
Abstract
Overnutrition causes hyperactivation of mTORC1-dependent negative feedback loops leading to the downregulation of insulin signaling and development of insulin resistance. In osteoblasts (OBs), insulin signaling plays a crucial role in the control of systemic glucose homeostasis. We utilized mice with conditional deletion of Rptor to investigate how the loss of mTORC1 function in OB affects glucose metabolism under normal and overnutrition dietary states. Compared to the controls, chow-fed Rptorob−/− mice had substantially less fat mass and exhibited adipocyte hyperplasia. Remarkably, upon feeding with high-fat diet, mice with pre- and post-natal deletion of Rptor in OBs were protected from diet-induced obesity and exhibited improved glucose metabolism with lower fasting glucose and insulin levels, increased glucose tolerance and insulin sensitivity. This leanness and resistance to weight gain was not attributable to changes in food intake, physical activity or lipid absorption but instead was due to increased energy expenditure and greater whole-body substrate flexibility. RNA-seq revealed an increase in glycolysis and skeletal insulin signaling pathways, which correlated with the potentiation of insulin signaling and increased insulin-dependent glucose uptake in Rptor-knockout osteoblasts. Collectively, these findings point to a critical role for the mTORC1 complex in the skeletal regulation of whole-body glucose metabolism and the skeletal development of insulin resistance.
Similar content being viewed by others


Introduction
An appropriate supply of glucose is fundamental for efficient cellular function and requires complex inter-tissue communication networks. The skeleton plays an important role in the regulation of systemic glucose metabolism, with deletion of the insulin receptor (INSR) in the bone-forming osteoblast (OB) (INSROB−/− mice) leading to elevated blood glucose, reduced serum insulin, and insulin resistance.1,2 Conversely, mice with OB-specific deletion of either FoxO1 or ATF4, negative regulators of insulin signaling, display the opposite phenotype to INSROB−/− mice with improved glucose disposal and insulin sensitivity.3,4 Apart from hepatocytes, OBs are the only cell type in which disruption of insulin signaling hampers glucose metabolism in mice fed a normal chow diet (NCD),1 further highlighting the importance of the skeleton in regulating glucose homeostasis.
Mechanistically, insulin signaling in OBs regulates whole-body glucose metabolism via a feed forward loop involving the bone-specific protein osteocalcin (OCN). Undercarboxylated OCN (unOCN) acts as a hormone that improves glucose handling by directly stimulating pancreatic β-cell proliferation5 and insulin secretion6 and indirectly stimulating glucagon-like peptide-1 secretion by the gut.7 unOCN also increases insulin sensitivity in liver, muscle and adipose tissue by increasing adiponectin,6,8 an adipokine that modulates glucose homeostasis independently of insulin. Consistent with these observations, daily administration of OCN to insulin-resistant high-fat diet (HFD)-fed mice partially restores their insulin sensitivity and glucose tolerance.8,9
The mammalian target of rapamycin complex 1 (mTORC1) is a critical mediator of insulin signaling and a primary nutrient-sensing pathway that coordinates anabolic and catabolic processes to control cellular growth and metabolism. Insulin signaling is negatively regulated by mTORC1, which becomes hyper-activated in response to nutrient overload leading to an insulin-resistant state (review in10). Accumulating evidence suggests that in addition to being vital for insulin action at an intracellular level, mTORC1 plays an important role in controlling whole-body glucose metabolism. Indeed, conditional deletion of Rptor (encoding RAPTOR, an essential component of mTORC1) in insulin-responsive tissues results in systemic alterations in metabolism, albeit in a tissue-specific manner.11,12,13
Previous loss-of-function studies by our group14 and others15,16 have revealed an important role for OB-mTORC1 in the control of pre- and post-natal skeletal development. Loss of mTORC1 function in pre-OBs (Rptorob−/− mice) results in osteopenia and skeletal fragility due, in part, to a decrease in the protein synthetic properties of OBs.14 Conversely, mice with OB-specific deletion of Tsc2, a negative regulator of mTORC1 (∆Tsc2 mice), exhibit increased bone mass and accumulation of poorly differentiated OBs.17 With advanced age, ∆Tsc2 mice develop a metabolic phenotype similar to INSROB−/− mice despite having high levels of circulating OCN.17 Collectively, these data suggest a potential role for the mTORC1 complex in osteoblasts (OB-mTORC1) in the skeletal regulation of glucose metabolism.
To investigate this question, we examined the metabolic phenotype of Rptorob−/− mice under both NCD and HFD conditions and found that OB-specific deletion of Rptor has a profound effect on whole-body metabolism, as evidenced by reduced fat mass, increased insulin sensitivity and glucose disposal and elevated serum adiponectin levels. Moreover, both pre- and post-natal deletion of Rptor in osteoprogenitor cells led to protection from HFD-induced weight gain and metabolic dysfunction supporting a central role for the mTORC1 complex in bone in the development of systemic insulin resistance.
Results
Loss of OB-mTORC1 function results in lean mice with increased energy expenditure and a metabolic shift towards fat oxidation
Mice with conditional deletion of Rptor in osteoprogenitor cells were generated as previously described14 and owing to previously observed gene dosage effects in these mice,14,18 both heterozygous (Rptorob+/−) and homozygous (Rptorob−/−) knockout animals analyzed. As our preliminary studies revealed that body weight adjusted fat mass, lean mass adjusted total energy expenditure (TEE), serum OCN levels (both total and undercarboxylated), fasting glucose levels and glucose metabolism (glucose tolerance and insulin sensitivity) are unaltered in Osx1-GFP:Cre mice, relative to age/sex matched wild-type mice, wild-type littermates were used as the control group (Supplementary Fig. 1a–n). It is also important to note that we failed to detect Cre-mediated deletion of Rptor in iWAT and no differences in RAPTOR protein levels in metabolic organs including adipose tissue, muscle, or liver was observed in heterozygous or homozygous knockout animals, relative to controls, suggesting no off-target Rptor deletion (Supplementary Fig. 2a–c). Furthermore, no detectable off-target Cre recombinase activity in metabolic tissues including spleen, liver, brain, and intestine has previously been observed in Osx1-GFP:Cre mice.19
From weaning, NCD-fed Rptorob−/− mice weighed significantly less than control and Rptorob+/− littermates (Fig. 1a). An assessment of body composition by dual X-ray absorptometer (DXA) revealed a significant reduction in %fat mass and body weight-adjusted white adipose tissue (WAT) depots in both Rptorob+/− and Rptorob−/− mice, relative to controls (Fig. 1b, c), while %lean mass and normalized weights of major lean organs were unchanged (Supplementary Fig. 3a, b). Histological examination of WAT depots revealed a significant shift toward adipocyte hypotrophy in gonadal and inguinal WAT (gWAT and iWAT, respectively) in Rptorob−/− mice compared to the controls (Fig. 1d–f). Consistent with this, the expression levels of key adipogenic genes (Pparg, Cebpa, and Lpl) were significantly increased in fat depots of Rptorob−/− mice (Fig. 1g, h). The levels of Fasn mRNA, a regulator of lipogenesis, was decreased in gWAT but increased in iWAT. In contrast, the expression of both lipolytic genes (Atgl and Plin) was increased and unchanged in iWAT and gWAT, respectively, in Rptorob−/− mice compared to the control (Fig. 1g, h).

Loss of mTORC1 function in pre-OBs results in a lean phenotype. a Temporal change in body weight (n ≥ 15/genotype). b %Fat normalized to total body weight. c Body weight-adjusted gonadal and inguinal white adipose tissue (gWAT and iWAT) and interscapular brown adipose tissue (iBAT) mass (9 weeks of age; n = 10/group). d Representative H&E section of gWAT and iWAT, scale bar = 100 µm. e, f Size distribution of gWAT and iWAT adipocytes calculated from H&E stained sections using Image J (n = 5 sections/mouse, n = 3–6/genotype). g, h Gene expression levels of adipogenesis, lipogenesis, and lipolysis genes, normalized to Actb, in gWAT and iWAT (n = 3–5/genotype). i Serum triglyceride levels (n = 8–10/genotype). j Serum-free fatty acid levels (n = 8–10/genotype). k Total energy expenditure, normalized to lean mass, over 48 h and l per light/dark cycle. m Average respiratory quotient (RQ) over 48 h and n per light/dark cycle. Data are expressed as mean ± SEM from n = 7/group, unless indicated otherwise. *P < 0.05, **P < 0.01, ***P < 0.001, one-way ANOVA with Tukey’s post hoc test. k, m Shaded regions represent dark cycle
The reduced adiposity of Rptorob+/− and Rptorob−/− mice was independent of changes in food intake (despite reduced leptin levels (Supplementary Fig. 3c)), total physical activity (XYZ counts) or percentage of fecal lipid content (Supplementary Fig. 3d–g). To account for genotypic differences in body weight, measurements of food intake, oxygen consumption (VO2), carbon dioxide production (VCO2), and TEE data were normalized to lean mass. TEE was significantly increased in Rptorob−/− mice during both the light and dark periods (Fig. 1k, l). Furthermore, Rptorob−/− mice exhibited a significantly higher rate of VO2, with transiently higher rate of VCO2 (Supplementary Fig. 3h-k). Consequently, the respiratory quotient values (RQ, Fig. 1m, n) were significantly lower in Rptorob−/− mice compared to the controls, suggesting a preference for fat oxidation. The elevated TEE and lower RQ observed in the Rptorob−/− mice were associated with an increase in expression of Pcg1a and Ucp1 in interscapular brown adipose tissue (iBAT) (Supplementary Fig. 3l). However, no consistent changes in iBAT UCP1 protein levels were observed suggesting the elevated TEE and lower RQ was not due to increased iBAT thermogenesis (Supplementary Fig. 3m). Measurements of circulating triglyceride (TG) and free fatty acids (FFA) levels revealed a significantly lower TG levels in the serum of Rptorob−/− mice compared to controls, whereas no significant difference was observed in levels of FFAs (Fig. 1i, j).
Rptor ob −/− mice display improved glucose metabolism and increased insulin sensitivity
Fasting glucose levels were significantly lower in Rptorob−/− mice (−22.8% and −14.9% compared to control and Rptorob+/− mice, respectively, Fig. 2a), while no difference was observed between Rptorob+/− and control mice. When fasted animals were challenged with a bolus of glucose, Rptorob−/− mice exhibited enhanced glucose clearance compared to controls, whereas no change in glucose tolerance was observed in Rptorob+/− mice (Fig. 2b, c). Hypoglycemia was not due to hyperinsulinemia, as fasting insulin levels were significantly reduced in both Rptorob−/− and Rptorob+/− mice compared to controls (Fig. 2d). Consistent with lower circulating insulin levels, histological analysis of pancreatic tissue revealed a significant decrease in the β-cell mass and a trend toward a significant decrease in average islet area (P = 0.061) in Rptorob−/− mice, with no significant change in pancreatic islet number (Fig. 2e). Despite their reduced insulin levels, the ability to secrete insulin in response to a glucose bolus [glucose-stimulated insulin secretion (GSIS) tests] in both Rptorob+/− and Rptorob−/− mice remained functional as demonstrated by a significant increase in insulin levels at 30 min post glucose bolus compared to the basal levels (Fig. 2f). Moreover, the relative insulin levels at 30 min (i.e., insulin fold induction relative to basal levels to account for lower basal insulin levels) in response to glucose bolus were significantly higher in Rptorob+/− and Rptorob−/− mice relative to controls (Fig. 2g).

NCD-fed Rptorob−/− mice display an improvement in glucose metabolism. a Fasting blood glucose levels in 9-week-old mice (n = 15/genotype). b GTT blood glucose levels in 9-week-old mice, and c incremental area under the curve analysis (n = 11–14/genotype). d Fasting serum insulin levels (n = 10/genotype). e Representative images of insulin immunostaining in pancreas samples from 9-week-old mice. Scale bars = 100 µm and histomorphometric analysis of β-cell area, islet number, and islet size (n = 6/genotype). f Glucose-stimulated insulin levels and g fold change in insulin levels at 30 min relative to basal levels (n = 9–12/genotype). h ITT blood glucose levels in 8-week-old mice and i area under the curve analysis (n = 11–15/group). All panels except b and h: data are expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, one-way ANOVA with Tukey’s post hoc test. b, h *P < 0.05 between Rptorob−/− and Ctrl, #P < 0.05 between Rptorob−/− and Rptorob+/− and $P < 0.05 between Rptorob+/− and Ctrl, two-way ANOVA with Tukey’s post hoc test
The reduced fasting insulin and glucose levels observed in Rptorob−/− mice, coupled with their improved glucose disposal and ability to maintain lower levels of circulating insulin after glucose bolus, suggest that these mice were more sensitive to insulin compared to their Rptorob+/− and control littermates. As expected, insulin tolerance tests (ITTs) indicated a significant increase in insulin sensitivity in Rptorob−/− mice compared to both controls and Rptorob+/− mice, while no difference was observed in Rptorob+/− mice relative to controls (Fig. 2h, i). Of note, an increased sensitivity to insulin was not attributable to increased insulin sensitivity in muscle and liver, as evidenced by the lack of changes in gene expression for markers of insulin sensitivity (Pcg1α and its target genes, Mcad and Nrf1, in muscle and Foxa2 in liver: Supplementary Fig. 4a, b). No significant changes in the expression levels of gluconeogenesis genes, Pepck and G6p, was observed (P = 0.55 and P = 0.57, respectively, controls vs. Rptorob−/− mice). Conversely, we found that the expression levels of glucokinase, the first-rate limiting enzyme in glycolysis, was upregulated in the Rptorob−/− mice (Supplementary Fig. 4a). Moreover, no significant difference in insulin-stimulated AKT phosphorylation was observed in muscle and liver tissue lysates isolated from Rptorob−/− mice relative to control mice (Supplementary Fig. 4c, d).
The metabolic improvements observed in Rptor ob −/− mice occur independently of osteocalcin
As shown in Fig. 3a–c, OCN gene (Bglap) expression and circulating levels of total and unOCN were significantly reduced in Rptorob+/− and Rptorob−/− mice compared to controls (consistent with the previously reported osteopenia observed in these mice14), suggesting that OCN is unlikely to be mediating the metabolic improvements observed in Rptorob−/− mice. In addition to OCN, more recent studies have shown that the skeleton can regulate appetite and glucose homeostasis via lipocalin-2 (LCN2),20 an OB-enriched, secreted protein. To determine if increased LCN2 levels may be compensating for the decreased unOCN levels observed in Rptorob+/− and Rptorob−/− mice, serum LCN2 levels were measured; however, no differences were found across the three genotypes (Fig. 3d).

Improvements in glucose metabolism observed in NCD-fed Rptorob−/− mice occur independently of osteocalcin. a Bglap gene expression, normalized to Actb, in flushed long bones (femur/tibia, n = 3–4/genotype) from 9-week-old mice. b Serum OCN (total) levels (n = 10–13/genotype). c Serum unOCN levels (n = 8–13/genotype). d Serum lipocalcin-2 levels (n = 7–10/genotype). e Serum adiponectin levels (n = 13/genotype). f, g Western immunoblot analysis and quantification of high molecular weight (HMW), low molecular weight (LMW), and total adiponectin levels in serum (n = 4/genotype). h Adipoq gene expression, normalized to Actb, in gWAT, iWAT, and whole long bone (tibia/femur) samples (n = 3–5/genotype). i Levels of total adiponectin protein expression in gWAT, iWAT, and tibia/femur samples (n = 3–4/genotype) and j Quantitative analysis of adiponectin protein levels, relative to ACTIN, from i. Data are expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, one-way ANOVA with Tukey’s post hoc test
In contrast, the increase in insulin sensitivity in Rptorob−/− mice was associated with a significant (Approximately twofold) elevation in circulating adiponectin levels (Fig. 3e). In particular, levels of the high molecular weight (HMW) form of adiponectin, the most bio-active form which negatively correlates with insulin resistance,21 were significantly higher in Rptorob−/− mice (Fig. 3f, g). Analysis of adiponectin mRNA (Adipoq) and protein levels revealed WAT (iWAT and gWAT) was not the source (Fig. 3h–j). In addition to WAT, a distinct population of adipocytes that reside within the bone marrow adipose tissue (BMAT) has also been shown to contribute to hyperadiponectinemia,22 and we have previously reported markedly elevated BMAT levels in Rptorob−/− mice.14 Consistent with an increase in BMAT, levels of Adipoq mRNA were increased (~2.3-fold) in long bones (combined Tibia/femur: Tib/fem) from Rptorob−/− mice, and a ~2.5-fold increase in adiponectin protein levels was detected by western blot (Fig. 3h–j).
Rptor ob −/− mice are protected from diet-induced obesity and insulin resistance
In light of the metabolic phenotype of NCD-fed Rptorob−/− mice and previous data demonstrating that nutrient overload leads to hyperactivation of mTORC1 signaling and the development of insulin resistance,23 we next examined the response of Rptorob−/− mice to an obesogenic diet. Initial studies, performed on 8-week-old animals maintained on HFD from weaning (i.e., ~4 weeks), showed that this timeframe was insufficient to induce insulin resistance in the controls (Supplementary Fig. 5a–d), thus all subsequent HFD studies were performed on 16-week-old mice following 12 weeks of HFD. As shown in Fig. 4a, b, Rptorob−/− mice gained weight at a similar rate to control and Rptorob+/− mice; however, after 6 weeks of HFD, their weight gains plateaued. End-of-study body composition analyses revealed that Rptorob−/− mice remained relatively lean with fat mass 50% and 66% lower compared to control and Rptorob+/− mice, respectively (Fig. 4c). Fat mass (as percentage of body weight) was significantly lower in Rptorob−/− mice whilst the percentage of lean mass was significantly higher in both Rptorob+/− and Rptorob−/− mice compared to the controls (Fig. 4d). Consistent with this, the major fat pads were significantly smaller in Rptorob−/− mice compared to both control and Rptorob+/− mice, whereas no differences in the weights of other lean organs were observed (Fig. 4e and Supplementary Fig. 6a).

Pre- and post-natal deletion of Rptor protects against high-fat diet-induced obesity and insulin resistance. a Temporal change in body weight (n ≥ 15/genotype). b % of body weight gained in response to high-fat diet (HFD). c Body analysis by DXA scanning of total mass and d composition as percentage of body weight in 18-week-old HFD-fed mice (n = 5/genotype). e Body weight adjusted fat depot (n = 7–10/genotype). f GTT blood glucose levels in 17-week-old mice. g ITT blood glucose levels in 16-week-old mice. h Incremental AUC analysis of GTT from f and AUC analysis of ITT from g (n = 12–15/genotype). i Fasting blood glucose levels (n = 13/genotype). j Fasting serum insulin levels (n = 8–10/genotype). k Schematic showing use of doxycycline (DOX) administration to suppress the tTA:Osx:cre transgene until the pups are 4 weeks of age. DOX chow was replaced with a high-fat diet to induce obesity and insulin resistance (upper panel). Body composition at 4 weeks (HFD day 0; n = 4–6/genotype) and 18 weeks (HFD week 14; n = 7–8/genotype) of age (lower panels). l Body composition as percentage of total body weight n = 7–8/genotype). m GTT blood glucose levels in 17-week-old mice. n ITT blood glucose levels. o Incremental AUC analysis of GTT from m and AUC analysis of ITT from n (n = 12–15/genotype). Data presented as mean ± SEM. a–e *P < 0.05, ***P < 0.001, One-way ANOVA with Tukey’s post hoc test. f, g *P < 0.05 between Rptorob−/− and Ctrl, #P < 0.05 between Rptorob−/− and Rptorob+/− and $P < 0.05 between Rptorob+/− and Ctrl, two-way ANOVA with Tukey’s post hoc test. k–o *P < 0.05, **P < 0.01, Student’s t-test
Strikingly, the metabolic improvements observed in NCD-fed Rptorob−/− mice were also observed in HFD-fed Rptorob−/− mice. Compared to control and Rptorob+/− mice, Rptorob−/− mice exhibited improved glucose clearance and increased insulin sensitivity (Fig. 4f–h), with these improvements accompanied by a reduction in fasting glucose and insulin levels and a trend toward increased GSIS (P = 0.07) (Fig. 4i, j and Supplementary Fig. 6b, c). Moreover, smaller β-cell islet hypertrophy were observed in Rptorob−/− mice (Supplementary Fig. 6d) which is likely to be a secondary effect of persistently low glucose levels.
Rptorob−/− mice have significantly lower body weight adjusted fat mass, under both normal chow and high-fat feeding, which is likely to play an important role in the metabolic phenotype of these mice. The lower %fat mass is potentially a compensatory mechanism as a consequence of deleting Rptor during early development (from E13.524). To rule out this possibility, we utilized the tetracycline-repressible element of the tTA:Osx:Cre transgene24 to examine the metabolic effects of inhibiting mTORC1 function in skeletally mature mice. Deletion of Rptor was suppressed (via doxycycline-supplemented chow diet) until 4 weeks of age and mice were then challenged with a HFD (Fig. 4k). In contrast to the pre-natal Rptor deletion model, no differences in body weight and body composition was observed between dox-treated control littermates and dox-treated Rptorob−/− mice (+dox Rptorob−/−) at 4 weeks of age (Fig. 4k). However, after 14 weeks on a HFD (at 18 weeks of age), +dox Rptorob−/− mice recapitulated several key protective phenotypes observed in the HFD-fed Rptorob−/− mice including resistance to weight gain, increased glucose tolerance and increased insulin sensitivity (Fig. 4k–o).
To further investigate insulin sensitivity in Rptorob−/− mice, we examined the acute response to insulin in liver, muscle and adipose tissue. An increase in insulin-stimulated phosphorylation of INSR (p-INSR) was observed in all the tissues examined. Furthermore, significantly higher levels of p-INSR were observed in the gWAT of Rptorob−/− mice compared to the controls (Fig. 5d, h). No significant differences in insulin-stimulated AKT activation was observed between genotypes in these tissues (Fig. 5a–h). We also examined the levels of total IRS1 and the activation status of rpS6 (Ser240/244), an effector protein modulated by S6K1, an mTORC1 substrate. mTORC1 negatively regulates insulin signaling leading to phosphorylation of IRS1 and subsequent ubiquitin-mediated degradation.25,26 A significant increase in insulin-stimulated rpS6 phosphorylation (p-rpS6) was observed in the gWAT of Rptorob−/− mice relative to controls (Fig. 5d, h). An increase in p-rpS6 was also observed in iWAT of Rptorob−/− mice, however, this did not reach significance (P = 0.19) (Fig. 5c, g). A significantly higher level of total IRS1 were observed in the gWAT of Rptorob−/− mice, both basal and insulin-stimulated, compared to controls (Fig. 5d, h) while an increase total IRS1 was also observed in iWAT, under insulin stimulation, however, this did not reach significance (P = 0.09) (Fig. 5c, g). Decreased levels of total IRS1 in adipocytes has previously been reported in insulin-resistant subjects27 and led to impaired glucose uptake in a type 2 diabetes animal model.28 Collectively, these results suggest that an enhanced insulin signaling in the fat depots of Rptorob−/− mice may account for the higher peripheral sensitivity to insulin (Fig. 4g). Consistent with higher insulin sensitivity, serum adiponectin levels (both the HMW and LMW forms) were elevated in HFD-fed Rptorob−/− mice relative to both Rptorob+/− and control mice (Fig. 5i–k) whereas serum leptin levels where significantly lower in Rptorob−/− mice relative to Rptorob+/− and control mice, concordant with reduced fat mass in these mice (Fig. 5l).

Increased insulin signaling in adipose tissue depots and elevated serum adiponectin in HFD-fed Rptorob−/− mice. Western immunoblot analysis of indicated protein levels in skeletal muscle (a), liver (b), iWAT (c), and gWAT (d) tissues from 18-week-old HFD-fed mice collected after a 6-h fast, under either basal (6-h fast + PBS; n = 2/genotype) or insulin-stimulated (6-h fast + insulin stimulation; n = 2–3/genotype) conditions. e–h Quantitative analysis of indicated protein levels from a–d, relative to ACTIN. i Serum adiponectin levels (n = 10–15/genotype). j Western immunoblot analysis of HMW, LMW forms and total adiponectin levels in sera from 18-week-old HFD-fed mice (n = 4/genotype). k Quantitative analysis of protein levels from j. l Serum leptin levels (n = 11–14/genotype). All panels except e–h: data are expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, one-way ANOVA with Tukey’s post hoc test. e–h P < 0.05, two-way ANOVA with Tukey’s post hoc test
Increased energy expenditure, greater substrate flexibility, and browning of white adipose tissue in Rptor ob −/− mice
As shown in Fig. 6, TEE in HFD-fed Rptorob−/− mice remained similar to control and Rptorob+/− mice during the light cycle but was significantly higher during the dark cycle, while food intake, total physical activity and fecal lipid content were equivalent across all genotypes (Fig. 6a, b and Supplementary Fig. 7a–e). Interestingly, while 24-h RQ values were similar, the hourly RQ patterns were noticeably different (Fig. 6c, d). To interrogate the hourly RQ patterns further, we calculated the food quotient (FQ), where FQ is the theoretical RQ produced by the diet29 and 24-h RQ ≈ FQ indicates a state of energy and macronutrient balance (for the HFD diet used herein, FQ = 0.8081; Supplementary Fig. 7f). Both Rptorob−/− and Rptorob+/− mice exhibited RQ > FQ during the dark cycle, relative to controls, whereas only in Rptorob−/− mice was RQ < FQ during the light cycle culminating in a 24-h RQ close to the FQ (Fig. 6d). This suggests that Rptorob−/− mice maintain their constant weight and energy balance through greater substrate flexibility, with greater utilization of fat storage during the light cycle which provide a glycerol substrate for increased gluconeogenesis, thus resulting in higher carbohydrate oxidation in the dark cycle. Conversely, in Rptorob+/− mice, higher carbohydrate oxidation and lipid synthesis in the dark cycle was not offset by greater utilization of fat storage during the light cycle (Fig. 6d). Thus, even with grater substrate flexibility, as is evident in Rptorob−/− mice, higher energy storage during the dark cycle surpassed fat oxidation during the light cycle, resulting in overall continuous weight gain (Fig. 4a, b). In control mice, the average phasic RQ values remained relatively constant (Fig. 6d), which is indicative of fewer shifts in their fat storage and oxidation and a more constant accumulation of adipose tissue mass (Fig. 4a, b).

HFD-fed Rptorob−/− mice exhibit greater substrate flexibility and browning of white adipose tissue. a Total energy expenditure (TEE), normalized to lean mass, over 48 h and b per light/dark cycle. c Average RQ over 48 h and d per light/dark cycle. e Serum triglyceride levels (n = 8–10/genotype). f Hepatic triglyceride levels and g hepatic free fatty acid levels in 18-week-old HFD-fed mice (n = 3–4/genotype). h Representative images of H&E and Oil Red O-stained sections of liver, gWAT, and iWAT, scale bar = 100 µm. i, j Size distribution of gWAT and iWAT adipocytes calculated from H&E stained sections using Image J (n = 5 sections/mouse, n = 5–6/genotype). k, l Gene expression levels of adipogenesis, de novo lipogenesis, and lipolysis markers in gWAT and iWAT tissues, normalized to Actb (n = 3–5/genotype). m Gene expression levels of brown adipose tissue markers in iWAT tissue, normalized to Actb (n = 3–5/genotype). n Representative images of H&E and UCP1 immunostaining section of iWAT from HFD-fed mice, scale bar = 100 µm. o Levels of UCP1 protein expression in iWAT (top) and quantitative analysis of protein levels relative to β-actin (bottom) (n = 4/genotype). Data are expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, one-way ANOVA with Tukey’s post hoc test
The increased ability to effectively switch metabolism between carbohydrate and fat oxidation in Rptorob−/− mice was associated with lower circulating TG levels in these mice (Fig. 6e), while FFA levels were higher in Rptorob+/− mice relative to control and Rptorob−/− mice (Supplementary Fig. 7g). Histology revealed less steatosis in the liver of Rptorob−/− mice compared to the controls (Fig. 6h) and significantly lower levels of TG and FFA content (Fig. 6f, g). Furthermore, a significant shift towards adipocyte hypotrophy associated with an upregulation of adipogenic and de novo lipogenic (DNL) genes while no significant changes in the lipolytic genes was observed in both gWAT and iWAT depots from Rptorob−/− mice in response to the HFD (Fig. 6h–l). An increase adipose DNL has been shown to be beneficial for systemic lipid metabolism by sequestering excess TG and thus prevent lipotoxicity in other tissues30 suggesting that increased adipose DNL in the Rptorob−/− mice could contribute to the lower levels of circulating and hepatic TG observed in these mice.
Strikingly, we observed an increased multilocularity of lipid droplets, a characteristic of brown adipocytes, in iWAT of Rptorob−/− mice suggesting “browning” of adipocytes (Fig. 6h). Consistent with these changes in morphology, a strong upregulation of Ucp1 mRNA along with other genes (Pcg1a, Cidea and Cox7a), typically expressed in “browning” WAT, were observed in iWAT of Rptorob−/− mice (Fig. 6m). Immunohistochemistry and Western blot confirmed a significant increase in UCP1 protein levels in Rptorob−/− mice (Fig. 6n, o). We also observed upregulation of Ucp1 and Pcg1a mRNA expression, but not protein, in gWAT of Rptorob−/− mice, whereas no changes in the brown adipose tissue were observed (Supplementary Fig. 8a–d).
Transcriptional changes associated with deletion of Rptor in pre-OBs
RNA sequencing and Gene set enrichment analysis (GSEA) were used to identify transcriptional networks that are differentially regulated in the bones of HFD-Rptorob−/− mice. GSEA of the whole transcriptome revealed positively enriched gene sets for glucose uptake and metabolism and insulin signaling pathways (Fig. 7a, b). After data processing and filtering, 986 genes were found to be differentially expressed with 868 genes significantly upregulated and 118 genes significantly downregulated in the bones of Rptorob−/− mice. These differentially expressed genes (DEGs) were then mapped to specific pathways using KEGG pathway enrichment analysis. Consistent with GSEA, DEGs were highly clustered in glucose metabolism and insulin-responsive pathways including the insulin signaling pathway (15/140 genes; enrichment FDR = 7.95E−04), glycolysis pathway (9/67 genes; enrichment FDR = 1.87E−03) and PI3K-Akt signaling pathway (25/357 genes; enrichment FDR = 8.65E−03). A complete list of upregulated and downregulated gene sets from GSEA and KEGG are detailed in Supplementary Table 1. Gene expression analyses revealed significantly higher levels of genes involved in glycolysis, including HkII, Pgk1, Ldha, Pdk1, and Pfkm1 (Fig. 7c). The expression levels of Glut4 in the bone of Rptorob−/− mice were twofold higher than the controls, while no differences in Glut1 expression levels were observed (Fig. 7c). Of note, an increase in glycolytic gene and Glut4 expression was also observed in NCD-fed Rptorob−/− mice relative to controls (Supplementary Fig. 9a). The higher expression of Glut4, together with the upregulation of glycolysis and insulin signaling pathways suggests a potential increase in insulin signaling and insulin-stimulated glucose uptake in the OBs of the Rptorob−/− mice.

Increased glycolysis and insulin signaling pathways in the bone of HFD-fed Rptorob−/− mice. a Gene set enrichment analysis (GSEA) of positively enriched pathways (upregulated) in RNA isolated from combined flushed femur and tibia (n = 5; controls versus Rptorob−/−). b Enrichment plots of glycolysis and insulin-like growth factor receptor signaling pathways. c Gene expressions of glycolysis (HkII, Pgk1, Ldha, Pdk1, and Pfkm1) and glucose transporters (Glut1 and Glut4) in the flushed femur/tibia from HFD-fed mice (n = 3–5/genotype). d Gene expression of Bglap in the flushed femur/tibia, normalized to Actb (n = 3–4/genotype). e Serum levels of total osteocalcin (OCN) and f undercarboxylated osteocalcin (unOCN) levels in 18-week-old HFD-fed mice (n = 7–15/genotype). g Whole-body bone mineral density (BMD) measured by DXA (n = 5/genotype). Data are expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, one-way ANOVA with Tukey’s post hoc test. a An orange bar indicates pathways relating to glucose metabolism or insulin signaling, a green bar indicates pathways relating to lipid metabolism, and a black bar indicates pathways relating to bone development. Gray bar (ns) = not significant
Increased insulin signaling and glycolysis in bone of Rptor ob −/− mice
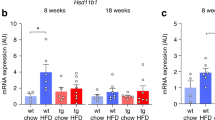
We next interrogated if the systemic changes in glucose metabolism in HFD-fed Rptorob−/− mice was associated with an increase in insulin sensitivity in bone. Gene expression analysis revealed a marked upregulation of Bglap in the bone of Rptorob−/− mice (Fig. 7d). However, circulating levels of both total and unOCN were equivalent across all genotypes (Fig. 7e, f). An assessment of bone mass in HFD-fed Rptorob−/− mice revealed a significant reduction (8.7%) in bone mineral density (BMD) as assessed by DEXA (Fig. 7g).
We next directly assessed insulin responsiveness in bone (calvaria) of Rptorob−/− mice. Initially, Rptor deletion was confirmed in calvaria by qRT-PCR, using primers specific to floxed exon 6. A significant reduction in Rptor gene expression was observed in Rptorob−/− mice consistent with Cre-mediated excision (Fig. 8a). Phosphorylation of INSR at Tyr1150/1151 was significantly higher in Rptorob−/− mice (Fig. 8b) whereas phosphorylation of rpS6 at Ser240/244 was blunted consistent with reduced mTORC1 activity. Importantly, both basal and insulin-stimulated phosphorylation levels of AKT (Thr308 and Ser473) were significantly increased in Rptorob−/− mice (Fig. 8b). This was associated with increased phosphorylation of AKT substrates, glycogen synthase kinase (GSK)-3β Ser,9 and AS160 Thr642 (which regulates insulin-stimulated GLUT4 trafficking) (Fig. 8b). This data, in combination with the RNA-seq data, suggests that loss of mTORC1 function in OBs increases basal and insulin-dependent glucose uptake in Rptorob−/− mice.

In vivo and in vitro loss of OB-mTORC1 lead to potentiation of OB insulin signaling. a Gene expression of Rptor in the calvarial bone tissue, normalized to Actb (n = 3–4/genotype). Data are expressed as mean ± SEM. *P < 0.05, unpaired Student’s t-test. b Western immunoblot analysis of indicated protein levels in calvarial bone tissue from 18-week-old HFD-fed mice collected after a 6 h fast under either basal (6-h fast + PBS; n = 2–3/genotype) or insulin-stimulated (6-h fast + insulin stimulation; n = 2/genotype) conditions. c Western immunoblot analysis of indicated protein levels in wildtype (WT) and Rptor knockout (KO) cultured primary osteoblasts collected under normal growth and a 4-h serum starved conditions before or after stimulation with 100 nmol·L−1 insulin. d Gene expression analysis of glycolysis and glucose transporters in WT or Rptor KO OBs (n = 3). e 2-Deoxy-D-glucose uptake (measured as units of luminescence) of WT or Rptor KO OBs under either basal or insulin-stimulated conditions. Data are expressed as mean ± SD. ***P < 0.001, unpaired Student’s t-test
To address this question, we generated Rptor knockout OBs (RptorKO) in vitro.14 Deletion of Rptor caused a reduction in basal and insulin-stimulated phosphorylation of the key mTORC1 substrates, p70S6K and 4E-BP1 and rpS6, findings consistent with reduced mTORC1 activity (Fig. 8c). Deletion of Rptor was also associated with an increase in basal and insulin-stimulated AKT activation and phosphorylation of GSK-3β at Ser,9 (Fig. 8c), consistent with the in vivo insulin stimulation experiments (Fig. 8b). Furthermore, qRT-PCR analysis revealed an increase in the expression of glycolytic genes in RptorKO cells relative to wildtype control cells mirroring in vivo findings (Fig. 8d). Finally, using the accumulation of 2-deoxyglucose as a surrogate measure of glucose uptake, loss of mTORC1 function in OBs increased both basal and insulin-stimulated glucose uptake into OBs (Fig. 8e). Taken together, our findings support a central role for the mTORC1 complex in OBs in the control of systemic glucose metabolism and highlight the importance of this complex in bone in the development of systemic insulin resistance.
Discussion
In this study, we show that NCD-fed Rptorob−/− mice displayed a marked metabolic phenotype characterized by low fat mass, hypoglycemia, enhanced glucose tolerance, and increased insulin sensitivity. These beneficial metabolic effects were maintained under excess nutrient conditions resulting in protection against HFD-induced obesity and insulin resistance. Furthermore, we provide evidence that these phenotypes are, at least in part, attributable to an enhanced responsiveness to insulin and insulin-dependent glucose uptake in OBs (Fig. 9).

Schematic: Suppression of mTORC1 activity in pre-OBs results in relaxation of mTORC1-dependent negative feedback control (both directly and indirectly via activation of p70S6K) of insulin signaling by disrupting the interaction between IRS1 and INSR. This lead to an enhanced and prolonged insulin signaling as indicated by hyperactivation of AKT at Ser473 and Thr308 in both basal and insulin-stimulated states. AKT hyperactivation attenuates the inhibitory effects of the AS160 on RAB leading to GLUT4 membrane translocation. These molecular changes are associated with an increased cellular glucose uptake in the Rptor KO OBs and upregulation of an insulin-dependent glucose transporter 4 (Glut4) and glycolysis-promoting enzymes in the skeleton of Rptorob−/− mice. Systemically, Rptorob−/− mice showed persistently low fasting glucose levels and significantly increased tolerance to glucose; the metabolic phenotypes which are, at least in part, attributable to an enhanced responsiveness to insulin and insulin-dependent glucose uptake in OBs
Skeletal mTORC1, insulin signaling, and glucose uptake
Previous studies have shown that modulation of insulin signaling in OBs, by genetically decreasing or increasing the levels of INSR expression, either worsens or enhances glucose tolerance and insulin resistance in HFD-fed animals, respectively.31 Furthermore, mice with OB-specific deletion of FoxO1 (FoxO1OB−/−), a negative regulator of insulin signaling, are protected from HFD-induced obesity.3 Consistent with these data, suppression of skeletal mTORC1 activity, a downstream mediator and negative regulator of insulin signaling, protects mice from diet-induced obesity. Of note, the lower fasting glucose levels, enhanced glucose tolerance and increased insulin sensitivity displayed by Rptorob−/− mice mimics the metabolic phenotype of mice that overexpress the human INSR in the OBs (Col1a1-INSRTg mice).31 At the molecular level, we did not observe enhanced insulin-stimulated activation of AKT, an important effector of insulin signaling, in any peripheral insulin-targeted tissues, however, insulin signaling in the bone of Rptorob−/− mice was significantly enhanced. Similarly, improved glucose tolerance and insulin sensitivity in Col1a1-INSRTg mice were only associated with enhanced insulin signaling in bone.31
Several lines of evidence point to the potentiation of insulin signaling as being an important mechanism in the metabolic phenotype of Rptorob−/− mice. Firstly, using bones isolated from HFD-fed mice, RNA-seq and GSEA revealed an upregulation of genes involving in glycolysis pathway. This gene expression pattern was also observed in the bones of NCD-fed Rptorob−/− mice and Rptor KO calvarial cells in vitro. Secondly, basal and insulin-stimulated levels of AKT and AS160 were evident in calvarial cells of HFD-fed Rptorob−/− mice in vivo. Hyperphosphorylation of AKT at Ser473 and Thr308 was also observed in vitro in Rptor KO OBs, in both basal and insulin-stimulated states. Thirdly, glucose uptake was significantly increased in Rptor KO cells in vitro in both basal and insulin-stimulated states, suggesting that constitutive activation of AKT leads to an increase in glucose transport in the absence of mTORC1 activity. Several studies have now recognized bone as a significant site of glucose uptake accounting for up to 15% of steady state levels and the magnitude of glucose uptake by OBs is sufficient to impact systemic glucose disposal in mice.19,32,33 The higher basal phosphorylation levels of AKT and AS160 in vivo suggests that an increase in basal skeletal uptake of glucose could account for the low glycemic phenotype of Rptorob−/− mice (Fig. 9).
Metabolic phenotypes independent of OCN
The improved metabolic phenotype of NCD-fed Rptorob−/− mice appears to contradict the widely held view that low unOCN levels are associated with poor metabolic outcomes. Circulating OCN levels are inversely correlated with plasma glucose levels, fat mass and the extent of metabolic impairment in both animal and human studies.6,34 Furthermore, Ocn-deficient mice (Ocn−/−) exhibit higher adiposity, hyperglycemia, hypoinsulinemia, impaired insulin secretion and sensitivity and are glucose intolerant,6 a metabolic phenotype opposite to that observed in Rptorob−/− mice. The defects in glucose homeostasis in Ocn−/− mice are accompanied by impaired pancreatic β-cell proliferation and insulin secretion and the development of insulin resistance in other insulin-target tissues.6 While the hypoinsulinemia observed in our Rptorob−/− mice may be attributed to low serum levels of unOCN, GSIS tests indicated maintenance of ability to secrete insulin in response to glucose in the Rptorob−/− mice. The low serum levels of OCN observed in NCD-fed Rptorob−/− mice are likely secondary to the stall in osteogenesis that occurs in response to reduced mTORC1 activity in pre-OBs.14,35 While we cannot rule out the possibility that the metabolic improvements observed in Rptorob−/− mice are due to low levels of OCN priming its target tissues to become highly sensitive to OCN, this is unlikely to be the case, as serum unOCN levels were reduced in both Rptorob+/− and Rptorob−/− mice, but only Rptorob−/− mice displayed an improved metabolic phenotype. Furthermore, in contrast to other animal models (i.e., FoxO1OB−/− and Col1a1-INSRTg mice) where protection from HFD-induced glucose intolerance and insulin resistance were attributed to an insulin-mediated increase in bone resorption leading to an increase in OCN activity and serum levels (although no difference in Ocn mRNA expression was observed),31 both RNA-seq and qRT-PCR analysis of bone samples from HFD-fed Rptorob−/− mice revealed a downregulation of genes involved in bone resorption and osteoclast differentiation pathways, and upregulation of genes associated with bone formation and OB differentiation. Intriguingly, despite detection of significantly higher Bglap mRNA expression levels in HFD-fed Rptorob−/− mice, circulating levels of both total and unOCN protein were equivalent to those of the controls. At this stage, the mechanisms underlying this disparity remain to be determined. OCN is expressed exclusively from mature OBs and low serum levels are used as a marker of reduced OB activity and bone mass.36 Low serum OCN levels correlated with the low bone mass phenotype of normal chow-fed Rptorob−/− mice,14 while no difference in BMD was observed in HFD-fed Rptorob−/− mice relative to controls (Fig. 7g). Previous studies have shown that HFD-fed wild-type mice have reduced total and unOCN levels,31,37 suggesting that the apparent “normalization” of OCN levels in HFD-fed Rptorob−/− mice may simply reflect this reduction. Another possibility is that OB-mTORC1 is a negative regulator of Bglap gene expression and possibly required for Bglap translation. Notwithstanding this, an important finding of this present study is the observation that the beneficial metabolic effects associated with OB-mTORC1 inhibition occur independently of OCN. Several studies have implicated OCN-independent mechanisms in the bone-metabolism interplay. For example, genetic ablation of OBs, or conditional OB-specific inactivation of GSK-3β, β-catenin or Vhl, a regulator of Hypoxia-inducible factors (HIFs), leads to systemic metabolic alterations that cannot be fully rescued or explained by congruent changes in serum OCN.19,38,39,40 These studies suggest the existence of additional, as yet unidentified, bone secretagogues, amongst other factors, that influence global glucose homeostasis and energy metabolism.
Energy metabolism and whole-body metabolic flexibility
In NCD-fed Rptorob−/− mice, the failure to build up their energy store (as evident by a marked reduction in body fat and accumulation of small adipocytes in the fat depots) is likely due to their increased energy expenditure, with unaltered caloric intake, and the preference for fat oxidation (as indicated by the relatively lower RQ). This lipoatrophy could represent an adaptive response to the concurrent low insulin and glucose levels which, together, increase the production of ketone bodies,41 as an alternative energy source, by increased mobilization of fatty acid from the fat storage to the liver, in order to prevent hypoglycemic death. In support of this, recent studies have shown that during short-term starvation in lean rats, both hypoleptinemia and insulinopenia are necessary to maintain euglycemia and thus promote survival.42 Hypoleptinemia stimulates secretion of corticosterone which, in the presence of hypoinsulinemia, leads to WAT lipolysis and the shift from whole-body carbohydrate to fat/ketone oxidation.42 Furthermore, in NCD-fed Rptorob−/− mice, the observed changes in body composition (i.e., a significant reduction in total body weight and fat mass) while paradoxically increasing BMAT are commonly observed under states of starvation including dietary restricted mouse models43 and in human anorexia nervosa patients.44 A growing number of studies now attribute the beneficial effects of BMAT expansion to adiponectin production under restricted caloric conditions22,43,45 and, as such, elevated serum adiponectin levels could contribute to the improved metabolic phenotype of Rptorob−/− mice. Adiponectin has been reported to inhibit gluconeogenesis and increase fatty acid beta-oxidation in the liver to facilitate its insulin-sensitizing effects.46,47 Of note, the upregulation of genes involved in fatty acid beta-oxidation (Cpt1a, Acox, and Acc2) was observed in the liver of Rptorob−/− mice (Supplementary Fig. 3a). The lack of differences in the Pepck and G6p expression levels in the Rptorob−/− mice may be attributable to their persistently high circulating adiponectin levels (both in the fed (Fig. 3e) and fasting states (data not shown)). Furthermore, the collection of tissues during fasting could potentially mask any potential differences as fasting would lead to the upregulation of Pepck in the control mice.48 Adiponectin has been reported to increase fatty acid beta-oxidation in liver to facilitate its insulin-sensitizing effects.46,47 These increases in fatty acid oxidation genes in the liver were indeed consistent with the lower respiratory quotient (Fig. 1m, n) observed in these mice.
Rptorob−/− mice are resistant to diet-induced weight gain and maintained a constant body weight, after 6 weeks of HFD, despite consuming a similar amount of food to the controls. Importantly, the reduced adiposity (Fig. 4c, d) and lack of adipocyte hypertrophy (Fig. 6h–j) in the Rptorob−/− mice were observed despite an upregulation of lipogenic genes in these tissues. Downregulation of lipogenic genes upon HFD feeding or in obesity is associated with the development of adipocyte insulin resistance49,50,51,52 and thus the upregulation of these genes in the Rptorob−/− mice is likely a secondary effect of their ability to retain sensitivity to insulin. Both lipogenesis and adipogenesis are well-known insulin-regulated processes in adipose tissues53 and consistent with this, a significant upregulation of Pparg, an important regulator of adipogenesis, was also observed in the adipose tissue of Rptorob−/− mice (Fig. 6k, l). No significant changes in the expression levels of Atgl, an important lipolytic gene, was observed the adipose tissue of Rptorob−/− mice which is consistent with a higher lipogenesis-to-lipolysis ratio as previously reported in smaller insulin-sensitive adipocytes.51 Finally, an increased sensitivity to insulin in adipose tissue of Rptorob−/− mice is evidenced by a significant, or a trend toward significant, increase in insulin-stimulated rpS6 phosphorylation (Fig. 5c, d, g, h) and the upregulation of Glut4 transcripts (Supplementary Fig. 10).
The mechanism(s) underlying the browning of iWAT in Rptorob−/− mice appears to be independent of previously reported browning agents including fibroblast growth factor 21 (FGF21)54 and muscle-secreted irisin55 as we observed no changes in the expression levels of these factors (data not shown). It is interesting to note that we did observe an upregulation of two bone-secreted factors, bone morphogenetic protein 7 and Wnt-signaling inhibitor, sclerostin (Supplementary Fig. 9b, c), both of which have been implicated in the browning of WAT.56,57 With regards to sclerostin, mTORC1 has been shown to negatively regulate SOST in osteocytes via activation of SIRT1, a negative regulator of Sost gene expression58 linking mTORC1 function in OBs/osteocytes with maintaining systemic energy balance.
We propose that the protection from diet-induced weight gain is partially attributable to their ability to switch substrate oxidation, while maintaining energy balance. Rptorob−/− mice exhibited a greater capacity to switch between oxidizing fatty acids and carbohydrates during the light and dark cycles, under ad libitum feeding, suggesting they are capable of maintaining metabolic flexibility (defined as the increase in RQ between fasting and postprandial states). In humans, impairments in metabolic flexibility have been associated with obesity, the development of insulin resistance and diabetes (reviewed in ref. 59). Furthermore, clinical studies report lower fat oxidation and higher RQ in subjects with a family history of diabetes,60 and, in particular, a smaller decrease in RQ was observed during the sleeping period in these subjects.61 These results, combined with the impaired glucose metabolism observed in the control mice, indicate a correlation between metabolic inflexibility and abnormality in glucose homeostasis. As such, the flexibility in substrate switching in Rptorob−/− mice could produce the systemic metabolic protection phenotype observed in these mice.
Inhibition of mTORC1 as a novel therapeutic approach to treat T2DM
To our knowledge, this is the first report to describe the metabolic phenotype associated with loss of mTORC1 function in OBs. In other insulin-responsive tissues, loss of mTORC1 function has positive and negative effects on whole-body metabolism. In WAT, deletion of Rptor using Ap2-Cre (raptorad−/−) resulted in lean mice that were resistant to the negative effects of HFD-induced obesity on glycemic control and HFD-induced hyperphosphorylation of S6K1.12 In contrast, deletion of Rptor in mature adipocytes (AdipoQ-Cre) led to lipodystrophy and insulin resistance that was associated with a significant increase in liver mass and ectopic lipid accumulation.13 Inactivation of Rptor in muscle (RAmKO mice) results in protection from HFD-induced weight gain and enhanced glucose tolerance without improvement in insulin sensitivity.62 Reduced S6K1 activation and increased phosphorylation of AKT and AS160 were also observed in the muscle of RAmKO mice. However, this was associated with a significant decrease in the expression of glucose transporters and glycolytic genes and an increase in glycogen synthesis and muscle glycogen levels.11,62 Conversely, liver-specific deletion of Rptor (RaptorΔhep mice) resulted in severe liver damage with augmented inflammation and fibrosis while glucose tolerance was improved only in NCD-fed mice.63 These tissue-specific functions and responses to mTORC1 inhibition are further highlighted in studies where administration of rapamycin has been shown to have both beneficial64,65 and detrimental effects on metabolism66,67,68,69 demonstrating that sustained systemic suppression of mTORC1 function has significant limitations.
Skeletal involvement in T2DM is well established with T2DM patients displaying an increased risk of fracture despite higher femoral neck and lumbar spine BMD.70,71 At the microarchitecture level, an increase in cortical porosity and bone micro-fractures has been observed in T2DM patients which may reflect disrupted bone remodeling.72 Indeed, a decrease in the number and differentiation of OBs, leading to diminished quantities of osteoid is considered an important driver of skeletal change in T2DM.73 While a role for OB-mTORC1 in these changes requires further investigation, hyperactivation of mTORC1 via deletion of Tsc2 in OBs (∆Tsc2) results in mice with a high bone mass phenotype and insulin resistance.17 These phenotypes are associated with an accumulation of poorly differentiated OBs and disorganized bone structure. Similarly, an accumulation of poorly organized sclerotic bone occurs in patients with tuberous sclerosis, an autosomal dominant disorder caused by the mutation of TSC1 or TSC2 leading to hyperactivation of mTORC1.74 While further evaluation of the role of mTORC1 in OBs in the development of diet-induced insulin resistance is required, the protective phenotypes observed in HFD-fed mice with either pre- or post-natal deletion of Rptor in OBs suggest OB-mTORC1 is a potential target for the development of antidiabetic drugs, whereby localized inhibition of skeletal mTORC1 could have favorable metabolic outcomes in patients with T2DM.
In summary, our studies have demonstrated that suppression of skeletal mTORC1 signaling in mice leads to a dramatic improvement in glucose metabolism and protection from diet-induced obesity and insulin resistance. Collectively, these results point to a critical role for the mTORC1 complex in OBs in integrating whole-body nutrient status and local insulin signaling in order to maintain systemic glucose homeostasis.
Materials and methods
Transgenic mice and diet
All mice were bred and group-housed (maximum five mice/cage) in pathogen-free conditions at the SAHMRI Bioresources Facility (Adelaide, Australia) under a 12 h light–dark cycle (lights on at 06:00) and constant temperature (20–23 °C) and ad libitum access to a standard chow diet (Teklad Global Diet #2918: 18.6% protein, 6.2% fat; Harlan, IN, USA) and water. For diet-induced obesity studies, mice were fed a HFD (Specialty Feeds #SF16-096: 19.4% protein, 23% fat (43.4% kcal from fat); WA, Australia) from weaning to 18 weeks of age. For postnatal deletion studies, pregnant dams and pups were maintained on doxycycline chow (Specialty Feeds #SF08-026: 600 mg Doxycycline per kg; WA, Australia). At 4 weeks of age, pups were placed on a HFD (Specialty Feeds #SF16-096; WA, Australia) until 18 weeks of age. FQ was estimated by linear regression analysis of changes in body mass (∆M: x-axis) and 24-h RQ (y-axis); the y-intercepts were the 24-h RQ when ∆M equaled zero, or theoretical FQ29,75 (Supplementary Fig. 5). All studies were performed with Institutional Ethics approval (SAHMRI Animal Ethics Committee, #SAM164). Male conditional knockout mice in which Rptor was disrupted in early osteoprogenitor cells were generated using Osx1-GFP::Cre mice,24 R26eYFP mice,76 and Rptorfl/fl mice11 as previously described.14 Animals were weighed twice weekly for the duration of the study and at the end of the study, body length and whole-body lean and fat mass were measured postmortem using a dedicated mouse DXA (Lunar Piximus II, GE Medical Systems, Madison WI, USA).
Metabolic phenotyping
ITTs and glucose tolerance tests (GTTs) were performed following intraperitoneal injection of 0.75 U·kg−1 insulin (Novo Nordisk Pharmaceuticals, Australia) or 2 g·kg−1 glucose, respectively after a 6-h fast (07:00–13:00). Blood glucose levels were measured at indicated time points using a handheld glucometer (Accu-Chek, Roche, Australia). At 0- and 30-min time points during the GTT, ~50 µL of whole blood was collected, serum isolated and then immediately frozen at −80 °C for later assessment of glucose-stimulated insulin levels by ELISA (EZRMI-13K, Millipore, MA, USA) as per manufacturer’s instructions.
Indirect calorimetry and body composition analyses
Indirect calorimetry assessments were performed using the Promethion metabolic cage system (Sable Systems, NV, USA). Mice were single-housed in Promethion cages for 72–96 h with ad libitum access to food and water. Monitoring was performed for 48 h following an initial 24-h acclimatization period. RQ was calculated as the ratio of VCO2/VO2.
Fecal lipid assessment
Dried feces (1 g) were collected from single-housed mice over a 72-h period, pulverized using a mortar and pestle and rehydrated in 5 ml of normal saline. A 2:1 chloroform: methanol solution was used to extract lipids from the suspension as previously described.77 After lipid extraction and a 4-d evaporation of the organic phase, the remaining lipid mass was weighed using analytical balance. The lipid content was calculated as the percentage of the total fecal weight.
ELISA
Non-fasted (for chow studies) and fasted (for HFD studies) blood samples were collected (12:00–14:00) via cardiac puncture into Minicollect tubes (Greiner Bio One, Kremsmünster, Austria), centrifuged at 845 × g for 10 min at room temperature and extracted serum stored at −80 °C. Commercial ELISA kits were used for the measurement of: TGs and FFA (ab65336 and ab65341, respectively, Abcam, MA, USA), leptin and adiponectin (EZML-82K and EZMADP-60K, respectively, Millipore, MA, USA), OCN (BT-470, Alfa Aesar, Lancashire, UK), unOCN (MK129, Takara, Japan), and LCN2 (RDSMLCN20, R&D Systems, Minneapolis, MN, USA) as per manufacturer’s instructions.
Histology and Immunohistochemistry
Tissues were dissected, fixed in 10% formalin and embedded in paraffin. Hematoxylin and eosin staining was performed on 5 μm adipose tissue sections. Image J was used to measure adipocyte cell size and distribution and was presented as a percentage of total cells from n = 5 images per mouse. Immunohistochemistry was performed on iBAT using rabbit anti-UCP1 (ab209483, Abcam, Cambridge, MA, USA, 1:5 000). For pancreatic tissues, immunohistochemistry was performed using rabbit anti-insulin (ab209483, Abcam, 1:1 000). Total pancreatic surface and β-cell area (area positive for insulin immunostaining) were quantified from at least five sections per pancreas (each 50 µm apart) using a DP72 camera (Olympus, Tokyo, Japan) attached to a Leica microscope (Wetzlar, Germany) and Osteomeasure software (Osteometrics, GA, USA). For Oil red O staining, liver samples were mounted in Tissue-Tek Optimum Cutting Temperature compound (Sakura Finetek, The Netherlands). Cryosections were cut at 10 μm and Oil red O staining was performed as previously described.78
Protein isolation and western blotting
For insulin signaling studies, mice were fasted for 6 h (07:00–13:00) prior to administration of insulin (150 mU·g−1 BW, i.p) or PBS control. After 20 min, mice were euthanized and tissues were harvested, snap-frozen in liquid nitrogen and stored at −80 °C. Tissue samples were lysed in modified RIPA buffer with addition of protease inhibitors (cOmplete™, Roche, Basel, Switzerland) and homogenized using a TissueRuptor (QIAGEN, Victoria, Australia). Equal amounts of protein (50 µg,) were resolved by SDS-PAGE and transferred to Immobilon-FL PVDF Membrane (MerckMillipore, Darmstadt, Germany). Antibodies for immunoblotting are listed in Supplementary Table 2. After incubation with fluorescently tagged secondary antibody, membranes were scanned using a Li-Cor Odyssey imaging system (LI-COR Biosciences, NE, USA). Quantitative analysis was performed using Image Studio software LI-COR Biosciences. Protein levels were quantified and levels normalized to a loading control. The analysis of adiponectin multimers in serum was performed as previously described.79
Full images of western blots are included in Supplementary Fig. 11.
RNA isolation and quantitative RT-PCR
All RNA extractions were carried out using TRIzol reagent (Sigma) according to manufacturer’s instructions. Total RNA (1.5 µg) was reverse transcribed into cDNA using Superscript IV Reverse Transcriptase (Invitrogen, CA, USA). Real-time PCR reactions were performed using RT2 SYBR Green ROX reagent (QIAGEN, Hilden, Germany) in a CFX Connect™ Real-Time PCR machine (Bio-Rad). Forward and reverse primer pairs are listed in Supplementary Table 3. Relative mRNA expression was determined using the 2–∆∆Ct method.80
RNA high-throughput sequencing and gene expression analysis
RNA isolated from marrow-flushed combined femur/tibia were processed and sequenced through the low-input RNA-seq pipeline at The David Gunn Genomics Facility, SAHMRI (Illumina Nextseq, San Diego, CA, USA) with a 75 cycle v2 High Output sequencing kit. Analyses were then conducted by SAHMRI Bioinformatics Facility. Initial raw read processing was performed using an in-house pipeline developed at SAHMRI. Raw 75 bp single-end FASTQ reads were assessed for quality using FastQC and results aggregated using R/Bioconductor package ngsReports.81 Reads were then trimmed for sequence adapters using AdapterRemoval82 and aligned to the GRCh38/mm10 version of the mouse genome using the RNA-seq alignment algorithm STAR.83 After alignment, mapped sequence reads were summarized to the mm10 gene intervals using featureCounts,84 and count table transferred to the R statistical programming environment for expression analysis.
Gene counts were filtered for low expression counts by removing genes with less than 1 count per million in more than four samples and then normalized by the method of trimmed mean of M-values.85 Differential gene expression was carried out on log-CPM counts and precision weights available from the limma voom function86 and edgeR.87,88 GSEA was performed from Molecular Signature Database and other gene sets available from the limma package.89 Gene sets returning an FDR adjusted P value < 25% were accepted as statistically enriched.
Primary cell culture and insulin pulse
Primary calvarial OB cultures were established from new born (P4) Rptorfl/fl mice and Rptor knockout cells generated as previously described.14 For insulin pulse experiments, cells were incubated under serum-free conditions for 4 h then pulsed with 100 nmol·L−1 insulin (ProSpec-Tany TechnoGene Ltd, Ness-Ziona, Israel) for 10 min. RNA and protein was extracted from wild-type and Rptor KO cells, under normal growth and serum starved conditions before or after stimulation with 100 nmol·L−1 insulin, as described above. qRT-PCR and western blotting were performed as described above.
Glucose uptake assays
Basal and insulin-stimulated glucose uptake was performed using the Glucose Uptake-GloTM assay (Promega, Madison, WI, USA). Briefly, wildtype and Rptor knockout cells (1 × 104/well) in clear bottom 96 wells plates (Corning, New York, USA) were allowed to adhere overnight then serum deprived for 4 h. Cells were then stimulated with 100 nmol·L−1 insulin in glucose- and serum-free DMEM (ThermoFisher) supplemented with 10 nmol·L−1 Hepes, pH 7.4, and 1 nmol·L−1 sodium pyruvate for 1 h at 37 °C/5% CO2. Media was replaced with 50 µL/well of 2-deoxyglucose (1 nmol·L−1) for 10 min at 25 °C. Subsequent steps were performed according to the manufacturer’s instructions. Luciferase activity was detected using a GloMax® Luminometer (Promega).
Statistical analysis
All data are presented as mean ± standard error of the mean. Statistical analyses were performed using a one-way or two-way ANOVA with a Tukey’s post hoc test or an unpaired Students’s t-test using GraphPad Prism (GraphPad Software Inc, CA, USA). Significance was accepted at P < 0.05, with asterisks denoting P value levels: *P < 0.05; **P < 0.01; ***P < 0.001.
References
Ferron, M. et al. Insulin signaling in osteoblasts integrates bone remodeling and energy metabolism. Cell 142, 296–308 (2010).
Fulzele, K. et al. Insulin receptor signaling in osteoblasts regulates postnatal bone acquisition and body composition. Cell 142, 309–319 (2010).
Rached, M. T. et al. FoxO1 expression in osteoblasts regulates glucose homeostasis through regulation of osteocalcin in mice. J. Clin. Invest. 120, 357–368 (2010).
Yoshizawa, T. et al. The transcription factor ATF4 regulates glucose metabolism in mice through its expression in osteoblasts. J. Clin. Invest. 119, 2807–2817 (2009).
Wei, J., Hanna, T., Suda, N., Karsenty, G. & Ducy, P. Osteocalcin promotes beta-cell proliferation during development and adulthood through Gprc6a. Diabetes 63, 1021–1031 (2014).
Lee, N. K. et al. Endocrine regulation of energy metabolism by the skeleton. Cell 130, 456–469 (2007).
Mizokami, A. et al. Oral administration of osteocalcin improves glucose utilization by stimulating glucagon-like peptide-1 secretion. Bone 69, 68–79 (2014).
Ferron, M., Hinoi, E., Karsenty, G. & Ducy, P. Osteocalcin differentially regulates beta cell and adipocyte gene expression and affects the development of metabolic diseases in wild-type mice. Proc. Natl Acad. Sci. USA 105, 5266–5270 (2008).
Ferron, M., McKee, M. D., Levine, R. L., Ducy, P. & Karsenty, G. Intermittent injections of osteocalcin improve glucose metabolism and prevent type 2 diabetes in mice. Bone 50, 568–575 (2012).
Um, S. H., D’Alessio, D. & Thomas, G. Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cell Metab. 3, 393–402 (2006).
Bentzinger, C. F. et al. Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell Metab. 8, 411–424 (2008).
Polak, P. et al. Adipose-specific knockout of raptor results in lean mice with enhanced mitochondrial respiration. Cell Metab. 8, 399–410 (2008).
Lee, P. L., Tang, Y., Li, H. & Guertin, D. A. Raptor/mTORC1 loss in adipocytes causes progressive lipodystrophy and fatty liver disease. Mol. Metab. 5, 422–432 (2016).
Fitter, S. et al. mTORC1 plays an important role in skeletal development by controlling preosteoblast differentiation. Mol. Cell. Biol. 37, e00668–00616 (2017).
Chen, J. & Long, F. mTORC1 signaling promotes osteoblast differentiation from preosteoblasts. PLoS One 10, e0130627 (2015).
Dai, Q. et al. mTOR/Raptor signaling is critical for skeletogenesis in mice through the regulation of Runx2 expression. Cell Death Differ. 24, 1886–1899 (2017).
Riddle, R. et al. Tsc2 is a molecular checkpoint controlling osteoblast development and glucose homeostasis. Mol. Cell. Biol. 34, 1850–1862 (2014).
Martin, S. K. et al. mTORC1 plays an important role in osteoblastic regulation of B-lymphopoiesis. Sci. Rep. 8, 14501 (2018).
Dirckx, N. et al. Vhl deletion in osteoblasts boosts cellular glycolysis and improves global glucose metabolism. J. Clin. Invest. 128, 1087–1105 (2018).
Mosialou, I. et al. MC4R-dependent suppression of appetite by bone-derived lipocalin 2. Nature 543, 385–390 (2017).
Hara, K. et al. Measurement of the high-molecular weight form of adiponectin in plasma is useful for the prediction of insulin resistance and metabolic syndrome. Diabetes Care 29, 1357–1362 (2006).
Cawthorn, W. P. et al. Bone marrow adipose tissue is an endocrine organ that contributes to increased circulating adiponectin during caloric restriction. Cell Metab. 20, 368–375 (2014).
Shah, O. J., Wang, Z. & Hunter, T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr. Biol. 14, 1650–1656 (2004).
Rodda, S. J. & McMahon, A. P. Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development 133, 3231–3244 (2006).
Um, S. H. et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 431, 200–205 (2004).
Harrington, L. S. et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J. Cell Biol. 166, 213–223 (2004).
Carvalho, E. et al. Low cellular IRS 1 gene and protein expression predict insulin resistance and NIDDM. FASEB J. 13, 2173–2178 (1999).
Wang, Y., Nishina, P. M. & Naggert, J. K. Degradation of IRS1 leads to impaired glucose uptake in adipose tissue of the type 2 diabetes mouse model TALLYHO/Jng. J. Endocrinol. 203, 65–74 (2009).
Flatt, J. P. Dietary fat, carbohydrate balance, and weight maintenance. Ann. N. Y. Acad. Sci. 683, 122–140 (1993).
Solinas, G., Borén, J. & Dulloo, A. G. De novo lipogenesis in metabolic homeostasis: More friend than foe? Mol. Metab. 4, 367–377 (2015).
Wei, J. et al. Bone-specific insulin resistance disrupts whole-body glucose homeostasis via decreased osteocalcin activation. J. Clin. Investig. 124, 1781–1793 (2014).
Zoch, M. L., Abou, D. S., Clemens, T. L., Thorek, D. L. J. & Riddle, R. C. In vivo radiometric analysis of glucose uptake and distribution in mouse bone. Bone Res. 4, 16004 (2016).
Sulston, R. J. et al. Increased circulating adiponectin in response to thiazolidinediones: investigating the role of bone marrow adipose tissue. Front Endocrinol. 7, 128 (2016).
Pittas, A. G., Harris, S. S., Eliades, M., Stark, P. & Dawson-Hughes, B. Association between serum osteocalcin and markers of metabolic phenotype. J. Clin. Endocrinol. Metab. 94, 827–832 (2009).
Singha, U. K. et al. Rapamycin inhibits osteoblast proliferation and differentiation in MC3T3-E1 cells and primary mouse bone marrow stromal cells. J. Cell Biochem. 103, 434–446 (2008).
Kuo, T.-R. & Chen, C.-H. Bone biomarker for the clinical assessment of osteoporosis: recent developments and future perspectives. Biomark. Res. 5, 18 (2017).
Cao, J. J., Sun, L. & Gao, H. Diet-induced obesity alters bone remodeling leading to decreased femoral trabecular bone mass in mice. Ann. N. Y. Acad. Sci. 1192, 292–297 (2010).
Yoshikawa, Y. et al. Genetic evidence points to an osteocalcin-independent influence of osteoblasts on energy metabolism. J. Bone Min. Res. 26, 2012–2025 (2011).
Gillespie, J. R. et al. GSK-3β function in bone regulates skeletal development, whole-body metabolism, and male life span. Endocrinology 154, 3702–3718 (2013).
Yao, Q. et al. Wnt/beta-catenin signaling in osteoblasts regulates global energy metabolism. Bone 97, 175–183 (2017).
Laffel, L. Ketone bodies: a review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes/Metab. Res. Rev. 15, 412–426 (1999).
Perry, R. J. et al. Leptin mediates a glucose-fatty acid cycle to maintain glucose homeostasis in starvation. Cell 172, 234–248 (2018). e217.
Devlin, M. J. et al. Caloric restriction leads to high marrow adiposity and low bone mass in growing mice. J. Bone Min. Res. 25, 2078–2088 (2010).
Bredella, M. A. et al. Increased bone marrow fat in anorexia nervosa. J. Clin. Endocrinol. Metab. 94, 2129–2136 (2009).
Cawthorn, W. P. et al. Expansion of bone marrow adipose tissue during caloric restriction is associated with increased circulating glucocorticoids and not with hypoleptinemia. Endocrinology 157, 508–521 (2016).
Yamauchi, T. et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 423, 762–769 (2003).
Liu, Q. et al. Adiponectin regulates expression of hepatic genes critical for glucose and lipid metabolism. Proc. Natl Acad. Sci. 109, 14568–14573 (2012).
Zhang, F., Xu, X., Zhou, B., He, Z. & Zhai, Q. Gene expression profile change and associated physiological and pathological effects in mouse liver induced by fasting and refeeding. PLoS One 6, e27553 (2011).
Ortega, F. J. et al. The gene expression of the main lipogenic enzymes is downregulated in visceral adipose tissue of obese subjects. Obesity 18, 13–20 (2010).
Jiang, L. et al. Leptin contributes to the adaptive responses of mice to high-fat diet intake through suppressing the lipogenic pathway. PLoS One 4, e6884 (2009).
Roberts, R. et al. Markers of de novo lipogenesis in adipose tissue: associations with small adipocytes and insulin sensitivity in humans. Diabetologia 52, 882–890 (2009).
McLaughlin, T. et al. Adipose cell size and regional fat deposition as predictors of metabolic response to overfeeding in insulin-resistant and insulin-sensitive humans. Diabetes 65, 1245–1254 (2016).
Herman, M. A. et al. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature 484, 333–338 (2012).
Fisher, F. M. et al. FGF21 regulates PGC-1alpha and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. 26, 271–281 (2012).
Bostrom, P. et al. A PGC1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 481, 463–468 (2012).
Brun, J. et al. Bone regulates browning and energy metabolism through mature osteoblast/osteocyte PPARgamma expression. Diabetes 66, 2541–2554 (2017).
Fulzele, K. et al. Osteocyte-secreted Wnt signaling inhibitor sclerostin contributes to beige adipogenesis in peripheral fat depots. J. Bone Min. Res 32, 373–384 (2017).
Liu, W. et al. Osteocyte TSC1 promotes sclerostin secretion to restrain osteogenesis in mice. Open Biol. 9, 180262 (2019).
Goodpaster, B. H. & Sparks, L. M. Metabolic flexibility in health and disease. Cell Metab. 25, 1027–1036 (2017).
De Pergola, G. et al. Fuel metabolism in adult individuals with a wide range of body mass index: effect of a family history of type 2 diabetes. Diabetes Nutr. Metab. 16, 41–47 (2003).
Ukropcova, B. et al. Family history of diabetes links impaired substrate switching and reduced mitochondrial content in skeletal muscle. Diabetes 56, 720–727 (2007).
Guridi, M. et al. Alterations to mTORC1 signaling in the skeletal muscle differentially affect whole-body metabolism. Skelet. Muscle 6, 13 (2016).
Umemura, A. et al. Liver damage, inflammation, and enhanced tumorigenesis after persistent mTORC1 inhibition. Cell Metab. 20, 133–144 (2014).
Tremblay, F. & Marette, A. Amino acid and insulin signaling via the mTOR/p70 S6 kinase pathway. A negative feedback mechanism leading to insulin resistance in skeletal muscle cells. J. Biol. Chem. 276, 38052–38060 (2001).
Chang, G. R. et al. Rapamycin protects against high fat diet-induced obesity in C57BL/6J mice. J. Pharm. Sci. 109, 496–503 (2009).
Liu, Y. et al. Rapamycin-induced metabolic defects are reversible in both lean and obese mice. Aging 6, 742–754 (2014).
Chang, G. R. et al. Long-term administration of rapamycin reduces adiposity, but impairs glucose tolerance in high-fat diet-fed KK/HlJ mice. Basic Clin. Pharm. Toxicol. 105, 188–198 (2009).
Lamming, D. W. et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 335, 1638–1643 (2012).
Houde, V. P. et al. Chronic rapamycin treatment causes glucose intolerance and hyperlipidemia by upregulating hepatic gluconeogenesis and impairing lipid deposition in adipose tissue. Diabetes 59, 1338–1348 (2010).
Moayeri, A. et al. Fracture risk in patients with type 2 diabetes mellitus and possible risk factors: a systematic review and meta-analysis. Ther. Clin. Risk Manag. 13, 455–468 (2017).
Vestergaard, P. Discrepancies in bone mineral density and fracture risk in patients with type 1 and type 2 diabetes-a meta-analysis. Osteoporos. Int. 18, 427–444 (2007).
Oei, L. et al. High bone mineral density and fracture risk in type 2 diabetes as skeletal complications of inadequate glucose control: the Rotterdam Study. Diabetes Care 36, 1619–1628 (2013).
Leite Duarte, M. E. & da Silva, R. D. Histomorphometric analysis of the bone tissue in patients with non-insulin-dependent diabetes (DMNID). Rev. Hosp. Clin. 51, 7–11 (1996).
Boronat, S., Barber, I. & Thiele, E. A. Sclerotic bone lesions in tuberous sclerosis complex: a genotype-phenotype study. Am. J. Med. Genet. 173, 1891–1895 (2017).
Longo, K. A. et al. The 24-hour respiratory quotient predicts energy intake and changes in body mass. Am. J. Physiol.-Regulatory, Integr. Comp. Physiol. 298, R747–R754 (2010).
Srinivas, S. et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 1, 4 (2001).
Kraus, D., Yang, Q. & Kahn, B. B. Lipid extraction from mouse feces. Bio-Protoc. 5, e1375 (2015).
Mehlem, A., Hagberg, C. E., Muhl, L., Eriksson, U. & Falkevall, A. Imaging of neutral lipids by oil red O for analyzing the metabolic status in health and disease. Nat. Protoc. 8, 1149 (2013).
Fitter, S. et al. Plasma adiponectin levels are markedly elevated in imatinib-treated chronic myeloid leukemia (CML) patients: a mechanism for improved insulin sensitivity in type 2 diabetic CML patients? J. Clin. Endocrinol. Metab. 95, 3763–3767 (2010).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408 (2001).
Ward, C. M., Thu-Hien, T. & Pederson, S. M. ngsReports: a bioconductor package for managing FastQC reports and other NGS related log files. Bioinformatics 36, 2587–2588 (2019).
Schubert, M., Lindgreen, S. & Orlando, L. AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res Notes 9, 88 (2016).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2012).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2013).
Zhou, Y. et al. A statistical normalization method and differential expression analysis for RNA-seq data between different species. BMC Bioinforma. 20, 163 (2019).
Law, C. W., Chen, Y., Shi, W. & Smyth, G. K. Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29 (2014).
McCarthy, D. J., Chen, Y. & Smyth, G. K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 40, 4288–4297 (2012).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Smyth, G. K. Limma: Linear Models for Microarray Data. In: Gentleman R., Carey V. J., Huber W., Irizarry R. A., Dudoit S. (eds) Bioinformatics and Computational Biology Solutions Using R and Bioconductor. Statistics for Biology and Health. Springer, New York, NY (2005).
Acknowledgements
The authors gratefully acknowledge Mrs Vicki Wilczek for assisting with mouse genotyping. This work was supported by grants from the National Health and Medical Research Council of Australia (APP1109207, awarded to ACWZ, PMB, and CGP), Australian Research Council (DP160100454, awarded to ACWZ and PMB), Diabetes Australia Research Program (awarded to ACWZ, SF and SM) and an Australia Postgraduate Award (PT).
Author information
Authors and Affiliations
Contributions
S.F., S.K.M., and A.C.W.Z. conceived and designed the study. P.T., S.K.M., P.Y.C., and S.F. acquired the data. J.B. and C.Y.M. analyzed the RNA-seq data. P.T., S.K.M., S.F., and A.C.W.Z. analyzed and interpreted the data with assistance from P.M.B., G.A.W., A.J.P., and C.G.P. P.T., S.K.M., S.F., and A.C.W.Z. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tangseefa, P., Martin, S.K., Chin, P.Y. et al. The mTORC1 complex in pre-osteoblasts regulates whole-body energy metabolism independently of osteocalcin. Bone Res 9, 10 (2021). https://doi.org/10.1038/s41413-020-00123-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41413-020-00123-z
