
Abstract—
Neutrophils are the “first line” of defense against pathogens in the locus of inflammation, where they use effector functions such as phagocytosis, degranulation, and formation of reactive oxygen species (ROS). In 2004, Artuto Zychlinsky characterized one more neutrophil effector function—the release of neutrophil extracellular traps (or NETs). NETs are a modified chromatin “decorated” by bactericidal proteins of granules, nucleus, and cytoplasm. The release of NETs can be activated by diverse physiological and pharmacological stimuli and depends on ROS, for which NADPH oxidase is the main source. In the process of NET formation, the release of bactericidal components of granules into the cytoplasm, modification of histones leading to chromatin decondensation, destruction of the nuclear envelope and cytoplasmic membrane with the involvement of gasdermin D protein, and, finally, the release of chromatin outside the cell occurs. At the same time, uncontrolled formation of NETs is a provoking factor in the development of many inflammatory and autoimmune diseases. NETs were found at autoimmune diseases such as systemic lupus erythematosus, rheumatoid arthritis, psoriasis, and vasculitis; NETs are involved in the pathogenesis of cardiovascular, pulmonary, and oncological diseases. In this review, the main ideas about the mechanisms of NET formation, as well as their role in physiological processes and pathogenesis of a number of diseases (including COVID-19), are discussed.
Similar content being viewed by others
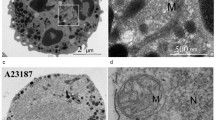


TABLE OF CONTENTS
1. Introduction………………………………………………….
2. Mechanisms of NET formation………………………...
2.1. NETosis involving NADPH oxidase…………….
2.2. NETosis involving mitochondrial ROS………..
2.3. Role of histones in NET formation………………
2.4. Vital release of chromatin…………………………..
3. Role of NETs in host protection and pathology……
3.1. NETs and inflammatory lung diseases………….
3.2. NETs and thrombosis……..………………………..
3.3. NETs and cancer……………………………………..
3.4. NETs and autoimmune diseases………………….
3.5. Sepsis…………………………………………………….
Conclusions……………………………………………………..
References…………………………………………………...
1 INTRODUCTION
Since the decoding of the genetic code in the mid-1960s and until 2004, it was considered that DNA serves exclusively for the storage of genetic information. However, it was found that chromatin is also a part of innate immunity and performs important effector functions against a large variety of pathogens. This discovery belongs to a group of scientists working under the guidance of Arturo Zychlinsky and who published their “revolutionary” work in Science journal in 2004 [1]. The release of chromatin was for the first time observed in human and murine neutrophils, and it was called neutrophil extracellular traps (NETs). NETs consist of modified chromatin “decorated” with the proteins of granules, nucleus, and cytoplasm. Since it was initially demonstrated that NET formation is accompanied by the death of neutrophils, this process was called NETosis [2], and it differs in a number of traits from apoptosis, necrosis, necroptosis, and autophagy.
NET formation can be activated by a large amount of diverse physiological stimuli, such as bacteria, fungi, protozoa, viruses, and components of bacterial cell wall (lipopolysaccharides, LPS). The release of NETs can be induced by antibodies and immune complexes, cytokines, and chemokines (IL-8, TNF-α, IFN-γ), cholesterol, stearylamine, and microcrystals as well as such pharmacological stimuli as phorbol 12-myristate 13-acetate (PMA), calcium (ionomycin, A23187), and potassium (nigericin) ionophores [3].
The release of chromatin was also found in other types of granulocytes, including in eosinophils [4], basophils [5], and mast cells [6] as well as in lymphocytes (T lymphocytes, B lymphocytes, natural killer cells) [7], monocytes [8], and macrophages [9]. It is interesting that decondensed chromatin is used for protection from pathogens not only by animals but also by lower eukaryotes (for example, Dictyostelium discoideum soil amoeba [10]) as well as by plants. Thus, the release of chromatin by plant roots in response to pathogenic fungi was found in the work of Hawes et al. [11].
In addition to involvement in protecting the host from pathogens, NETs play a significant role in the pathogenesis of many inflammatory and autoimmune diseases. NETs were found in severe pulmonary, cardiovascular, and oncological diseases. NET components are triggers for the formation of autoantibodies, and NET-mediated immune complexes were found in autoimmune pathologies such as systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), psoriasis, vasculitis, etc. In this regard, understanding the signaling pathways underlying NET formation is extremely important for creating drugs to fight with the listed diseases. Currently, a lot of data on the mechanisms of NET formation have been accumulated, but their understanding is far from complete. In this review, basic ideas about the mechanisms of formation of neutrophil traps are considered, and their role in the pathogenesis of a number of diseases is highlighted.
2 MECHANISMS OF NET FORMATION
2.1 NETosis Involving NADPH Oxidase
NETosis involving NADPH oxidase and resulting in the death of neutrophil was studied in the first works of A. Zychlinsky and was conditionally named classic or suicidal [1]. Many microorganisms and pharmacological stimuli, including PMA, are agonists of classic NETosis. The mechanism of this type of NETosis is currently studied in detail (Figs. 1, 2). Imitating the effect of diacylglycerol, PMA activates protein kinase C (PKC) isoforms involved in phosphorylation of NADPH oxidase subunits [12]. Subsequent assembly and activation of NADPH oxidase lead to the conversion of molecular oxygen into superoxide anion radical (\({\text{O}}_{2}^{{\centerdot - }}\)). Pharmacological inhibition of NADPH oxidase or neutralization of ROS suppress the formation of NETs [13]. It is important to note that neutrophils in patients with mutations in NADPH oxidase subunits are not able to form NETs in response to pharmacological stimuli and microorganisms, which confirms the important role of ROS at the genetic level [14]. Superoxide anion radical dismutes spontaneously or with the involvement of superoxide dismutase with the formation of hydrogen peroxide, which then transforms into haloid acids with the involvement of myeloperoxidase (MPO). It was demonstrated that MPO inhibitors also block NETosis, and neutrophils of patients with mutations of this enzyme do not form NETs [15].

Schematic representation of classic NETosis. Phorbol 12-myristate 13-acetate (PMA), a stimulator of classical NETosis, activates protein kinase C (PKC) isoforms involved in phosphorylation of NADPH oxidase subunits. Subsequent assembly and activation of NADPH oxidase on the membranes of specific granules (SG) and on the cytoplasmic membrane lead to the conversion of molecular oxygen to superoxide anion radical (\({\text{O}}_{2}^{{\centerdot - }}\)). Then superoxide dismutes spontaneously or with the involvement of superoxide dismutase with the formation of hydrogen peroxide (Н2О2). NADPH oxidase can also be activated by a chemoattractant N-formyl-methionyl-leucyl-phenylalanine (fMLP), which, binding to a specific receptor, activates phospholipase C (PLC) stimulating the formation of diacylglycerol (DAG) and inositol triphosphate (IP3) from phosphatidylinositol diphosphate. For reasons still unclear, fMLP does not induce NETosis. Hydrogen peroxide stimulates dissociation of “azurosomes” located in the membranes of azurophilic granules (AG) and consisting of eight types of proteins, including myeloperoxidase (MPO) and serine proteases (neutrophilic elastase (NE), cathepsin G, and azurocidin). Serine proteases are released into the cytosol and cleave F-actin, contributing to the dissociation of the actin cytoskeleton and immobilization of neutrophil. Then serine proteases migrate from the cytosol to the nucleus, where they (mainly NE) cleave histones, contributing to chromatin decondensation. MPO is also moving to the nucleus, where it acts synergistically with NE. Peptidyl arginine deaminase 4 (PAD4), which provides citrullination of histones, also comes from the cytoplasm to the nucleus; this leads to a weakening of the connection of histones with chromatin and its subsequent decondensation. Simultaneously, nuclear membrane vesiculation and destruction of granule membranes with the involvement of pore-forming protein gasdermin D occur; this leads to subsequent electrostatic binding of the granule content with decondensed chromatin. At the final stage of the process, pores also formed by gasdermin D are generated in the cytoplasmic membrane; through these pores, chromatin is released into the extracellular space (NETosis occurs). NETosis caused by calcium ionophore А23187 begins with mobilization of Ca2+ from the endoplasmic reticulum (ER). Subsequent accumulation of Ca2+ in the mitochondrial matrix (M) leads to the activation of nonselective mitochondrial pore (mitochondrial permeability transition pore, mPTP) and formation of mitochondrial reactive oxygen species (mtROS). mtROS then leave the mitochondria into the cytosol, where they activate NADPH oxidase, apparently, with the involvement of protein kinase C [16, 18, 31]. Dotted arrows indicate a hypothetical way of the information transmission.

Neutrophil extracellular traps (NETs). Immunofluorescent staining. Different stages of NET formation after the stimulation of human neutrophils with PMA can be seen in the photo (A). Solid arrows indicate NETotic neutrophils; dotted, activated cells with delobulated nucleus; wavy, intact neutrophils. The chromatin is stained with DAPI (blue color); myeloperoxidase, with FITC-labeled antibodies to MPO (green color). Magnification: 40×. Scale 25 µm. Neutrophil trap obtained when stimulating neutrophils with PMA is presented in the photo (B). DNA skeleton is stained with DAPI (blue color); cationic antimicrobial protein cathelicidin hCAP18, with FITC-labeled antibodies to LL-37 (green color). Magnification 100×. Scale 5 µm. Photos were obtained by N.V. Vorobjeva.
MPO is a part of a protein complex called “azurosome” that is located in azurophilic granules. The azurosome includes eight types of proteins, such as MPO, neutrophil elastase (NE), azurocidin, cathepsin G, lactoferrin, proteinase 3, lysozyme, and eosinophilic cationic protein, and three of them are highly homologous serine proteases (NE, cathepsin G, and azurocidin) [16]. It was demonstrated that hydrogen peroxide causes dissociation of the azurosome, which contributes to the release of serine proteases from granules into the cytosol [16]. In cytosol, NE cleaves F-actin, which leads to dissociation of cytoskeleton and immobilization of neutrophil [16]. Serine proteases then migrate from the cytosol to the nucleus, apparently, passively diffusing through nuclear pores. In the nucleus, NE cleaves histones, contributing to chromatin decondensation. MPO also moves to the nucleus, where it acts synergistically with NE, although the exact function of this enzyme in NETosis is unknown. Peptidyl arginine deaminase 4 (PAD4) [17], which provides citrullination of histones (deamination of positively charged arginine residues with the formation of electrically neutral citrullines) is also transferred from the cytoplasm to the nucleus; this leads to a weakening of the connection of histones with chromatin and its subsequent decondensation. After the destruction of nuclear and granular membranes with the involvement of pore-forming protein gasdermin D (GSDMD) [18], electrostatic binding of granule contents to decondensed chromatin occurs. At the final stage of the process, pores also formed by gasdermin D [18] are generated in the cytoplasmic membrane; through them, chromatin is released into the extracellular space (NETosis occurs) [19].
It was demonstrated that kinases such as c-Raf, MEK, Akt, and ERK (also involved in the activation of NADPH oxidase [13]) are induced in the process of classical signaling pathway.
It should be noted that data concerning the involvement of PAD4 in PMA-induced NETosis are contradictory. On the one hand, it was demonstrated that murine neutrophils having a knockout for the PAD4 do not form NETs in response to PMA. In addition, pharmacological suppression of PAD4 with chloramidine GSK484 also led to the inhibition of NETosis induced by phorbol ester [20]. On the other hand, using a confocal microscopy, it was established that PMA does not cause a significant histone deamination during NETosis [21] (the main function of PAD4).
A possible involvement of PAD4 in classical PMA-induced NETosis raises another interesting question. Since PAD4 is a calcium-dependent enzyme, an increase in cytosolic Са2+ is required during the activation of NETosis with PMA. However, as was demonstrated in early studies, PMA does not stimulate the release of Са2+ from endoplasmic reticulum [22] or extracellular Са2+ influx into the cytosol [23]. In this regard, the involvement of PAD4 in PMA-induced NETosis becomes unlikely. At the same time, it was established in our work [24] that PMA-induced NETosis is suppressed efficiently and in a dose-dependent way by cytosolic Са2+ chelator BAPTA-AM, which indicates a latent increase in the content of cytosolic Са2+, sufficient for the activation of PAD4 and induction of NETosis.
The activation of specific receptors can not only stimulate but also suppress NETosis. For example, prostaglandin E2 inhibited NETosis due to the stimulation of the corresponding receptors (ER2 and ER4) associated with the G protein, leading to an increase in the content of intracellular cyclic AMP and, further, to the suppression of PKC and subsequent stages of NET formation [25]. In addition, activated C protein (serine proteinase possessing the anticoagulant and anti-inflammatory effect) also inhibited NETosis due to binding to specific receptor (EPCR) or cooperative interaction with protease-activated receptor 3 (PAR3) and integrins CD11b/CD18 (Mac-1) [26]. These facts indicate that NETosis can be regulated by both pro- and anti-inflammatory mediators.
Recently, it was demonstrated that NADPH oxidase-dependent NETosis occurs upon activation of cyclin-dependent kinases (CDK) that promote the transfer of neutrophils from the stage G0 back to the cell cycle [27]. Since neutrophils are terminally differentiated cells, the involvement of cell cycle proteins in NETosis is an amazing fact. In neutrophils induced to NETosis, cell cycle events such as expression of a proliferation marker (Ki-67 nuclear antigen), phosphorylation of retinoblastoma protein and nuclear lamins, and centrosome separation were found; however, no DNA replication and transcription of histone genes occurred in this case [27]. The above-described facts indicate that neutrophils use a part of the cell cycle apparatus to destroy the nuclear membrane in the process of NETosis.
2.2 NETosis Involving Mitochondrial ROS
Previously, it was demonstrated that calcium ionophores А23187 and ionomycin, as well as potassium ionophore nigericin, stimulate NETosis occuring without NADPH oxidase [28, 29]. However, it was found that ROS are still needed for this form of NETosis, and mitochondrial ROS (mtROS) [30] or ROS released by the pathogen itself [28] can be their source.
In our recent work [31] using mitochondria-targeted antioxidant SkQ1 and specific NADPH oxidase inhibitors, we demonstrated that both mitochondria and mtROS and NADPH oxidase are involved in NETosis induced by А23187, and cross interaction occurs between them. In addition, using inhibitory analysis, it was demonstrated that the formation of mtROS is caused by a reversible opening of the mitochondrial pore, mPTP [31] (Fig. 1).
At the same time, neutrophils isolated from the blood of patients with chronic granulomatous disease (CGD) and possessing nonfunctional NADPH oxidase (X-linked CGD) formed NETs in response to A23187 without the involvement of oxidase [31]. We assume that mtROS in NADPH oxidase-deficient neutrophils are formed with increased intensity, and their number is enough for the activation of NETosis [31]. Such increased formation of mtROS is probably associated with the absence of electrogenic function of the enzyme, which normally provides membrane depolarization [32], and thus stops the uncontrolled entrance of extracellular Ca2+ into the cytosol. Recently, we demonstrated increased concentration of cytosolic Са2+ in neutrophils isolated from the blood of patients with CGD when activating NETosis by А23187 [24]. Apparently, the excess inflow of Ca2+ into mitochondria causes aberrantly high production of mtROS with the involvement of mPTP [33], which stimulates NETosis during the activation by A23187, but, at the same time, it can cause many inflammatory and autoimmune diseases in such patients.
Thus, in our opinion, it would be more correct to name NETosis induced by calcium ionophores and frequently named “NADPH oxidase-independent” as “mitochondria-dependent” NETosis.
2.3 Role of Histones in NET Formation
Histones are small proteins involved in the structural organization of chromatin neutralizing negatively charged phosphate groups of DNA due to positive charges of amino-acid residues, which enables a dense packing of DNA in the nucleus. Histones have both powerful antimicrobial and toxic effects in animal cells. At present, the cytotoxicity of histones is well understood, and it was demonstrated that antihistone antibodies prevent the pathogenesis of different diseases in murine models [34]. In the process of NET formation, a huge number of histones are released into the tissues that, on the one hand, are toxic for the pathogens and, on the other hand, cause tissue damage. Apparently, the antimicrobial function of NETs (at least partially) is caused by the effect of histones.
It was established that posttranslational modification of histones regulates not only gene expression but also chromatin structure [35]. Irreversible cleavage by serine proteases is one of such modifications occurring during NETosis; this contributes to chromatin decondensation. In the process of NETosis, histones can also be citrullinated with the involvement of peptidyl arginine deaminase (PAD) [20, 36]. At the same time, data concerning the involvement of PAD4 in NETosis are contradictory, and this question requires more thorough study. It was established that acetylation of histones also contributes to the formation of NETs [37], apparently, due to neutralization of positively charged histone groups.
2.4 Vital Release of Chromatin
Lifetime (or vital) release of extracellular chromatin, in which the cells maintain their viability and natural effector functions, is an alternative form of suicidal NETosis. It was found that this process develops much faster than suicidal NETosis and can occur with the release of both nuclear and mitochondrial DNA.
In the model developed by Clark et al. [38], vital release of chromatin induced by a cell wall component of gram-negative bacteria (LPS) was described. This type of NETosis was induced by the interaction of TLR4-activated platelets with neutrophils and occurred with the release of nuclear chromatin but without the involvement of NADPH oxidase. Yipp et al. [39] also demonstrated in vivo the stimulation of vital release of chromatin by opsonized gram-positive bacteria, which was mediated by TLR2 and a complement. It is interesting that such neutrophils were capable of chemotaxis and phagocytosis of bacteria after the formation of neutrophil traps [39].
In 2010, Pilsczek et al. [40] described the release of chromatin as a part of vesicles in response to Staphylococcus aureus toxin (Panton–Valentine leucotoxin) without the activation of NADPH oxidase.
In works performed under the guidance of Simon [4, 41], lifetime release of chromatin by neutrophils and eosinophils, primed by proinflammatory cytokines GM-CSF or IL-5/IFN-γ, respectively, and stimulated by LPS, was described. At the same time, both types of granulocytes released chromatin of exclusively mitochondrial origin, while the process itself depended on the activity of NADPH oxidase. It is interesting that eosinophilic chromatin (as well as neutrophilic) possessed bactericidal activity.
It is amazing that vital release of chromatin was also found in lymphocytes. Thus, it was demonstrated for the first time in the original work of Ingelsson et al. [7] that B lymphocytes, T lymphocytes, and natural killer cells isolated from the blood of healthy donors, as well as B cells of patients with a chronic lymphocytic leukemia, performed a rapid release of mitochondrial DNA in the form of long fragments in response to C class oligodeoxynucleotides. Filaments formed by B lymphocytes were studied in detail, and it was found that they are not associated with the formation of ROS or cell death [7]. Mass spectrometric analysis of DNA filaments demonstrated that they differ from NETs by a protein composition and have no bactericidal properties. In addition, the filaments induced the synthesis of type I IFN by peripheral blood mononuclear cells, acting as signaling molecules but not bactericidal traps.
3 ROLE OF NETS IN HOST PROTECTION AND PATHOLOGY
The biological role of NETs became apparent after the diseases associated with the violation of their formation were found. The significance of NETs in the host protection was demonstrated for the first time when studying neutrophils isolated from the blood of patients with CGD possessing a mutant NADPH oxidase and unable to form ROS. Such patients suffer from recurrent infections, among which invasive aspergillosis is the most common cause. Neutrophils of such patients are also unable to form NETs; however, gene therapy leads to a complete restoration of this function of neutrophils [14].
Initially, it was assumed that neutrophils form NETs when faced with pathogens that they are unable to phagocytose [42]. However, data concerning the ability of NETs to kill pathogens are highly contradictory [28]. It was found that the ability of NETs to destroy a particular pathogen largely depends on the pathogen itself faced by neutrophil. Thus, many pathogens synthesize virulence factors, for example, capsules [43] or endonucleases [44], allowing them to avoid the effect of neutrophil traps. At present, it is generally accepted that NETs prevent the spread of infection from the focus of inflammation, contributing to their subsequent destruction by antimicrobial proteins and professional phagocytes.
After the completion of the infectious process, NETs must be eliminated. As demonstrated by Farrera and Fadeel [45] in vitro, NET degradation occurs first with the involvement of DNase I, after which the debris is endocytosed and lysed by macrophages.
However, inadequate formation of NETs or, as they sometimes say, aberrant NETosis, can lead to inflammatory and autoimmune pathology as well as to blockage of blood vessels. Such states occur both in the case of overproduction of NETs and in the case of violation of the mechanisms of their elimination (for example, in the absence of DNase I).
3.1 NETs and Inflammatory Lung Diseases
In lung diseases, NETs can play both a positive and a negative role. Thus, it was demonstrated that NETs increase the viscosity of mucus protecting the organism from infection. NETs were found in sputum and other lung secretions at bacterial, fungal, and viral infections [14, 46, 47]. It was also established that NETs are involved in the destruction of Aspergillus nidulans causing severe invasive pulmonary aspergillosis [14].
At the same time, the formation of NETs with chronic obstructive pulmonary disease (COPD) worsened respiratory function and led to the blockade of the respiratory tract, and the amount of NETs in the sputum of patients with COPD correlated with the severity of the disease and the composition of microflora [48].
Increased production of NETs was also found at cystic fibrosis. Cystic fibrosis is a severe hereditary disease caused by a mutation in the CFTR gene, which results in violation of the structure and function of the protein named “cystic fibrosis transmembrane conductance regulator.” The mutations of this gene lead to the violation of normal transport of Cl– ions through the cell epithelium and, as a consequence, cause dehydration, concentration of secretions of exocrine glands, and difficulty of their outflow. In the case of the pulmonary form of cystic fibrosis, increased viscosity of sputum contributes to colonization of the lungs with bacterial microflora. Further, bacterial colonization attracts neutrophils and stimulates them to form NETs, which further increases the viscosity of sputum and decreases the respiration function in such patients. It is interesting that inhalation with recombinant DNase improved lung function at cystic fibrosis, while neutrophil elastase contributed to the dissolution of sputum, making it more accessible to DNase [49].
Acute lung injury (ALI) and more severe acute respiratory distress syndrome (ARDS) cause the rapid development of respiratory failure and can be caused by various etiology. In patients with transfusion-related ALI, the content of NETs in the blood plasma was higher than in individuals without ALI [50]. In animal models, it was also demonstrated that NETs are formed in response to stimuli-inducing ALI, while NET inhibitors reduce the severity of the disease and increase the survival [50]. Neutrophils of patients with ARDS caused by pneumonia were primed to the formation of NETs, while the degree of priming and the content of NETs in the blood correlated with the severity of the disease and mortality [51]. In patients with ARDS, increased content of histones in bronchoalveolar lavage and plasma was also found [52], which is probably partly due to the formation of NETs.
Severe acute respiratory infection COVID-19 (corona virus disease-19), which was for the first time registered in December 2019 in the Chinese city of Wuhan and subsequently developed into a pandemic, has affected to date more than 35 million individuals from 250 countries. The disease was caused by a new coronavirus SARS-CoV-2 (severe acute respiratory syndrome coronavirus-2) and was accompanied by influenza-like symptoms and viral pneumonia frequently transforming into ARDS and multiple organ failure [53].
Increased level of peripheral neutrophils is one of the traits of a severe course of COVID-19 [54]. It is assumed that the NET activation can underlie the pathogenesis of this disease. Increased level of NET markers (free DNA, MPO–DNA complexes, and citrullinated H3 histone) in the sera of patients infected with SARS-CoV-2 was for the first time demonstrated in the original work of Zuo et al. [55]. Moreover, the concentration of free DNA correlated with the level of acute phase protein (C-reactive protein), D-dimer (thrombosis marker), and lactate dehydrogenase (cell death marker), as well as with the absolute number of neutrophils. It is interesting that sera of patients with COVID-19 induced NET formation in neutrophils of healthy donors in vitro. As suggested by the authors, epithelial cells affected by the virus, activated platelets, and endothelial cells, as well as proinflammatory cytokines (IL-1β, IL-8, G-CSF), could be activators of NETosis.
3.2 NETs and Thrombosis
NETs play an important role in thrombosis [56] activating platelets and inducing coagulation [57]. It is yet unknown whether NET-induced thrombosis/coagulation play a role in innate immunity providing the organism homeostasis or are a pathological consequence of aberrant NETosis. However, it was demonstrated in multiple models of thrombotic diseases that the addition of DNase significantly decreases the blockage of blood vessels [58]. Recent analysis of cardiovascular human diseases demonstrated that the prognosis and severity of such diseases correlates with the presence of free DNA [59]. In humans, NETs were found at a stroke [60], thrombotic microangiopathies [61], atherosclerosis [62], and antiphospholipid syndrome [63]. And although the role of NETs in the pathogenesis of listed diseases is not completely understood, it was established in the model of atherosclerosis that serine protease deficiency or introduction of DNase decrease the size of atherosclerotic plaques [62].
Aggregated NETs were induced by microcrystals of calcium carbonate and change in pH [64], which led to the blockage of pancreatic duct and pancreatitis. The addition of DNase in the murine pancreatitis model alleviated the severity of the disease [65].
Gout develops as a result of accumulation of crystals of sodium salts of uric acid (urates) causing temporary arthritis. Urate crystals induce the formation of aggregated NETs with a high concentration of proteases capable of degrading proinflammatory cytokines and chemokines [66]. The degradation of cytokines in gout with the involvement of NETs prevented the development of chronic disease [66].
3.3 NETs and Cancer
Cancer is a heterogeneous disease, and the role of NETs in oncology depends on the type of tumor. In breast cancer models, it was demonstrated that NETs contribute to metastasis since this process is suppressed by the introduction of DNase [67]. It is considered that NET-induced coagulation is a complication of a number of oncological diseases [68]. In an intestinal cancer model, DNase has been shown to reduce coagulation and carcinogenesis [69]. On the other hand, therapeutic viral infection of tumors induced neutrophil-induced intratumoral coagulation and the destruction of cancer cells. However, it remains to be seen whether this process is indeed associated with the formation of NETs [70].
3.4 NETs and Autoimmune Diseases
NETs play an important role in autoimmunity [19, 71]. Thus, autoantibodies that recognize NET components such as double-stranded DNA [72], citrullinated proteins [73], and the components of azurophilic granules [74] were found in patients with SLE, RA, and antineutrophil cytoplasmic antibodies-associated vasculitis (ANCA-associated vasculitis, AAV), respectively. These antibodies can appear due to the long-term presence of NETs caused by their excessive formation or reduced ability of their degradation by DNase I. It was demonstrated that genetically determined deficiencies in DNase lead to the development of juvenile form of SLE [75]. In addition, it was established in an RA model that the uptake of NETs by antigen-presenting cells controls the autoimmune process, since fibroblasts loaded with NETs stimulated the formation of antibodies against citrullinated peptides [76]. It is interesting that antibodies against NETs were also able to inhibit the degradation of traps, thus exacerbating the disease [77]. The immune complexes with NET components were found at glomerulonephritis (glomerular nephritis), a common complication of SLE and AAV [77, 78]. It was demonstrated that antihistone antibodies protect mice from the development of renal failure on the model of necrotizing glomerulonephritis [79].
3.5 Sepsis
Sepsis is an acute complication of a severe infectious process characterized by high mortality. The pathology of this systemic disease is complex, but, as it was found out, NETs can contribute to the survival in sepsis. It was found that neutrophils of patients that survived after sepsis produce more NETs in vitro than neutrophils of subsequently deceased patients [80]. This can be partly caused by a bactericidal effect of NETs at early stages of the disease. Thus, it was demonstrated in a multimicrobial murine model that the introduction of DNase accelerated the development of sepsis [81]. However, as the disease progresses, NETs can damage the lungs (ALI and ARDS) and liver [82, 83]. It is interesting that direct injections of histones to mice simulated sepsis, while antihistone antibodies protected mice on multiple infectious models [84]. Thrombosis can also contribute to organ damage in sepsis. The same as in thrombotic diseases, the presence of free DNA correlated with the severe course of sepsis in patients and with organ damage in mice [85]. The introduction of DNase reduced the severity of organ damage and increased the survival of mice but only in combination with antibiotic therapy [85]. These facts indicate a dual role of NETs in sepsis: positive at early stages of the disease and negative associated with organ pathology at later stages.
CONCLUSIONS
To date, many signaling pathways leading to the formation of NETs were deciphered. It was demonstrated that NETs can be induced by a large number of both physiological (microorganisms and their components) and pharmacological stimuli. The existence of two mechanisms of NET formation was established: classic (or suicidal) leading to cell death (NETosis) and vital, in which the cells retain not only their viability but also all natural effector functions. It was established that the enzyme complex NADPH oxidase and ROS formed with its help are a necessary participants of classic NETosis. However, mitochondrial ROS “come into play” when activating NETosis by other stimuli (for example, Са2+ ionophores).
After the discovery of NETs, it was found that these structures are a source of antigens and support (and often activate) autoimmune processes [71, 72]. Thus, NETs were found in sera of patients with almost all autoimmune diseases, including SLE, RA, psoriasis, etc. NETs were found in many inflammatory noninfectious diseases, for example, Alzheimer’s disease, pancreatitis, cancer (Table 1). In this regard, the creation of medicines for the treatment of the listed diseases largely depends on the decoding of signaling pathways leading to the formation of NETs and their targeted mediators. And although many mechanisms and mediators were deciphered over the past 16 years since the first description of NETs, there is still a sufficient number of unresolved questions, the answers to which are expected in the near future.
REFERENCES
Brinkmann, V., Reichard, U., Goosmann, C., Fauler, B., Uhlemann, Y., Weiss, D.S., Weinrauch, Y., and Zychlinsky, A., Neutrophil extracellular traps kill bacteria, Science, 2004, vol. 303, no. 5663, pp. 1532–1535.
Steinberg, B.E. and Grinstein, S., Unconventional roles of the NADPH oxidase: Signaling, ion homeostasis, and cell death, Sci. STKE, 2007, vol. 2007, no. 379, pe11.
Papayannopoulos, V., Neutrophil extracellular traps in immunity and disease, Nat. Rev. Immunol., 2018, vol. 18, no. 2, pp. 134–147.
Yousefi, S., Gold, J.A., Andina, N., Lee, J.J., Kelly, A.M., Kozlowski, E., Schmid, I., Straumann, A., Reichenbach, J., Gleich, G.J., and Simon, H.U., Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense, Nat. Med., 2008, vol. 14, no. 9, pp. 949–953.
Morshed, M., Hlushchuk, R., Simon, D., Walls, A.F., Obata-Ninomiya, K., Karasuyama, H., Djonov, V., Eggel, A., Kaufmann, T., Simon, H.U., and Yousefi, S., NADPH oxidase-independent formation of extracellular DNA traps by basophils, J. Immunol., 2014, vol. 192, no. 11, pp. 5314–5323.
von Köckritz-Blickwede, M., Goldmann, O., Thulin, P., Heinemann, K., Norrby-Teglund, A., Rohde, M., and Medina, E., Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation, Blood, 2008, vol. 111, no. 6, pp. 3070–3080.
Ingelsson, B., Söderberg, D., Strid, T., Söderberg, A., Bergh, A.C., Loitto, V., Lotfi, K., Segelmark, M., Spyrou, G., and Rosén, A., Lymphocytes eject interferogenic mitochondrial DNA webs in response to CpG and non-CpG oligodeoxynucleotides of Class C, Proc. Natl. Acad. Sci. U.S.A., 2018, vol. 115, no. 3, pp. E478–E487.
Granger, V., Faille, D., Marani, V., Noël, B., Gallais, Y., Szely, N., Flament, H., Pallardy, M., Chollet-Martin, S., and de Chaisemartin, L., Human blood monocytes are able to form extracellular traps, J. Leukoc. Biol., 2017, vol. 102, no. 3, pp. 775–781.
Chow, O.A., von Köckritz-Blickwede, M., Bright, A.T., Hensler, M.E., Zinkernagel, A.S., Cogen, A.L., Gallo, R.L., Monestier, M., Wang, Y., Glass, C.K., and Nizet, V., Statins enhance formation of phagocyte extracellular traps, Cell Host Microbe, 2010, vol. 8, no. 5, pp. 445–454.
Zhang, X., Zhuchenko, O., Kuspa, A., and Soldati, T., Social amoebae trap and kill bacteria by casting DNA nets, Nat. Commun., 2016, vol. 7, 10938.
Hawes, M., Allen, C., Turgeon, B.G., Curlango-Rivera, G., Minh, TranT., Huskey, D.A., and Xiong, Z., Root border cells and their role in plant defense, Annu. Rev. Phytopathol., 2016, vol. 54, pp. 143–161.
Belambri, S.A., Rolas, L., Raad, H., Hurtado-Nedelec, M., Dang, P.M., and El-Benna, J., NADPH oxidase activation in neutrophils: Role of the phosphorylation of its subunits, Eur. J. Clin. Invest., 2018, vol. 48, suppl. 2, e12951.
Hakkim, A., Fuchs, T.A., Martinez, N.E., Hess, S., Prinz, H., Zychlinsky, A., and Waldmann, H., Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation, Nat. Chem. Biol., 2011, vol. 7, no. 2, pp. 75–77.
Bianchi, M., Hakkim, A., Brinkmann, V., Siler, U., Seger, R.A., Zychlinsky, A., and Reichenbach, J., Restoration of NET formation by gene therapy in CGD controls aspergillosis, Blood, 2009, vol. 114, no. 13, pp. 2619–2622.
Metzler, K.D., Fuchs, T.A., Nauseef, W.M., Reumaux, D., Roesler, J., Schulze, I., Wahn, V., Papayannopoulos, V., and Zychlinsky, A., Myeloperoxidase is required for neutrophil extracellular trap formation: Implications for innate immunity, Blood, 2011, vol. 117, no. 3, pp. 953–959.
Metzler, K.D., Goosmann, C., Lubojemska, A., Zychlinsky, A., and Papayannopoulos, V., A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis, Cell Rep., 2014, vol. 8, no. 3, pp. 883–896.
Anzilotti, C., Pratesi, F., Tommasi, C., and Migliorini, P., Peptidylarginine deiminase 4 and citrullination in health and disease, Autoimmun. Rev., 2010, vol. 9, no. 3, pp. 158–160.
Sollberger, G., Choidas, A., Burn, G.L., Habenberger, P., Di Lucrezia, R., Kordes, S., Menninger, S., Eickhoff, J., Nussbaumer, P., Klebl, B., Kruger, R., Herzig, A., and Zychlinsky, A., Gasdermin D plays a vital role in the generation of neutrophil extracellular traps, Sci. Immunol., 2018, vol. 3, eaar6689.
Pinegin, B., Vorobjeva, N., and Pinegin, V., Neutrophil extracellular traps and their role in the development of chronic inflammation and autoimmunity, Autoimmun. Rev., 2015, vol. 14, no. 7, pp. 633–640.
Li, P., Li, M., Lindberg, M.R., Kennett, M.J., Xiong, N., and Wang, Y., PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps, J. Exp. Med., 2010, vol. 207, no. 9, pp. 1853–1862.
Douda, D.N., Khan, M.A., Grasemann, H., and Palaniyar, N., SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx, Proc. Natl. Acad. Sci. U.S.A., 2015, vol. 112, no. 9, pp. 2817–2822.
Mahomed, A.G. and Anderson, R., Activation of human neutrophils with chemotactic peptide, opsonized zymosan and the calcium ionophore A23187, but not with a phorbol ester, is accompanied by efflux and store-operated influx of calcium, Inflammation, 2000, vol. 24, no. 6, pp. 559–569.
Hu, T.H., Bei, L., Qian, Z.M., and Shen, X., Intracellular free calcium regulates the onset of the respiratory burst of human neutrophils activated by phorbolmyristate acetate, Cell. Signal, 1999, vol. 11, no. 5, pp. 355–360.
Vorobjeva, N.V. and Chernyak, B.V., NADPH oxidase modulates Ca2+-dependent formation of neutrophil extracellular traps, Moscow Univ. Biol. Sci. Bull., 2020, vol. 75, no. 3, pp. 104–109.
Shishikura, K., Horiuchi, T., Sakata, N., Trinh, D.A., Shirakawa, R., Kimura, T., Asada, Y., and Horiuchi, H., Prostaglandin E2 inhibits neutrophil extracellular trap formation through production of cyclic AMP, Br. J. Pharmacol., 2016, vol. 173, no. 2, pp. 319–331.
Healy, L.D., Puy, C., Fernandez, J.A., Mitrugno, A., Keshari, R.S., Taku, N.A., Chu, T.T., Xu, X., Gruber, A., Lupu, F., Griffin, J.H., and McCarty, O.J.T., Activated protein C inhibits neutrophil extracellular trap formation in vitro and activation in vivo, J. Biol. Chem., 2017, vol. 292, no. 21, pp. 8616–8629.
Amulic, B., Knackstedt, S.L., Abu Abed, U., Deigendesch, N., Harbort, C.J., Caffrey, B.E., Brinkmann, V., Heppner, F.L., Hinds, P.W., and Zychlinsky, A., Cell-cycle proteins control production of neutrophil extracellular traps, Dev. Cell, 2017, vol. 43, no. 4, pp. 449–462.
Kenny, E.F., Herzig, A., Krüger, R., Muth, A., Mondal, S., Thompson, P.R., Brinkmann, V., Bernuth, H.V., and Zychlinsky, A., Diverse stimuli engage different neutrophil extracellular trap pathways, eLife, 2017, vol. 6, e24437.
Neeli, I. and Radic, M., Opposition between PKC isoforms regulates histone deimination and neutrophil extracellular chromatin release, Front. Immunol., 2013, vol. 4, 38.
Lood, C., Blanco, L.P., Purmalek, M.M., Carmona-Rivera, C., De Ravin, S.S., Smith, C.K., Malech, H.L., Ledbetter, J.A., Elkon, K.B., and Kaplan, M.J., Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease, Nat. Med., 2016, vol. 22, no. 2, pp. 146–153.
Vorobjeva, N., Galkin, I., Pletjushkina, O., Golyshev, S., Zinovkin, R., Prikhodko, A., Pinegin, V., Kondratenko, I., Pinegin, B., and Chernyak, B., Mitochondrial permeability transition pore is involved in oxidative burst and NETosis of human neutrophils, Biochim. Biophys. Acta Mol. Basis Dis., 2020, vol. 1866, no. 5, 165664.
Geiszt, M., Kapus, A., and Ligeti, E., Chronic granulomatous disease: More than the lack of superoxide?, J. Leukocyte Biol., 2001, vol. 69, no. 2, pp. 191–196.
Bernardi, P., Rasola, A., Forte, M., and Lippe, G., The mitochondrial permeability transition pore: Channel formation by F-ATP synthase, integration in signal transduction, and role in pathophysiology, Physiol. Rev., 2015, vol. 95, pp. 1111–1155.
Wildhagen, K.C., García de Frutos, P., Reutelingsperger, C.P., Schrijver, R., Aresté, C., Ortega-Gómez, A., Deckers, N.M., Hemker, H.C., Soehnlein, O., and Nicolaes, G.A., Nonanticoagulant heparin prevents histone-mediated cytotoxicity in vitro and improves survival in sepsis, Blood, 2014, vol. 123, no. 7, pp. 1098–1101.
Bannister, A.J. and Kouzarides, T., Regulation of chromatin by histone modifications, Cell Res., 2011, vol. 21, no. 3, pp. 381–395.
Lewis, H.D., Liddle, J., Coote, J.E., et al., Inhibition of PAD4 activity is sufficient to disrupt mouse and human net formation, Nat. Chem. Biol., 2015, vol. 11, no. 3, pp. 189–191.
Hamam, H.J., Khan, M.A., and Palaniyar, N., Histone acetylation promotes neutrophil extracellular trap formation, Biomolecules, 2019, vol. 9, no. 1, 32.
Clark, S.R., Ma, A.C., Tavener, S.A., et al., Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood, Nat. Med., 2007, vol. 13, no. 4, pp. 463–469.
Yipp, B.G., Petri, B., Salina, D., Jenne, C.J., Scott, B.N.V., Zbytnuik, L.D., Pittman, K., Asaduzzaman, M., Wu, K., Meijndert, H.C., Malawista, S.E., de Boisfleury Chevance, A., Zhang, K., Conly, J., and Kubes, P., Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo, Nat. Med., 2012, vol. 18, no. 9, pp. 1386–1393.
Pilsczek, F.H., Salina, D., Poon, K.K., Fahey, C., Yipp, B.G., Sibley, C.D., Robbins, S.M., Green, F.H.Y., Surette, M.G., Sugai, M., Bowden, M.G., Hussain, M., Zhang, K., and Kubes, P., A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus, J. Immunol., 2010, vol. 185, no. 12, pp. 7413–7425.
Yousefi, S., Mihalache, C., Kozlowski, E., Schmid, I., and Simon, H.U., Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps, Cell. Death Differ., 2009, vol. 16, no. 11, pp. 1438–1444.
Branzk, N., Lubojemska, A., Hardison, S.E., Wang, Q., Gutierrez, M.G., Brown, G.D., and Papayannopoulos, V., Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens, Nat. Immunol., 2014, vol. 15, no. 11, pp. 1017–1025.
Wartha, F., Beiter, K., Albiger, B., Fernebro, J., Zychlinsky, A., Normark, S., and Henriques-Normark, B., Capsule and D-alanylatedlipoteichoic acids protect Streptococcus pneumoniae against neutrophil extracellular traps, Cell. Microbiol., 2007, vol. 9, no. 5, pp. 1162–1171.
Wilton, M., Halverson, T.W.R., Charron-Mazenod, L., Parkins, M.D., and Lewenza, S., Secreted phosphatase and deoxyribonuclease are required by Pseudomonas aeruginosa to defend against neutrophil extracellular traps, Infect. Immun., 2018, vol. 86, no. 9, e00403-18.
Farrera, C. and Fadeel, B., Macrophage clearance of neutrophil extracellular traps is a silent process, J. Immunol., 2013, vol. 191, no. 5, pp. 2647–2656.
Cortjens, B., de Boer, O.J., de Jong, R., Antonis, A.F., Sabogal Pineros, Y.S., Lutter, R., Van Woensel, J.B., Reinout, A., and Bem, R.A., Neutrophil extracellular traps cause airway obstruction during respiratory syncytial virus disease, J. Pathol., 2016, vol. 238, no. 3, pp. 401–411.
Hamaguchi, S., Seki, M., Yamamoto, N., Hirose, T., Matsumoto, N., Irisawa, T., Takegawa, R., Shimazu, T., and Tomono, K., Case of invasive nontypable Haemophilus influenzae respiratory tract infection with a large quantity of neutrophil extracellular traps in sputum, J. Inflamm. Res., 2012, vol. 5, pp. 137–140.
Dicker, A.J., Crichton, M.L., Pumphrey, E.G., et al., Neutrophil extracellular traps are associated with disease severity and microbiotadiversity in patients with chronic obstructive pulmonary disease, J. Allergy Clin. Immunol., 2018, vol. 141, no. 1, pp. 117–127.
Papayannopoulos, V., Staab, D., and Zychlinsky, A., Neutrophil elastase enhances sputum solubilization in cystic fibrosis patients receiving DNAse therapy, PLoS One, 2011, vol. 6, no. 12, e28526.
Caudrillier, A., Kessenbrock, K., Gilliss, B.M., Nguyen, J.X., Marques, M.B., Monestier, M., Toy, P., Werb, Z., and Looney, M.R., Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury, J. Clin. Invest., 2012, vol. 122, no. 7, pp. 2661–2671.
Bendib, I., de Chaisemartin, L., Granger, V., Schlemmer, F., Maitre, B., Hüe, S., Surenaud, M., Beldi-Ferchiou, A., Carteaux, G., Razazi, K., Chollet-Martin, S., Dessap, A.M., and de Prost, N., Neutrophil extracellular traps are elevated in patients with pneumonia-related acute respiratory distress syndrome, Anesthesiology, 2019, vol. 130, no. 4, pp. 581–591.
Lv, X., Wen, T., Song, J., Xie, D., Wu, L., Jiang, X., Jiang, P., and Wen, Z., Extracellular histones are clinically relevant mediators in the pathogenesis of acute respiratory distress syndrome, Respir. Res., 2017, vol. 18, no. 1, 165.
Pedersen, S.F. and Ho, Y.C., SARS-CoV-2: A storm is raging, J. Clin. Invest., 2020, vol. 130, no. 5, pp. 2202–2205.
Mehta, P., McAuley, D.F., Brown, M., Sanchez, E., Tattersall, R.S., Manson, J.J., and HLH Across Speciality Collaboration, UK, COVID-19: Consider cytokine storm syndromes and immunosuppression, Lancet, 2020, vol. 395, no. 10229, pp. 1033–1034.
Zuo, Y., Yalavarthi, S., Shi, H., Gockman, K., Zuo, M., Madison, J.A., Blair, C.N., Weber, A., Barnes, B.J., Egeblad, M., Woods, R.J., Kanthi, Y., and Knight, J.S., Neutrophil extracellular traps in COVID-19, JCI Insight, 2020, vol. 5, no. 11, e138999.
Martinod, K. and Wagner, D.D., Thrombosis: Tangled up in nets, Blood, 2014, vol. 123, no. 18, pp. 2768–2776.
Fuchs, T.A., Brill, A., Duerschmied, D., Schatzberg, D., Monestier, M., Myers, D.D., Wrobleski, S.K., Wakefield, T.W., Hartwig, J.H., and Wagner, D.D., Extracellular DNA traps promote thrombosis, Proc. Natl. Acad. Sci. U.S.A., 2010, vol. 107, no. 36, pp. 15880–15885.
Brill, A., Fuchs, T.A., Savchenko, A.S., Thomas, G.M., Martinod, K., De Meyer, S.F., Bhandari, A.A., and Wagner, D.D., Neutrophil extracellular traps promote deep vein thrombosis in mice, J. Thromb. Haemost., 2012, vol. 10, no. 1, pp. 136–144.
Jimenez-Alcazar, M., Kim, N., and Fuchs, T.A., Circulating extracellular DNA: Cause or consequence of thrombosis?, Semin. Thromb. Hemost., 2017, vol. 43, no. 6, pp. 553–561.
Novotny, J., Oberdieck, P., Titova, A., et al., Thrombus NET content is associated with clinical outcome in stroke and myocardial infarction, Neurology, 2020, vol. 94, no. 22, pp. e2346–e2360.
Gloude, N.J., Khandelwal, P., Luebbering, N., Lounder, D.T., Jodele, S., Alder, M.N., Lane, A., Wilkey, A., Lake, K.E., Litts, B., and Davies, S.M., Circulating dsDNA, endothelial injury, and complement activation in thrombotic microangiopathy and GVHD, Blood, 2017, vol. 130, no. 10, pp. 1259–1266.
Borissoff, J.I., Joosen, I.A., Versteylen, M.O., Brill, A., Fuchs, T.A., Savchenko, A.S., Gallant, M., Martinod, K., Ten, CateH., Hofstra, L., Crijns, H.J., Wagner, D.D., and Kietselaer, B.L.J.H., Elevated levels of circulating DNA and chromatin are independently associated with severe coronary atherosclerosis and a prothrombotic state, Arterioscler. Thromb. Vasc. Biol., 2013, vol. 33, no. 8, pp. 2032–2040.
Yalavarthi, S., Gould, T.J., Rao, A.N., Mazza, L.F., Morris, A.E., Nunez-Alvarez, C., Hernandez-Ramirez, D., Bockenstedt, P.L., Liaw, P.C., Cabral, A.R., and Knight, J.S., Release of neutrophil extracellular traps by neutrophils stimulated with antiphospholipid antibodies: a newly identified mechanism of thrombosis in the antiphospholipid syndrome, Arthritis Rheumatol., 2015, vol. 67, no. 11, pp. 2990–3003.
Leppkes, M., Maueroder, C., Hirth, S., et al., Externalized decondensed neutrophil chromatin occludes pancreatic ducts and drives pancreatitis, Nat. Commun., 2016, vol. 7, 10973.
Merza, M., Hartman, H., Rahman, M., Hwaiz, R., Zhang, E., Renstrom, E., Luo, L., Morgelin, M., Regner, S., and Thorlacius, H., Neutrophil extracellular traps induce trypsin activation, inflammation, and tissue damage in mice with severe acute pancreatitis, Gastroenterology, 2015, vol. 149, no. 7, pp. 1920–1931.
Schauer, C., Janko, C., Munoz, L.E., et al., Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines, Nat. Med., 2014, vol. 20, no. 5, pp. 511–527.
Park, J., Wysocki, R.W., Amoozgar, Z., et al., Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps, Sci. Transl. Med., 2016, vol. 8, no. 361, 361ra138.
Levi, M., Management of cancer-associated disseminated intravascular coagulation, Thromb. Res., 2016, vol. 140, suppl. 1, pp. S66–S70.
Guglietta, S., Chiavelli, A., Zagato, E., Krieg, C., Gandini, S., Ravenda, P.S., Bazolli, B., Lu, B., Penna, G., and Rescigno, M., Coagulation induced by C3aR-dependent NETosis drives protumorigenic neutrophils during small intestinal tumorigenesis, Nat. Commun., 2016, vol. 7, 11037.
Breitbach, C.J., De Silva, N.S., Falls, T.J., et al., Targeting tumor vasculature with an oncolytic virus, Mol. Ther., 2011, vol. 19, no. 5, pp. 886–894.
Gupta, S. and Kaplan, M.J., The role of neutrophils and NETosis in autoimmune and renal diseases, Nat. Rev. Nephrol., 2016, vol. 12, no. 7, pp. 402–413.
Moore, S., Juo, H.H., Nielsen, C.T., Tyden, H., Bengtsson, A.A., and Lood, C., Role of neutrophil extracellular traps regarding patients at risk of increased disease activity and cardiovascular comorbidity in systemiclupuserythematosus, J. Rheumatol., 2019, vol. 47, no. 10, 190875.
Pratesi, F., Dioni, I., Tommasi, C., Alcaro, M.C., Paolini, I., Barbetti, F., Boscaro, F., Panza, F., Puxeddu, I., Rovero, P., and Migliorini, P., Antibodies from patients with rheumatoid arthritis target citrullinated histone 4 contained in neutrophils extracellular traps, Ann. Rheum. Dis., 2014, vol. 73, no. 7, pp. 1414–1422.
Falk, R.J. and Jennette, J.C., Anti-neutrophil cytoplasmic autoantibodies with specificity for myeloperoxidase in patients with systemic vasculitis and idiopathic necrotizing and crescentic glomerulonephritis, N. Engl. J. Med., 1988, vol. 318, no. 25, pp. 1651–1657.
Al-Mayouf, S.M., Sunker, A., Abdwani, R., et al., Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus, Nat. Genet., 2011, vol. 43, no. 12, pp. 1186–1188.
Carmona-Rivera, C., Carlucci, P.M., Moore, E., et al., Synovial fibroblast-neutrophil interactions promote pathogenic adaptive immunity in rheumatoid arthritis, Sci. Immunol., 2017, vol. 2, eaag3358.
Hakkim, A., Furnrohr, B.G., Amann, K., Laube, B., Abed, U.A., Brinkmann, V., Herrmann, M., Voll, R.E., and Zychlinsky, A., Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis, Proc. Natl. Acad. Sci. U.S.A., 2010, vol. 107, no. 21, pp. 9813–9818.
O’Sullivan, K.M., Lo, C.Y., Summers, S.A., Elgass, K.D., McMillan, P.J., Longano, A., Ford, S.L., Gan, P.Y., Kerr, P.G., Kitching, A.R., and Holdsworth, S.R., Renal participation of myeloperoxidase in antineutrophil cytoplasmic antibody (ANCA)-associated glomerulonephritis, Kidney Int., 2015, vol. 88, no. 5, pp. 1030–1046.
Kumar, S.V., Kulkarni, O.P., Mulay, S.R., Darisipudi, M.N., Romoli, S., Thomasova, D., Scherbaum, C.R., Hohenstein, B., Hugo, C., Muller, S., Liapis, H., and Anders, H.J., Neutrophil extracellular trap-related extracellular histones cause vascular necrosis in severe GN, J. Am. Soc. Nephrol., 2015, vol. 26, no. 10, pp. 2399–2413.
Park, S.Y., Shrestha, S., Youn, Y.J., et al., Autophagy primes neutrophils for neutrophil extracellular trap formation during sepsis, Am. J. Respir. Crit. Care Med., 2017, vol. 196, no. 5, pp. 577–589.
Meng, W., Paunel-Görgülü, A., Flohé, S., Hoffmann, A., Witte, I., MacKenzie, C., Baldus, S.E., Windolf, J., and Lögters, T.T., Depletion of neutrophil extracellular traps in vivo results in hypersusceptibility to polymicrobial sepsis in mice, Crit. Care, 2012, vol. 16, no. 4, pp. R137.
Saffarzadeh, M., Juenemann, C., Queisser, M.A., Lochnit, G., Barreto, G., Galuska, S.P., Lohmeyer, J., and Preissner, K.T., Neutrophil extracellular traps directly induce epithelial and endothelial cell death: A predominant role of histones, PLoS One, 2012, vol. 7, no. 2, e32366.
Weber, C., Liver: Neutrophil extracellular traps mediate bacterial liver damage, Nat. Rev. Gastroenterol. Hepatol., 2015, vol. 12, no. 5, 251.
Xu, J., Zhang, X., Pelayo, R., Monestier, M., Ammollo, C.T., Semeraro, F., Taylor, F.B., Esmon, N.L., Lupu, F., and Esmon, C.T., Extracellular histones are major mediators of death in sepsis, Nat. Med., 2009, vol. 15, no. 11, pp. 1318–1321.
Czaikoski, P.G., Mota, J.M., Nascimento, D.C., Sonego, F., Castanheira, F.V., Melo, P.H., Scortegagna, G.T., Silva, R.L., Barroso-Sousa, R., Souto, F.O., Pazin-Filho, A., Figueiredo, F., Alves-Filho, J.C., and Cunha, F.Q., Neutrophil extracellular traps induce organ damage during experimental and clinical sepsis, PLoS One, 2016, vol. 11, no. 2, e0148142.
Pietronigro, E.C., Bianca, V.D., Zenaro, E., and Constantin, G., NETosis in Alzheimer’s disease, Front. Immunol., 2017, vol. 8, 211.
Lachowicz-Scroggins, M.E., Dunican, E.M., Charbit, A.R., et al., Extracellular DNA, neutrophil extracellular traps, and inflammasome activation in severe asthma, Am. J. Respir. Crit. Care Med., 2019, vol. 199, no. 9, pp. 1076–1085.
Zhang, T., Mei, Y., Dong, W., Wang, J., Huang, F., and Wu, J., Evaluation of protein arginine deiminase-4 inhibitor in TNBS-induced colitis in mice, Int. Immunopharmacol., 2020, vol. 84, 106583.
Dinallo, V., Marafini, I., Di Fusco, D., Laudisi, F., Franze, E., Di Grazia, A., Figliuzzi, M.M., Caprioli, F., Stolfi, C., Monteleone, I., and Monteleone, G., Neutrophil extracellular traps sustain inflammatory signals in ulcerative colitis, J. Crohns Colitis, 2019, vol. 13, no. 6, pp. 772–784.
Gupta, A.K., Hasler, P., Holzgreve, W., Gebhardt, S., and Hahn, S., Induction of neutrophil extracellular DNA lattices by placental microparticles and IL-8 and their presence in preeclampsia, Hum. Immunol., 2005, vol. 66, no. 11, pp. 1146–1154.
Vecchio, F., Lo Buono, N., Stabilini, A., et al., Abnormal neutrophil signature in the blood and pancreas of presymptomatic and symptomatic type 1 diabetes, JCI Insight, 2018, vol. 3, no. 18, e122146.
Zhou, J., Yang, Y., Gan, T., Li, Y., Hu, F., Hao, N., Yuan, B., Chen, Y., and Zhang, M., Lung cancer cells release high mobility group box 1 and promote the formation of neutrophil extracellular traps, Oncol. Lett., 2019, vol. 18, no. 1, pp. 181–188.
Short, K.R., von Köckritz-Blickwede, M., Langereis, J.D., Chew, K.Y., Job, E.R., Armitage, C.W., Hatcher, B., Fujihashi, K., Reading, P.C., Hermans, P.W., Wijburg, O.L., and Diavatopoulos, D.A., Antibodies mediate formation of neutrophil extracellular traps in the middle ear and facilitate secondary pneumococcal otitis media, Infect. Immun., 2014, vol. 82, no. 1, pp. 364–370.
Hwang, J.W., Kim, J.H., Kim, H.J., Choi, I.H., Han, H.M., Lee, K.J., Kim, T.H., and Lee, S.H., Neutrophil extracellular traps in nasal secretions of patients with stable and exacerbated chronic rhinosinusitis and their contribution to induce chemokine secretion and strengthen the epithelial barrier, Clin. Exp. Allergy, 2019, vol. 49, no. 10, pp. 1306–1320.
Tibrewal, S., Ivanir, Y., Sarkar, J., Nayeb-Hashemi, N., Bouchard, C.S., Kim, E., and Jain, S., Hyperosmolar stress induces neutrophil extracellular trap formation: implications for dry eye disease, Invest. Ophthalmol. Vis. Sci., 2014, vol. 55, no. 12, pp. 7961–7969.
Shan, Q., Dwyer, M., Rahman, S., and Gadjeva, M., Distinct susceptibilities of corneal Pseudomonas aeruginosa clinical isolates to neutrophil extracellular trap-mediated immunity, Infect. Immun., 2014, vol. 82, no. 10, pp. 4135–4143.
Jin, X., Zhao, Y., Zhang, F., Wan, T., Fan, F., Xie, X., and Lin, Z., Neutrophil extracellular traps involvement in corneal fungal infection, Mol. Vis., 2016, vol. 22, pp. 944–952.
Magán-Fernández, A., Al-Bakri, S.M., O’Valle, F., Benavides-Reyes, C., Abadia-Molina, F., and Mesa, F., Neutrophil extracellular traps in periodontitis, Cells, 2020, vol. 9, no. 6, e1494.
Menegazzo, L., Ciciliot, S., Poncina, N., Mazzucato, M., Persano, M., Bonora, B., Albiero, M., de Kreutzenberg, S.V., Avogaro, A., and Fadini, G.P., NETosis is induced by high glucose and associated with type 2 diabetes, Acta Diabetol., 2015, vol. 52, no. 3, pp. 497–503.
Funding
This work was performed within the project “Molecular and Cellular Bases of Immunity” (state budget, section 0110 (for topics on the state task), no. 21-1-16, CITIS no. AAAA-A16-116021660081-0).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
ADDITIONAL INFORMATION
ORCID: https://orcid.org/0000-0001-5233-9338
COMPLIANCE WITH ETHICAL STANDARDS
The author declares that has no conflict of interests. This article does not contain any studies involving human participants or animals performed by the author.
Additional information
Translated by A. Barkhash
About this article

Cite this article
Vorobjeva, N.V. Neutrophil Extracellular Traps: New Aspects. Moscow Univ. Biol.Sci. Bull. 75, 173–188 (2020). https://doi.org/10.3103/S0096392520040112
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.3103/S0096392520040112
Keywords:
Abbreviations:
- ANCA-associated vasculitis (AAV)—antineutrophil cytoplasmic antibodies-associated vasculitis
- ROS—reactive oxygen species
- dsDNA—double-stranded DNA
- LPS—lipopolysaccharide
- MPO—myeloperoxidase
- NETosis—process of NET formation accompanied by death of neutrophil
- NE—neutrophil elastase
- ALI—acute lung injury
- ARDS—acute respiratory distress syndrome
- RA—rheumatoid arthritis
- SLE—systemic lupus erythematosus
- PMA—phorbol 12-myristate 13-acetate
- CGD—chronic granulomatous disease
- COPD—chronic obstructive pulmonary disease
- PKC—protein kinase C
- citr. histone—citrullinated histone
- mPTP—mitochondrial permeability transition pore
- NADPH oxidase—nicotinamide adenine dinucleotide phosphate oxidase
- NET—neutrophil extracellular traps
- PAD4—peptidyl arginine deaminase 4