulsen SA (2004) Synthesis of cyclic oligomers of a modified sugar amino acid utilising dynamic combinatorial chemistry. Tetrahedron Lett 45:9281–9284. https://doi.org/10.1016/j.tetlet.2004.10.064
Chakraborty TK, Ghosh S, Jayaprakash S (2002) Sugar amino acids and their uses in designing bioactive molecules. Curr Med Chem 9:421–435. https://doi.org/10.2174/0929867023370941
Chan WC, White PD (2000) Fmoc solid phase peptide synthesis: a practical approach. Oxford University Press, Oxford
Chandrasekhar S, Reddy MS, Jagadeesh B, Prabhakar A, Rao MHV, Jagannadh B (2004) Formation of a stable 14-helix in short oligomers of furanoid cis-β-sugar-amino acid. J Am Chem Soc 126:13586–13587. https://doi.org/10.1021/ja0467667
Chen M, Patkar LN, Lu K, Lee AS, Lin C (2004) Chemoselective deprotection of acid labile primary hydroxyl protecting groups under CBr4-photoirradiation conditions. Tetrahedron 60:11465–11475. https://doi.org/10.1016/j.tet.2004.09.095
Csordás B, Nagy A, Harmat V, Zsoldos-Mády V, Leveles I, Pintér I, Farkas V, Perczel A (2016) Origin of problems related to staudinger reduction in carbopeptoid syntheses. Amino Acids 48:2619–2633. https://doi.org/10.1007/s00726-016-2289-x
Dauban P, Chiaroni A, Riche C, Dodd RH (1996) Synthesis of optically pure 3,4-disubstituted l-glutamates from a novel 2,3-aziridino-γ-lactone 4-carboxylate derivative. J Org Chem 61:2488–2496. https://doi.org/10.1021/jo951983z
Decultot L, Policarpo RL, Wright BA, Huang D, Shair D (2020) Asymmetric total synthesis of C9′-epi-Sinefungin. Org Lett 22:5594–5599. https://doi.org/10.1021/acs.orglett.0c01956
Fernandez-Bolanos JG, Lopez O (2007) Heterocycles from carbohydrate isothiocyanates. Top Heterocycl Chem 7:67–100. https://doi.org/10.1007/7081_2007_052
Ferreira JT, Pina J, Ribeiro CAF, Fernandes R, Joao PC, Tome JPC, Rodriguez-Morgade MS, Torres T (2020) Highly efficient singlet oxygen generators based in ruthenium phthalocyanines: synthesis, characterization and in vitro evaluation for photodynamic therapy. Chem Eur J 26:1789–1799. https://doi.org/10.1002/chem.201903546
Fleet GW, Smith GPW (1985) Enantiospecific syntheses of deoxymannojirimycin, fagomine and 2R,5R-dihydroxymethyl-3R,4R-dihydroxypyrrolidine from d-glucose. Tetrahedron Lett 26(11):1469–1472. https://doi.org/10.1016/S0040-4039(00)99073-7
Ganapati G, Arvind ASR (2020) Nicotinyl riboside compounds and their uses. PCT Int Appl WO 2020/131578, pp 136–168
Gasch C, Merino-Montiel P, Lopez O, Fernandez-Bolanos JG, Fuentes J (2010) Spiranic d-gluco-configured N-substituted thiohydantoins as potential enzymatic inhibitors. Tetrahedron 66:9964–9973. https://doi.org/10.1016/j.tet.2010.09.109
Gelin M, Paoletti J, Nahori M-A, Huteau V, Leseigneur C, Jouvion G, Dugue L, Clement D, Pons J-L, Assairi L, Pochet S, Labesse G, Dussurget O (2020) From substrate to fragments to inhibitor active in vivo against Staphylococcus aureus. ACS Infect Dis 6:422–435. https://doi.org/10.1021/acsinfecdis.9b00368
Gellman SH (1998) Foldamers: a manifesto. Acc Chem Res 31:173–180. https://doi.org/10.1021/ar960298r
Goldschmidt Gőz V, Pintér I, Harmat V, Perczel A (2018) Approaches to pyranuronic β-sugar amino acid building blocks of peptidosaccharide foldamers. Eur J Org Chem 3:355–361. https://doi.org/10.1002/ejoc.201701612
Goldschmidt Gőz V, Nagy A, Farkas V, Keszei E, Perczel A (2019) Unwanted hydrolysis or α/β-peptide bond formation: how long should the rate-limiting coupling step take? RSC Adv 9:30720–30728. https://doi.org/10.1039/c9ra06124j
Gruner SAW, Locardi E, Lohof E, Kessler H (2002a) Carbohydrate-based mimetics in drug design: sugar amino acids and carbohydrate scaffolds. Chem Rev 102(2):491–514. https://doi.org/10.1021/cr0004409
Gruner SAW, Truffault V, Voll G, Locardi E, Stockle M, Kessler H (2002b) Design, synthesis, and NMR structure of linear and cyclic oligomers containing novel furanoid sugar amino acids. Chem Eur J 8(19):4365–4376. https://doi.org/10.1002/1521-3765(20021004)8:19%3c4365:AID-CHEM4365%3e3.0.CO;2-U
Hetényi A, Mándity IM, Martinek TA, Tóth GK, Fülöp F (2005) Chain-length-dependent helical motifs and self-association of β-peptides with constrained side chains. J Am Chem Soc 127:547–553. https://doi.org/10.1021/ja0475095
Hetényi A, Tóth G, Somlai C, Vass E, Martinek T, Fülöp F (2009) Stabilisation of peptide foldamers in an aqueous medium by incorporation of azapeptide building blocks. Chem Eur J 15:10736–10741. https://doi.org/10.1002/chem.200900724
Hill DJ, Mio MJ, Prince RB, Hughes TS, Moore JS (2001) A field guide to foldamers. Chem Rev 101:3893–4011. https://doi.org/10.1021/cr990120t
Iwata M, Ohrui H (1981) A simple regioselective partial hydrolysis of di-O-isorpopylidene monosaccharides with copper(II) ion. Bull Chem Soc Jpn 54(9):2837–2838. https://doi.org/10.1246/bcsj.54.2837
Jablonski JM, Hudalla CJ, Fountain KJ (2012) Preparative scale chromatography of a hydrophilic peptide using hydrophilic interaction chromatography. Water Application Note Part number:720004283en
Kim KS, Song YH, Lee BH, Hahn CS (1986) Efficient and selective cleavage of acetals and ketals using ferric chloride adsorbed on silica gel. J Org Chem 51:404–407. https://doi.org/10.1021/jo00353a027
Ko YJ, Li JS, Lu M, Lee O, Cheng PF, Liu CM (2017) Antibody-drug conjugate (ADC) and method for forming the same. European Patent Application EP 3 165 237 A1, pp 1–25
Li Q, Levi S, Jacobsen EN (2020) Highly selective β-mannosylations and β-rhamnosylations catalyzed by a bis-thiourea. J Am Chem Soc 142:11865–11872. https://doi.org/10.1021/jacs.0c04255
Ma R, Zhao Y, Liu L, Zhu Z, Wang B, Wang Y, Yin X, Su J, Zhou Y (2020) Novel position-specific 18O/16O measurement of carbohydrates. II. The complete intramolecular 18O/16O profile of the glucose unit in a starch of C4 origin. Anal Chem 92:7462–7470. https://doi.org/10.1021/acs.analchem.9b05314
Maddani MR, Prabhu KR (2011) Metal-free deprotection of terminal acetonides by using tert-butyl hydroperoxide in aqueous medium. Synlett 6:821–825. https://doi.org/10.1055/s-0030-1259917
Martinek TA, Tóth GK, Vass E, Hollósi M, Fülöp F (2002) cis-2-aminocyclopentanecarboxylic acid oligomers adopt a sheetlike structure: switch from helix to nonpolar strand. Angew Chem Int Ed 41(10):1718–1721. https://doi.org/10.1002/1521-3773(20020517)41:10%3c1718::AID-ANIE1718%3e3.0.CO;2-2
Masamune H, Deninno MP, Scott RW (2001) Compounds for the treatment of ischemia. PCT Int Appl WO 2001023399 A1 20010405, pp 40–46
Miljkovic M (2009) Cyclic acetals and ketals. In: Carbohydrates: synthesis, mechanisms and stereoelectronic effects. Springer Science & Business Media, pp 143–167. https://doi.org/10.1007/978-0-387-92265-2_6
Nagy A, Csordás B, Zsoldos-Mády V, Pintér I, Farkas V, Perczel A (2017) C-3 epimers of sugar amino acids as foldameric building blocks: improved synthesis, useful derivatives, coupling strategies. Amino Acids 49:223–240. https://doi.org/10.1007/s00726-016-2346-5
Nagy A, Goldschmidt Gőz V, Pintér I, Farkas V, Perczel A (2019) α/β-chimera peptide synthesis with cyclic β-sugar amino acids: the efficient coupling protocol. Amino Acids 51:669–678. https://doi.org/10.1007/s00726-019-02702-9
Nikam RR, Gore KR (2020) A mild and convenient approach for selective acetonide cleavage involved in carbohydrate synthesis using PPA-SiO2. J Carbohydr Chem. https://doi.org/10.1080/07328303.2019.1708374
Pandey SK, Jogdand GF, Oliveira JCA, Mata RA, Rajamohanan PR, Ramana CV (2011) Synthesis and structural characterization of homochiral homo-oligomers of parent cis- and trans-furanoid-β-amino acids. Chem Eur J 17:12946–12954. https://doi.org/10.1002/chem.201101855
Piccini M, Leak DJ, Chuck CJ, Buchard A (2020) Polymers from sugars and unsaturated fatty acids: ADMET polymerisation of monomers derived from d-xylose, d-mannose and castor oil. Polym Chem 11:2681–2691. https://doi.org/10.1039/C9PY01809C
Pikas D, Riesendeld J, Leontein K, Koch E, Oscarson S (2020) Preparation of heparin fragments and immobilized biological entities. PCT Int Appl WO 2020/070258, pp 47–53
Pogula PK, Chatterjee AD, Chi M, VanKoten HW, Das S, Patterson SE (2020) Triazoxins: novel nucleosides with anti-Giardia activity. Bioorg Med Chem Lett. https://doi.org/10.1016/j.bmcl.2020.127175
Rauter AP, Xavier NM, Lucas SD, Santos M (2010) Zeolites and other silicon-based promoters in carbohydrate chemistry. Adv Carbohydr Chem Biochem 63:29–99. https://doi.org/10.1016/S0065-2318(10)63003-X
Ravn J, Rosenbohm C, Reddy P (2019) Novel process for making allofuranose from glucofuranose. PCT Int Appl WO 2019/224172, pp 1–35
Rbaa M, Abousalem AS, Rouifi Z, Lakhrissi L, Galai M, Zarrouk A, Lakhrissi B, Lakhrissi Y (2020) Selective synthesis of new sugars based on 8-hydroxyquinoline as corrosion inhibitors for mild steel in HCl solution—effect of the saturated hydrocarbon chain: theoretical and experimental studies. Inorg Chem Commun. https://doi.org/10.1016/j.inoche.2020.108019
Risseeuw MDP, Vandermarel GA, Overkleeft HS, Overhand M (2009) Pyranocyclopropanyl sugar amino acids, a new class of constrained (di)peptide isosteres. Tetrahedron Asymmetry 20:945–951. https://doi.org/10.1016/j.tetasy.2009.03.009
Risseeuw M, Overhand M, Fleet GWJ, Simone MI (2013) A compendium of cyclic sugar amino acids and their carbocyclic and heterocyclic nitrogen analogues. Amino Acids 45:613–689. https://doi.org/10.1007/s00726-013-1521-1
Rosenthal A, Cliff BL (1980) Synthesis of an analog of the nucleoside moiety of the polyoxins. Carbohydr Res 79:63–77. https://doi.org/10.1016/S0008-6215(00)85132-0
Schweizer F (2002) Glycosamino acids: building blocks for combinatorial synthesis—implications for drug discovery. Angew Chem Int Ed 41:230–253. https://doi.org/10.1002/1521-3773(20020118)41:2%3c230:AID-ANIE230%3e3.0.CO;2-L
Seebach D, Overhand M, Kuehnle FNM, Martinoni B (1996) β-peptides: synthesis by Arndt-Eistert homologation with concomitant peptide coupling. Structure determination by NMR and CD spectroscopy and by X-ray crystallography. Helical secondary structure of a β-hexapeptide in solution and its stability towards pepsin. Helv Chim Acta 79(4):913–941. https://doi.org/10.1002/hlca.19960790402
Shuai M, Wenhe Z, Wang Y, Zhongjun L (2020) Stereoselective phenylselenoglycosylation of glycals bearing a fused carbonate moiety toward the synthesis of 2-deoxy-β-galactosides and β-mannosides. Org Lett 22:2981–2986. https://doi.org/10.1021/acs.orglett.0c00732
Sorensen MH, Nielsen C, Nielsen P (2001) Synthesis of a bicyclic analogue of AZT restricted in an unusual O4′-endo conformation. J Org Chem 66:4878–4886. https://doi.org/10.1021/jo010299j
Suhara Y, Kurihara M, Kittaka A, Ichikawa Y (2006) Efficient synthesis of carbopeptoid oligomers: insight into mimicry of β-peptide. Tetrahedron 62:8207–8217. https://doi.org/10.1016/j.tet.2006.05.080
Sukumar M, Jacques V, Pendri Y, Falck JR (1986) Synthesis of 12(S),20-, 12(S),19(R)-, and 12(S),19(S)-dihydroxyeicosa-cis-5,8,14-trans-10-tetraenoic acids, metabolites of 12(S)-hete. Tetrahedron Lett 27(24):2679–2682. https://doi.org/10.1016/S0040-4039(00)84615-8
Sun J, Fang J, Xiao X, Cai L, Zhao X, Zeng J, Wan Q (2020) Total synthesis of tricolorin A via interrupted pummerer reaction mediated glycosylation and one-pot relay glycosylation. Org Biomol Chem 18:3818–3822. https://doi.org/10.1039/D0OB00513D
Swamy NR, Venkateswarlu Y (2002) A mild and efficient method for chemoselective deprotection of acetonides by bismuth(III) trichloride. Tetrahedron Lett 43:7549–7552. https://doi.org/10.1016/S0040-4039(02)01809-9
Tian G, Hu J, Qin C, Li L, Zou X, Cai J, Seeberger PH, Yin J (2020) Chemical synthesis and immunological evaluation of Helicobacter pylori Serotype O6 tridecasaccharide O-antigen containing a unique dd-heptoglycan. Angew Chem Int Ed 5:6. https://doi.org/10.1002/anie.202004267
Vijayasaradhi S, Singh J, Aidhen IS (2000) An efficient, selective hydrolysis of terminal isopropylidene acetal protection by Zn(NO3)2·6H2O in acetonitrile. Synlett 1:110–112. https://doi.org/10.1055/s-2000-6458
Vlahovicek-Kahlina K, Vazdar M, Jakas A, Smrecki V, Jeric I (2018) Synthesis of glycomimetics by diastereoselective passerini reaction. J Org Chem 83:13146–13156. https://doi.org/10.1021/acs.joc.8b01874
Weber JF, Talhouk JW, Nachman RJ, You T-P, Halaska RC, Williams TM, Mosher HS (1986) Methyl 2,3-dideoxy-3-nitro-d-eryt-phreontofuranoside, isomers and derivatives. J Org Chem 51:2702–2706. https://doi.org/10.1021/jo00364a015
Xiao X, Zeng J, Fang J, Sun J, Li T, Song Z, Cai L, Wan Q (2020) One-pot relay glycosylation. J Am Chem Soc 142:5498–5503. https://doi.org/10.1021/jacs.0c00447
Yadav JS, Chander MC, Reddy KK (1992) Stereoselective synthesis of 10(S), 11(R), 12(R)-trihydroxyeicosa-5(Z), 8(Z), 14(Z)-trienoic acid from d-mannose. Tetrahedron Lett 33(1):135–138. https://doi.org/10.1016/S0040-4039(00)77695-7
Yadav JS, Reddy BVS, Reddy KS (2001) Yb(OTf)3·H2O: a novel reagent for the chemoselective hydrolysis of isopropylidene acetals. Chem Lett 30:430–431. https://doi.org/10.1246/cl.2001.430
Yamamoto K, Sato T, Hikiji Y, Katsuyama A, Matsumaru T, Yakushiji F, Yokota S-I, Ichikawa S (2019) Synthesis and biological evaluation of a MraY selective analogue of tunicamycins. Nucleosides Nucleotides Nucleic Acids. https://doi.org/10.1080/15257770.2019.1649696
Yanaisaka H, Kinoshita M, Nakada S, Umezawa S (1970) Synthesis of cyclic α-amino Acids. IV. Syntheses of adenine nucleosides of 3-amino-3-C-carboxy-3-deoxy-d-ribopyranose. Bull Chem Soc Jpn 45:246–252. https://doi.org/10.1246/bcsj.43.246
Yoo J, Mashalidis EH, Kuk ACY, Yamamoto K, Kaeser B, Yamamoto K, Kaeser B, Ichikawa S, Lee S-Y (2018) GlcNAc-1-P-transferase–tunicamycin complex structure reveals basis for inhibition of N-glycosylation. Nat Struct Mol Biol 25:217–224. https://doi.org/10.1038/s41594-018-0031-y
Yoshida T (2004) Peptide separation by hydrophilic-interaction chromatography: a review. J Biochem Biophys Methods 60:265–280. https://doi.org/10.1016/j.jbbm.2004.01.006
Yuan C, Schmidt MA, Ortiz A, Rogers AJ, Zhu JJ, Zhongmin X, Yu M, Simmons EM, Wu S (2020) Synthesis of 1,2,5-tri-O-benzoyl-3-dibenzylamino-3-deoxyribose as intermediate for producing 3′-amino-3′-deoxyadenosine and 3′-amino-3′-deoxyguanosine and the protected derivatives thereof. PCT Int Appl WO 2020/163415, pp 1–13
Acknowledgements
The authors gratefully acknowledge Gitta Schlosser for her help in HILIC LC–UV–MS measurements and analysis. We also wish to thank Viktor Farkas for RP-HPLC support and István Varga for NMR measurements. This work was supported by Grant (VEKOP-2.3.2-16-2017-00014) and the purchase of the mass spectrometer was supported by Grant (VEKOP-2.3.3-15-2017-00020) from the European Union and the State of Hungary, co-financed by the European Regional Development Fund.
Funding
Open access funding provided by Eötvös Loránd University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Research involving human participants and/or animals
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
Informed consent was obtained from all individual participants included in this study.
Additional information
Handling Editor: F. Albericio.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Duong, K.H.Y., Goldschmidt Gőz, V., Pintér, I. et al. Synthesis of chimera oligopeptide including furanoid β-sugar amino acid derivatives with free OHs: mild but successful removal of the 1,2-O-isopropylidene from the building block. Amino Acids 53, 281–294 (2021). https://doi.org/10.1007/s00726-020-02923-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-020-02923-3