
Abstract
The prediction of friction under elastohydrodynamic lubrication (EHL) conditions remains one of the most important and controversial areas of tribology. This is mostly because the pressure and shear rate conditions inside EHL contacts are particularly severe, which complicates experimental design. Over the last decade, molecular dynamics (MD) simulation has played an increasingly significant role in our fundamental understanding of molecular behaviour under EHL conditions. In recent years, MD simulation has shown quantitative agreement with friction and viscosity results obtained experimentally, meaning that they can, either in isolation or through the use of multiscale coupling methods, begin to be used to test and inform macroscale models for EHL problems. This is particularly useful under conditions that are relevant inside machine components, but are difficult to obtain experimentally without uncontrollable shear heating.
Similar content being viewed by others
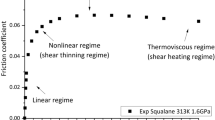

1 Introduction
Many lubricated machine components (such as gears, bearings, constant velocity joints, and cam/follower systems) contain non-conformal surfaces that can roll and slide together. In these components, the solid surfaces elastically deform, leading to elastohydrodynamic lubrication (EHL) conditions [1]. Here, the solid surfaces are generally separated by thin lubricant films (< 1 μm) subjected to very high pressures (> 1 GPa) and shear rates (> 105 s−1). The behaviour of lubricants under such extreme EHL conditions remains shrouded in mystery, which complicates the development of macroscale models capable of accurately predicting the frictional response that ultimately determines energy loss and component efficiency.
Numerical methods, analytical solutions, and experimental techniques have all advanced significantly over the last few decades [2]. However, the measurement and prediction of EHL friction remains a significant challenge and continues to attract much controversy [3,4,5]. In addition to the aforementioned methods, molecular dynamics (MD) simulations have emerged as tools capable of giving unique insights into the nanoscale behaviour of lubricants, including under EHL conditions [6]. Much of this work has been pioneered by Mark Robbins alongside his collaborators. In the remainder of this article, we give an overview of the state of the art in the use of MD simulations to help answer outstanding questions in EHL. We pay particular attention to the work of Robbins and co-workers, showing how his work has considerably advanced the field.
2 Shear Thinning
One of the key outstanding questions in EHL is the most suitable equation to describe the non-Newtonian response of lubricants at high shear rates. The two phenomenological models at the centre of this active debate are those due to Eyring [7] and Carreau [8]. The Eyring model [7] assumes that flow occurs by stress-augmented thermal activation (SATA) over a single characteristic energy barrier height. It can be used to describe the change in shear stress, \(\tau\), with shear rate, \(\dot{\gamma }\):
where \({\tau }_{e}\) is the Eyring stress, \({\eta }_{P}\) is the Newtonian shear viscosity at pressure \(P\). The Eyring model predicts that, at high \(\dot{\gamma }\) (when \(\tau\) exceeds \({\tau }_{e}\) by a factor of at least two), \(\tau\) increases linearly with \(\mathrm{log}(\dot{\gamma })\). The model has been used to describe a wide range of lubricant rheology data, mostly from tribometer experiments under EHL conditions rather than viscometers [3]. The related Ree–Eyring [9] model, which allows for multiple flow units (via different barrier heights), has also been applied to lubricant shear thinning under EHL conditions [3].
The Carreau model [8] can be used to describe the variation in shear stress, \(\tau\), with shear rate, \(\dot{\gamma }\):
where \(\lambda\) is a time constant and \(n <1\). The Carreau model predicts that, at high \(\dot{\gamma }\), \(\tau\) increases as a power law with \(\dot{\gamma }\). This equation was derived based on the network theory of Lodge [10], but it is now recognised that power-law behaviour can arise from several different physical pictures of flow, including SATA over a broad range of energy barrier heights. One immediate benefit of the Eyring equation over the Carreau equation is its simplicity, i.e. the fact that it contains fewer tuneable parameters [3]. The Carreau–Yasuda [11] equation contains modifications to improve fitting in the transition region between Newtonian and strong shear thinning regions [3].
The Carreau equation contains many other models as limiting cases [12], including the Eyring model [13]. While the Eyring and Carreau models predict similar shear stress (or viscosity) at low shear rates, they diverge at high shear rates [3]. In fact, the shear stresses from the models can differ by several order of magnitude at shear rates (> 107 s−1) encountered in lubricated components inside modern engines [14].
Testing models for shear thinning at such high shear rates and pressures represents a considerable experimental challenge. High-pressure viscometers can reach shear rates of only ~ 104 s−1 at EHL pressures (> 1 GPa) for low-viscosity fluids before shear heating becomes uncontrollable [15]. At similar pressures, tribometer experiments under thin-film EHL conditions can reach much higher shear rates (106 s−1) before shear heating becomes excessive [16], but it has been debated how useful the rheological data from such experiments are to predict EHL friction [3,4,5]. On the other hand, non-equilibrium molecular dynamics (NEMD) simulations are ideally suited to study the rheology of fluids at high shear rates, since thermostats can be used to suppress shear heating. In fact, NEMD simulations suffer from the opposite problem, in that to obtain a reasonable signal-to-noise ratio and a non-equilibrium steady state, high shear rates (usually > 107 s−1) must be used [6]. Consequently, through a combination of experiment and NEMD simulation, our understanding of shear thinning behaviour under EHL conditions has been substantially improved in recent years.
In 2002, Bair et al. [17, 18] conducted the first comparison between the non-linear rheology predicted by NEMD simulations with that obtained experimentally. They used the inexpensive united-atom force field developed by Siepmann et al. [19] to study the viscosity of squalane (C30H62), a model lubricant base oil. The SLLOD [20] equations of motion were applied to a bulk system of squalane molecules and NEMD simulations were conducted at shear rates between 107 and 1011 s−1 and pressures of up to 0.3 GPa. As in most bulk NEMD simulations, a global Nosé–Hoover thermostat was used to prevent shear heating [21]. The experiments were conducted at shear rates between 102 and 104 s−1 and pressures of up to 1.0 GPa. Although the experimental and NEMD data were separated by several orders of magnitude in shear rate, they collapsed onto the same time–temperature superposition (TTS) master curve. This master curve was successfully fit using the Carreau model [8].
Liu et al. [22] also successfully used the Carreau model [8] to describe the shear thinning behaviour of squalane, as well as several types of poly-α-olefin (PAO) molecules, from bulk NEMD simulations at high shear rate (108–1010 s−1). They correlated the onset of shear thinning with changes to the conformation of the molecules, quantified through the radius of gyration. Santak and Conduit [23] used the Carreau model [8] to estimate Newtonian viscosities from NEMD simulations preformed at high shear rate (109–1012 s−1) for 74 alkanes containing 5–10 carbon atoms with weighted least squares regression.
In 2017, Jadhao and Robbins [24] used bulk NEMD simulations to determine the non-equilibrium shear response of squalane over a much wider range of pressures (0.1–1.0 GPa) and shear rates (105–1010 s−1). They used the united-atom force field due to Mondello and Grest [25], which is an extension of the force field developed by Siepmann et al. [19]. This force field was previously shown to reproduce the experimental Newtonian viscosity of squalane within 15% [26]. The Newtonian viscosities obtained from the NEMD simulations were in good agreement with those from previous experiments performed at low shear rates (102–104 s−1) [15, 17].
At low pressures (and high temperatures), Jadhao and Robbins [24] found that shear thinning could be described using a power law, as in the Carreau model [8]. At higher pressures (and low temperatures), where the viscosity increased above ∼1 Pa.s, squalane began to exhibit glassy flow, and shear thinning was increasingly dominated by SATA processes. In this high-viscosity regime, the shear thinning behaviour was more accurately described using the Eyring model [7], as shown in Fig. 1. These observations have important implications in terms of predicting EHL friction since they imply that the Eyring model [7] is more suitable than the Carreau model [8] to describe the non-linear rheology in high-pressure EHL contacts.

Reproduced with permission from Ref. [24]. Copyright American National Academy of Sciences, 2017
This study by Jadhao and Robbins [24] was a significant breakthrough and was highlighted as a key study in our 2018 reviews on both SATA [27] and NEMD simulations [6] in tribology. Moreover, the authors made their large atomic/molecular massively parallel simulator (LAMMPS) [28] input files publically available in the Supporting Information, ensuring that their results were transparent, reproducible, usable by others, and extensible (TRUE) [29].
In 2019, Jadhao and Robbins [30] undertook a more detailed investigation of the transition between Eyring [7] and Carreau [8] shear thinning behaviour under EHL conditions using NEMD simulations. They showed that, in the high-viscosity regime (> 1 Pa.s), squalane viscosity data could be collapsed onto the Eyring model [7] over almost 30 orders of magnitude in shear rate using TTS. Conversely, the Carreau model [8] (with a power-law exponent of 0.5 as proposed by Bair et al. [17]) severely overestimated the viscosity from the NEMD simulations at high shear rate. The comparison is shown in Fig. 2a. For high-viscosity conditions, the response is dominated by SATA rearrangements over the lowest energy barrier, consistent with Eyring theory. For low-viscosity conditions, many different energy barriers contribute to molecular motion, leading to Carreau-like behaviour. It is noteworthy that modifications of the Eyring equation, such as the Ree–Eyring equation [9], can be used to account for multiple barrier heights. However, this increases the number of tuneable parameters and reduces the predictive power of the model [3, 30].

Reproduced with permission from Ref. [30]. Copyright Springer Nature, 2019
a Comparison of Eyring [7] and Carreau [8] models to viscosity data for squalane collapsed using TTS for values of P and T where η is high (> 1 Pa.s). Experimental and NEMD simulation data are, respectively, indicated by filled and open symbols. b Order parameters Sxx and Syy for the systems shown in a scale by their maximum magnitudes at high rates. Results for the order at all state points collapse when plotted against the same reduced rate that collapses viscosities. Note that alignment saturates at a reduced strain rate of about 10, where the viscosity has dropped by less than an order of magnitude.
Jadhao and Robbins [30] also studied the prevalence of different mechanisms that have been proposed to control shear thinning behaviour. One commonly invoked mechanism for power-law (e.g. Carreau) shear thinning is a change in some order parameter, more specifically alignment of chain-like molecules along the flow direction [4, 31]. This could perhaps lower the viscosity by decreasing the rate of collisions between molecules, as well as the associated momentum transfer and dissipation. In contrast, shear thinning in the Eyring model [7] only involves a change in the rate of molecular rearrangements through activated motion over fixed barriers. Jadhao and Robbins [30] showed that the alignment of squalane molecules in the NEMD simulations (Fig. 2b) saturates after the viscosity (Fig. 2a) has dropped by only about a factor of three due to shear thinning. On the other hand, the Eyring model [7] describes a reduction of six or more decades in viscosity resulting from shear thinning.
Using a binary atomic Lennard–Jones fluid [32], Jadhao and Robbins [30] also showed that the transition from Carreau to Eyring is generic, rather than specific to chain-like squalane molecules. Lin and Kedzierski [33] have recently shown that the Carreau to Eyring transition also occurs in a typical polyol ester lubricant, pentaerythritol tetrahexanoate (PEC6), using bulk NEMD simulations. They used the long-chain optimised potential for liquid simulations (L-OPLS) all-atom force field [34]. Prentice et al. [35] successfully employed the Eyring model to estimate the Newtonian viscosities from NEMD simulations of mixtures of hydrogenated 1-decene trimer and 1-decene tetramer as a model for PAO.
It is worth noting that some earlier work of Robbins and collaborators on glassy systems provided an initial guide to the existence of a crossover from power-law (Carreau) to log-rate (Eyring) behaviour driven by increasing Newtonian viscosity. For example, Rottler and Robbins [36] observed that, for bi-disperse Lennard–Jones fluids, a transition from Newtonian behaviour to a logarithmic shear rate dependence of shear stress (consistent with Eyring theory [7]) occurred as the temperature was lowered. Furthermore, Baljon and Robbins [37] studied the Kremer–Grest [38] model of short-chain molecules with 16 beads and observed a change from power-law to Eyring-like behaviour with decreasing temperature. Thompson et al. [39] also found similar changes in the behaviour of confined chains as the Newtonian viscosity was increased, due to decreasing film thickness.
Most of the bulk NEMD simulations performed by Jadhao and Robbins [24, 30] were performed at constant volume. This differs from viscometers and tribometer experiments that are performed at constant pressure. Jadhao and Robbins [24, 30] estimated the viscosity at each target pressure by interpolating between the constant volume results. Results from previous NEMD simulations have shown non-negligible differences between viscosities obtained in the constant-volume and constant-pressure ensembles [40]. Jadhao and Robbins [30] performed a subset of NEMD simulations at constant pressure (in the normal and shear directions) and found that this resulted in similar shear stress (or viscosity) up to shear rates of approximately 106 s−1, but somewhat lower shear stress at high shear rates.
Some potentially significant assumptions have been made in the bulk NEMD simulations that have been performed to date of lubricants under EHL conditions. The first assumption is that linear flow profiles develop between the sliding surfaces, which may not be the case in experiments (as will be discussed in detail in a subsequent section). This could be avoided by using a configurational thermostat [41, 42]; however, these are not yet implemented in open-source MD programs such as LAMMPS [28]. Another potential issue is that the NEMD equations of motion implemented in LAMMPS [28] are the so-called atomic SLLOD [20], rather than the molecular SLLOD [43]. The use of atomic SLLOD [20] for molecular systems results in enhanced orientational ordering and non-zero values for the antisymmetric part of the molecular stress tensor, indicating that a torque is applied to the molecules [44]. For a more detailed discussion of the potential importance of molecular SLLOD [44] in bulk NEMD simulations, see Ref. [45]. Finally, Bair has also recently questioned the ability of NEMD thermostats to maintain isothermal conditions at the high shear rates [46] employed by Jadhao and Robbins [24, 30]. The energy released in these NEMD simulations is certainly considerable; however, the thermostatting methods applied have been shown to maintain isothermal conditions up to even higher strain rates (1012 s−1) [47] than those considered by Jadhao and Robbins [24, 30].
In addition to bulk NEMD simulations, confined NEMD simulations have also shown good agreement with shear stress extrapolations from tribometer experiments conducted at lower shear rates for several molecular fluids, such as model lubricants squalane [48, 49] and diethylhexyl sebacate [48], traction fluids DM2H and Santotrac 2000 [48], as well as benzyl benzoate [49]. These studies were all performed at constant pressure (consistent with experiments) and used the L-OPLS force field [34]. Although, they showed good agreement with experimental trends, these studies did not directly compare shear thinning models.
3 High-Pressure Newtonian Viscosity
Accurate measurements of high-pressure viscosity are crucial for macroscale predictions of EHL film thickness and friction [3,4,5]. The viscosity in the inlet, where pressure is approximately 100 MPa, controls EHL film thickness, while viscosity in the central region of the contact, where pressure often exceeds 1 GPa, governs EHL friction. Through recent advances in computational performance, highly parallelised simulation software, and force field accuracy, MD simulations are now ideally suited to predicting the high-pressure viscosity of lubricant-like molecules [6].
In 2001, McCabe et al. [50] used NEMD simulations to study the high-pressure viscosity of 9-octylheptadecane. They used the united-atom force field developed by Mondello and Grest [25] to study the viscosity up to 1 GPa at high temperature (373 K). The Newtonian viscosity values from the simulations were significantly under-predicted (30–70%) compared to experiment; however, the pressure–viscosity coefficient, α, was within 10% of the experimental value. In calculating α, the authors assumed an exponential increase in viscosity over the entire pressure range studied (0.1–1.0 GPa), as predicted by the Barus equation [51].
More recently, Liu et al. [52] used the same force field to investigate the pressure dependency of viscosity of hydrogenated 1-decene trimer, as a model for PAO4. They found that, as previously observed experimentally, the pressure increase was slower than exponential up to pressures of 1 GPa, which is consistent with the McEwen model [53]. Liu et al. [52] observed a similar viscosity under-prediction as McCabe et al. [50] compared to experiment, and also noted that the pressure–viscosity coefficients they obtained under ambient (α0) and high-pressure (α*) conditions were in close agreement with experiment [15].
The 10th Industrial Fluid Properties Simulation Challenge (IFPSC) [54] involved determining the Newtonian viscosity of 2,2,4-trimethylhexane at 293 K and pressures between 0.1 and 1000 MPa using molecular simulations. New experimental data determined by Bair using a high-pressure viscometer [55] were kept secret from participants until they had all submitted their entries. First place in this competition was awarded to Galvani Cunha and Robbins [56], who used a similar approach to Jadhao and Robbins [24, 30]. At low pressure, the Newtonian viscosity could be obtained directly from the NEMD simulations [24, 30, 56], while at higher pressure this was estimated using extrapolations to lower shear rates with the Eyring equation [7]. Instead of the united-atom Modello and Grest force field [25], Galvani Cunha and Robbins [56] used the all-atom adaptive intermolecular reactive bond-order potential-Morse (AIREBO-M) [57] force field, developed by O’Connor and Robbins. Although they increase the computational cost by approximately an order of magnitude, all-atom force fields have previously been shown to be much more accurate than united-atom ones for the viscosity prediction of long-chain hydrocarbons [58]. Unlike the original AIREBO force field [59], AIREBO-M [57] uses a Morse potential, rather than the standard 12–6 Lennard–Jones potential for the non-bonded interactions. The exponent for the repulsive component in the 12–6 Lennard–Jones potential has never been confirmed from first principles or determined experimentally, and it is used mostly out of convention and convenience [60]. O’Connor and Robbins suggested that the unphysically divergent power-law repulsion causes AIREBO with 12-6 Lennard–Jones to fail when applied to systems at high pressure [57].
Galvani Cunha and Robbins [56] obtained 2,2,4-trimethylhexane viscosity values that deviated by less than 40% from the experimental values over the entire pressure range [55], as shown in Fig. 3a. The agreement was excellent in the 100–700 MPa range, but deviations were larger at lower and higher pressure. The viscosity increased at a slower than exponential rate at low pressures, reached an inflection point, and then increased at a faster-than-exponential rate. The authors fitted their pressure–viscosity data using a hybrid model of the McEwen model [53] and Paluch model [64], as proposed by Bair [62]. The McEwen model [53] is well suited to represent the slower than exponential increase in viscosity with pressure that is frequently observed at low pressure. It is equivalent to the Tait model [63] for ambient reference pressures. On the other hand, the Paluch model [64] can be used to fit the faster-than-exponential increase in viscosity with pressure that is commonly observed at high pressure. The Paluch model [64] assumes the existence of a finite pressure at which relaxation times diverge at a given temperature, which is analogous to the temperature where the viscosity diverges in the popular Vogel–Fulcher–Tammann (VFT) equation [65]. The hybrid model can be used to fit both types of pressure–viscosity response, including the inflection point that is often observed at intermediate pressure [62].

Pressure–viscosity relation for 2,2,4-trimethylhexane from experiment [55] and MD simulations by Galvani Cunha and Robbins [56] (a) and Kondratyuk and Pisarev [61] (b). Black dotted line in (a) is a fit to the MD data with the hybrid model [62] and red dotted line in (b) is a fit to the MD data with a Tait-like model [63].
Kondratyuk and Pisarev [61] were awarded second place in the 10th IFPSC. They employed the all-atom condensed-phase optimised molecular potentials for atomistic simulation studies (COMPASS) force field [66]. Instead of using NEMD simulations, they applied the Green–Kubo method [67, 68] in equilibrium molecular dynamics (EMD) simulations. Although EMD is a more direct route to the Newtonian viscosity, it is typically limited to low-viscosity systems (< 50 mPa.s) [69]. Systems with larger viscosities require longer integration times, which can lead to error accumulation. Kondratyuk and Pisarev [61] employed the time decomposition method (TDM) [70], which uses the time-averaged running integral from multiple independent, short MD trajectories to calculate the viscosity. Using the TDM, the authors were able to accurately predict the viscosity of 2,2,4-trimethylhexane up to 600 MPa. At higher pressure, the viscosity was significantly under-predicted compared to experiment [55], by up to around 70% at 1 GPa. Since they did not observe an inflection point or faster-than-exponential viscosity increase with pressure in the simulations, the authors fitted the data with a Tait-like model [63], which is equivalent to the McEwen model [53].
Messerly et al. [71] were awarded third place in the 10th IFPSC, using EMD simulations and Green–Kubo with the TDM [70]. They used a version of the united-atom transferable potentials for phase equilibria (TraPPE) force field [72] for branched alkanes with a 16-6 Mie potential [73]. Heyes [74] showed that varying the steepness of purely repulsive potentials has a dramatic effect on transport properties, while Gordon [75] showed that an n-6 Mie potential (where n = 14–20) could accurately describe the high-pressure viscosity of n-alkanes. The 16-6 Mie TraPPE force field [73] gave near-quantitative agreement with experimental viscosity data [55] (to within the combined uncertainties) from atmospheric pressure to 600 MPa. The force field also reproduced the inflection point and faster-than-exponential viscosity increase at high pressure that was observed experimentally [71]. However, the viscosity at high pressure was dramatically over-predicted (> 100%) compared to experiment at higher pressure. This suggests that the overly repulsive Mie 16-6 potential limits the pressure range over which the force field is reliable [71].
It is interesting to note that none of the entries used force fields with the standard 12-6 form of the Lennard–Jones potential for the non-bonded interactions. This is despite the recent observation that experimental data for a large number of molecular systems at high pressure may be described by a common repulsive potential \(U(r)\propto {r}^{-12}\) [60]. Thus, one area certainly worthy of further study is the identification of the most suitable potential to represent the non-bonded interactions for MD simulations of lubricant-sized hydrocarbons (C10–C40 [76]) at high pressures. This could perhaps be facilitated by re-parameterizing MD force fields using first principles calculations with accurate representations of van der Waals interactions [77].
Another interesting observation from the 10th IFPSC is that the majority of the successful entries used an all-atom force field. While accurate viscosities can be obtained for some hydrocarbons (e.g. squalane that contains multiple short methyl branches [78]) the viscosity of most hydrocarbons are severely under-predicted using united-atom force fields [58]. Although Messerly et al. [71] demonstrated that this can be overcome to some extent by using a more repulsive potential, this leads to large over-predictions of viscosity at high pressure. Thus, allowance for the atomic-scale roughness imposed by hydrogen atoms for systems of interacting molecules in MD simulations seems critical to accurately predict viscosity under high pressures. In most cases, the additional accuracy of all-atom force fields justifies the significantly associated computational cost.
Several subsequent studies have used MD simulations to predict the high-pressure viscosity of other systems. For example, Lu et al. [79] investigated the viscosity of traction fluid molecules containing linked cyclohexane rings using NEMD simulations and the united-atom TraPPE force field [80]. They found that the pressure–viscosity coefficient from NEMD was underestimated for these molecules by between 20 and 50% compared to experiment [79]. Using NEMD and the all-atom L-OPLS force field [34], Lin and Kedzierski [33] found that the McEwen model [53] accurately described the experimental viscosity of PEC6 up to 1 GPa.
Commercial lubricants are complicated mixtures rather than single well-defined molecules. Kondratyuk et al. [81] recently used the all-atom COMPASS [66] force field and Green–Kubo with the TDM [70] to study the high-pressure viscosity of mixtures. They studied the viscosity and density of methylcyclohexane, 1-methylnaphthalene, and their binary mixtures at 323 K and pressures of up to 500 MPa. The simulation results were in quantitative agreement with the previous experiments available up to 100 MPa for both pure compounds (200 MPa for 1-methylnaphthalene) and their mixtures. For 1-methylnaphthalene, the viscosity initially increased at a slower than exponential rate with pressure before it reached an inflection point and then increased faster than exponential. The inflection point (300 MPa) occurred at a pressure slightly below the one at which 1-methylnaphthalene is expected to enter the supercooled phase (400 MPa). For methylcyclohexane, the increase in viscosity with pressure was slower than exponential over the entire pressure range studied. The methylcyclohexane data were fitted using the McEwen model [53], while the 1-methylnaphthalene data were fitted using Bair’s hybrid model [62]. The binary mixtures showed intermediate pressure–viscosity responses between the two pure cases. The mixtures became increasingly non-ideal and the pressure was increased. The mixture viscosities were successfully predicted using the Grunberg–Nissan mixing rules [82] over the entire pressure range.
Despite significant recent progress, MD simulations of the high-pressure viscosity of lubricant-sized molecules (C10–C40 [76]) are still a computationally demanding and time-consuming task. An interesting recent development by Kajita et al. [83] has been to dramatically accelerate (by three orders of magnitude) the evaluation of viscosity using a femtosecond stress-tensor correlation. Using this method, they screened the viscosity–temperature behaviour of 55,000 lubricant-sized molecules. This enabled a Monte-Carlo tree search algorithm to be used to design molecules with a high-viscosity index. Kajita et al. [83] justified the use of femtosecond stress-tensor correlations through the shoving model developed by Dyre et al. [84]. Usually, nanosecond stress-tensor correlations are required in the Green–Kubo method to obtain accurate viscosities [69]. The shoving model was developed for glasses [84], and it is not yet clear whether the model is suitable to describe the viscosity of lubricant-sized molecules in the liquid phase. Some aspects of the model have been experimentally validated for some lubricant-like molecules, such as 5-phenyl-4-ether (5P4E) [85]. While this synthetic polyether has been used as a model lubricant [86], it has a much lower glass transition temperature than conventional lubricants [87]. Therefore, 5P4E is a more natural application of the shoving model than lubricant-sized molecules [84].
4 Flow and Phase Behaviour
A major assumption of most macroscale EHL models is that a linear velocity profile develops in the fluid between the sliding surfaces. The same assumption of planar Couette flow is made in bulk NEMD simulations that use the SLLOD [20] equations of motion alongside standard thermostats (e.g. Nosé-Hoover [21]). However, recent experiments have highlighted that the through-thickness velocity profiles in some systems can show significant deviations from linearity under EHL conditions. Some experiments have shown that the central region can become solid-like and flow more slowly than the liquid-like region close to the surfaces, which is often referred to as plug slip or plug flow [88,89,90]. Experiments for different systems and conditions have shown that the fluid close to the surfaces can become solid-like and flow more slowly than the liquid-like central region, which is known as central localisation or shear banding [91,92,93]. The different types of through-thickness flow profiles possible in experiments under EHL conditions are shown in Fig. 4.

Confined NEMD simulations in which the wall atoms (rather than the sheared fluid molecules) are thermostatted are more representative of tribometer experiments [94]. In these simulations, no assumptions are made regarding the flow profile that develops in the fluid between the sliding surfaces [95]. This enables deviations from Couette flow behaviour such as boundary slip [96] and shear banding [97] to be accurately captured. Moreover, temperature gradients are allowed to form within the fluid film, as in experiments [94]. The amount of shear heating can be controlled through the coupling time of the thermostat to the wall atoms [98].
In 1990, Thompson and Robbins [99] performed the first systematic study of the structure and flow of fluids near solid boundaries. They performed NEMD simulations of atomic Lennard–Jones fluids confined and sheared between solid surfaces. They observed a wide range in flow conditions, including slip, no-slip, and locking. They showed that the degree of slip was directly related to the amount of structure induced in the fluid by the solid walls. For weak wall–fluid interactions, there was little ordering and slip was observed. For large interactions, substantial epitaxial ordering was induced and the first few fluid layers became locked to the wall, leading to slip within the fluid (i.e. shear banding). Subsequent confined NEMD simulations of Lennard–Jones fluids by Butler and Harrowell [100], binary Lennard–Jones fluids by Varnik et al. [101], and short polymers by Thompson et al. [102] and Rottler and Robbins [36] have also shown various forms of shear banding. Washizu and Ohmori [103] performed the first confined NEMD simulations using a molecular fluid (n-hexane) with film thicknesses (10 nm) and shear rates (106 s−1) that were representative of EHL tribometer experiments.
Since 2012, Dini and co-workers have studied the friction and flow behaviour of Lennard–Jones and binary Lennard–Jones fluids under EHL conditions [104,105,106]. They observed that, above a certain pressure, the flow profiles transitioned from Couette-like to shear banding [104]. Non-equilibrium phase diagrams were constructed for Lennard–Jones fluids at different pressures and shear rates [105]. At low shear rate and high pressure, plug slip occurred where shear was localised close to the walls, while at higher shear rates, central localisation occurred where shear mainly occurred in the middle of the fluid film [104,105,106]. At very high shear rates, the central liquid region widened until eventually a Couette profile was recovered [104,105,106]. Unusual friction behaviour was noted in the central localisation and particularly plug slip regions [106]. Martinie and Vergne [107] verified the feasibility of mapping the lubricant phase diagram derived from the NEMD simulations to the results obtained from previous experiments. From this mapping, they suggested that the physical mechanism that initiates ‘limiting shear stress’ (LSS) behaviour in EHL contacts is shear banding [107]. The LSS describes the shear stress at which the friction coefficient becomes insensitive to pressure and shear rate. The LSS concept is commonly invoked when explaining and predicting EHL friction curves.
In recent years, a number of studies have investigated the potential links between friction and flow behaviour under EHL conditions for molecular fluids. Washizu et al. [108] observed shear banding in NEMD simulations on n-hexane confined between gold slabs at high pressure (0.1–8.0 GPa). They also observed LSS behaviour at the transition from Couette flow to CL. Ewen et al. [48] performed tribometer experiments and NEMD simulations of a range of lubricant (squalane, diethylhexyl sebacate) and traction fluid (Santotrac 2000, DM2H) molecules at pressures (0.5–2.0 GPa) and shear rates (104–1010 s−1) representative of EHL conditions. On increasing pressure, they observed a transition from Couette flow to CL for the lubricant molecules and from CL to PS for the traction fluids. While the lubricants showed a decreased dependence of the friction coefficient on shear rate at high pressure, they did not reach a LSS. Conversely, the traction fluids displayed a LSS at the same pressure as the CL-PS transition occurred. Porras-Vazquez et al. [49] used tribometer experiments and NEMD simulations to study the relationship between the LSS and flow behaviour for squalane and benzyl benzoate. They found similar results to Ewen et al. [48] on atomically smooth surfaces, but also found similar friction coefficients on rough surfaces, where the flow behaviour remained Couette-like even at high pressure. Based on these observations, they suggested that the LSS was caused by a pressure-induced liquid-glass transition in the lubricant [49]. This suggestion is consistent with the previous NEMD observations, since shear banding is well known to occur in glassy materials [101] and the LSS has been linked to the yield stress of glassy lubricants [109]. For n-alkanes, which crystallise more readily than branched lubricant molecules, LSS behaviour and non-linear flow were observed in NEMD simulations of solid-like C60, but not liquid-like C16 [110].
The glass transition in liquid lubricants has also been studied with MD simulations. Thompson et al. [39] showed that the dynamics of simple chain-like molecules became glassy when confined below around six molecular diameters. Recent MD simulations by Gao and Müser [111] of hydrogenated 1-decene trimer molecules between iron walls suggest that while confinement leads to drastically increased layering, load-bearing capacity, and effective viscosity, this does not always indicate crystallisation or a glass transition. However, under EHL conditions, lubricants are subjected not only to strong confinement (< 1 μm), but also very high pressures (> 1 GPa). As suggested by Robbins and Smith [112], there could be connections between lubricant behaviour under confinement and at high pressure.
The effect of the surface properties can also be important in determining flow under EHL conditions [113]. Ewen et al. [98] performed NEMD simulations to study the friction and flow of Santotrac 2000 and hydrogenated 1-decene trimer between surfaces with different thermal conductivities. Reducing the thermal conductivity of one of the surfaces led to large temperature rises close to that surface, resulting in asymmetric flow profiles and significant reductions in friction [98]. The traction fluid (Santotrac 2000) gave significantly higher friction and temperature rises compared to the lubricant (hydrogenated 1-decene trimer). Savio et al. [114] performed NEMD simulations of short n-alkanes (C3–C10) and showed that both regular and random slip patterns on solid surfaces can cause cavitation, particularly at small contact separations.
Although it is commonly observed in highly confined NEMD simulations with atomically smooth substrates [96, 115], slip directly at the solid–liquid interface (‘boundary slip’ in Fig. 4) is unlikely to be significant for lubricants on the rough, oleophilic steel surfaces usually found inside tribological contacts [116]. Gao et al. [117] showed that, for n-hexadecane molecules confined between gold surfaces, slip at the solid–liquid interface was eliminated in the presence of random nanoscale roughness. Furthermore, Washizu et al. [118] showed that, for n-hexane molecules confined between atomically smooth gold surfaces, the slip length decreased with increasing surface separation and was negligible for realistic EHL film thicknesses (400 nm).
Experimental studies have shown that boundary slip can occur inside EHL contacts when an oleophobic coating is applied to one of the surfaces [119]. Organic friction modifier (OFM) additives, such as fatty acids, are known to form oleophobic coatings on metal surfaces [120]. OFMs are generally believed to reduce friction in the boundary regime by forming strong monolayers that can prevent solid–solid asperity contact [121]. However, Choo and Spikes [122] showed experimentally that OFMs also reduce friction in the low-load hydrodynamic lubrication regime. They postulated that this was due to the OFMs forming oleophobic monolayers that promoted boundary slip [122]. Ewen et al. [123] confirmed this hypothesis with NEMD simulations of n-hexadecane lubricant confined between OFM monolayers adsorbed on iron oxide surfaces. The NEMD simulation results also suggested that larger reductions in friction might be possible in the high-pressure EHL regime, due to larger slip lengths [123]. This hypothesis was recently confirmed experimentally by Kalin and Kus [124], who showed that OFMs cause large reductions in friction compared to pure base oils under EHL conditions.
5 Hybrid Simulations
Our understanding of many problems in tribology, including EHL contacts, would benefit from multiscale modelling techniques [125]. The main limitation of MD simulations is the short time and length scales that can be accessed [6]. While coarse graining could help in this respect [126], parameterisation of coarse-grained models that can accurately describe lubricant rheology has proved a considerable challenge [127]. Macroscale techniques, such as continuum fluid dynamics (CFD), have been successfully applied to a range of EHL problems [128,129,130,131]. However, such techniques require several assumptions with respect to the rheology of the lubricant and flow boundary conditions. One approach to improve the accuracy of continuum models is to perform hierarchical coupling to smaller scales [6]. For example, the viscosity [132, 133] and slip length [134, 135] have been calculated in NEMD simulations and then used as inputs in continuum models.
A more significant challenge, which could transform the modelling of EHL contacts, would be to perform concurrent molecular-continuum coupling. The domain decomposition approach proposed by O’Connell and Thompson [136] defines atomic and continuum domains that are coupled through an overlap region in which the solutions in both domains are consistent. This limits the use of MD to regions where the atomistic scales need to be resolved, while employing a continuum-based solver for the remainder of the domain. This approach has been widely adopted to study a range of multiscale flow problems over the last few decades [137].
Robbins and co-workers applied the domain decomposition approach to perform hybrid molecular-continuum simulations of a range of factors important to EHL contacts including nanoscale roughness [138], cavity flow [139], and solid–liquid heat transfer [140]. Aspects linked to asperity interactions and elastic–plastic surface deformation have also been successfully considered [141,142,143], showing that hybrid coupling could be potentially used not only to capture the fluid behaviour and complex solid/fluid interactions, but also the evolution of solid surfaces.
One major limitation of concurrent coupling techniques is that the simulation time step is limited to the shortest timescale that needs to be modelled at the smallest scale [6]. Despite this, considerable performance enhancements can be achieved compared to modelling the entire system at the atomic level [137]. Moreover, Liu et al. [144] have developed a multi-timescale algorithm, which allows the molecular dynamics during one macro time step to be treated as in quasi-steady state, further decreasing the required computer time.
The majority of published modelling simulation research now utilises highly parallelised, open-source codes such as LAMMPS [28] and OpenFOAM [145]. In recent years, significant efforts have been made to enable hybrid molecular-continuum simulations using these software packages [146, 147] while building on the coupling algorithms and methods [137, 148] developed by Robbins and his collaborators [138,139,140,141,142]. The combined gains in computational efficiency from highly parallelised software and hybrid simulations could enable the study of EHL contacts with atomistic detail close to the surfaces in the near future. These multiscale methods could uncover many aspects of EHL behaviour that remain shrouded in mystery.
6 Summary
MD simulations are becoming integral to our fundamental understanding of lubricant behaviour inside EHL contacts. Both bulk and confined NEMD simulations have become promising tools to study the non-Newtonian rheology of lubricants under EHL conditions. Robbins and co-workers have shown that at relatively low pressure, many energy barriers contribute to molecular motion, leading to a power-law increase in shear stress with shear rate, as predicted with the Carreau equation. At high pressure, where the viscosity is much greater, the response is increasingly dominated by SATA rearrangements over the lowest energy barrier, leading to a logarithmic increase in shear stress with shear rate, consistent with Eyring theory.
MD simulations can now show quantitative agreement with experiments used to measure the high-pressure viscosity of lubricant-like molecules. Both EMD and NEMD simulations have been successfully applied to predict viscosities up to pressures of around 1 GPa. The most accurate predictions have used all-atom force fields that do not employ the standard 12-6 form of the Lennard–Jones potential for the non-bonded interactions. Since commercial lubricants are complicated multicomponent fluids, it will be interesting to investigate the predictive capability of MD simulations for the high-pressure viscosity of complex liquid mixtures.
The flow of thin, confined lubricant films under EHL conditions can deviate from the linear profile generally assumed in continuum models. Recent insights from NEMD simulations suggest that pressure-induced glass formation causes a transition in both the EHL friction and flow behaviour. Hybrid molecular-continuum modelling could give further insights into EHL problems by making larger domain sizes and timescales accessible than is possible through conventional MD simulations. We expect MD simulations and related methods, most of which build on Robbins’ pioneering ideas, to play an increasingly central role in both improving our understanding of lubricant behaviour under EHL conditions and improving models to predict macroscale behaviour.
References
Dowson, D., Higginson, G.R.: Elasto-Hydrodynamic Lubrication. Pergamon Press, Oxford (1966)
Lugt, P.M., Morales-Espejel, G.E.: A review of elasto-hydrodynamic lubrication theory. Tribol. Trans. 54, 470–496 (2011). https://doi.org/10.1080/10402004.2010.551804
Spikes, H., Jie, Z.: History, origins and prediction of elastohydrodynamic friction. Tribol. Lett. 56, 1–25 (2014). https://doi.org/10.1007/s11249-014-0396-y
Bair, S., Vergne, P., Kumar, P., Poll, G., Krupka, I., Hartl, M., Habchi, W., Larsson, R.: Comment on “History, origins and prediction of elastohydrodynamic friction” by Spikes and Jie. Tribol. Lett. 58, 16 (2015). https://doi.org/10.1007/s11249-015-0481-x
Spikes, H., Zhang, J.: Reply to the Comment by Scott Bair, Philippe Vergne, Punit Kumar, Gerhard Poll, Ivan Krupka, Martin Hartl, Wassim Habchi, Roland Larson on “History, origins and prediction of elastohydrodynamic friction” by Spikes and Jie in tribology letters. Tribol. Lett. 58, 17 (2015). https://doi.org/10.1007/s11249-015-0483-8
Ewen, J.P., Heyes, D.M., Dini, D.: Advances in nonequilibrium molecular dynamics simulations of lubricants and additives. Friction. 6, 349–386 (2018). https://doi.org/10.1007/s40544-018-0207-9
Eyring, H.: Viscosity, plasticity, and diffusion as examples of absolute reaction rates. J. Chem. Phys. 4, 283–291 (1936). https://doi.org/10.1063/1.1749836
Carreau, P.J.: Rheological equations from molecular network theories. Trans. Soc. Rheol. 16, 99–127 (1972). https://doi.org/10.1122/1.549276
Ree, T., Eyring, H.: Theory of non-Newtonian flow. I. Solid plastic system. J. Appl. Phys. 26, 793 (1955). https://doi.org/10.1063/1.1722098
Lodge, A.S.: A network theory of flow birefringence and stress in concentrated polymer solutions. Trans. Faraday Soc. 52, 120–130 (1956). https://doi.org/10.1039/TF9565200120
Yasuda, K., Armstrong, R.C., Cohen, R.E.: Shear flow properties of concentrated solutions of linear and star branched polystyrenes. Rheol. Acta. 20, 163–178 (1981). https://doi.org/10.1007/BF01513059
Müser, M.H.: Shear thinning in the Prandtl model and its relation to generalized newtonian fluids. Lubricants 8, 38 (2020). https://doi.org/10.3390/lubricants8040038
Heyes, D.M., Dini, D., Smith, E.R.: Incremental viscosity by non-equilibrium molecular dynamics and the Eyring model. J. Chem. Phys. 148, 194506 (2018). https://doi.org/10.1063/1.5027681
Taylor, R.I., de Kraker, B.R.: Shear rates in engines and implications for lubricant design. Proc. Inst. Mech. Eng. Part J. 231, 1106–1116 (2017). https://doi.org/10.1177/1350650117696181
Bair, S.: The high pressure rheology of some simple model hydrocarbons. Proc. Inst. Mech. Eng. Part J. 216, 139–149 (2002). https://doi.org/10.1243/1350650021543960
Zhang, J., Spikes, H.: Measurement of EHD friction at very high contact pressures. Tribol. Lett. 68, 42 (2020). https://doi.org/10.1007/s11249-020-1281-5
Bair, S., McCabe, C., Cummings, P.T.: Comparison of nonequilibrium molecular dynamics with experimental measurements in the nonlinear shear-thinning regime. Phys. Rev. Lett. 88, 058302 (2002). https://doi.org/10.1103/PhysRevLett.88.058302
Bair, S., McCabe, C., Cummings, P.T.: Calculation of viscous EHL traction for squalane using molecular simulation and rheometry. Tribol. Lett. 13, 251–254 (2002). https://doi.org/10.1023/a:1021011225316
Siepmann, J.I., Karaborni, S., Smit, B.: Simulating the critical behavior of complex fluids. Nature 365, 330–332 (1993). https://doi.org/10.1038/365330a0
Evans, D.J., Morriss, G.P.: Nonlinear-response theory for steady planar Couette flow. Phys. Rev. A. 30, 1528–1530 (1984). https://doi.org/10.1103/PhysRevA.30.1528
Evans, D.J., Holian, B.L.: The Nose-Hoover thermostat. J. Chem. Phys. 83, 4069 (1998). https://doi.org/10.1063/1.449071
Liu, P., Lu, J., Yu, H., Ren, N., Lockwood, F.E., Wang, Q.J.: Lubricant shear thinning behavior correlated with variation of radius of gyration via molecular dynamics simulations. J. Chem. Phys. 147, 084904 (2017). https://doi.org/10.1063/1.4986552
Santak, P., Conduit, G.: Enhancing NEMD with automatic shear rate sampling to model viscosity and correction of systematic errors in modeling density: application to linear and light branched alkanes. J. Chem. Phys. 153, 014102 (2020). https://doi.org/10.1063/5.0004377
Jadhao, V., Robbins, M.O.: Probing large viscosities in glass-formers with nonequilibrium simulations. Proc. Natl. Acad. Sci. USA 114, 7952–7957 (2017). https://doi.org/10.1073/pnas.1705978114
Mondello, M., Grest, G.S.: Molecular-dynamics of linear and branched alkanes. J. Chem. Phys. 103, 7156–7165 (1995). https://doi.org/10.1063/1.470344
Moore, J.D., Cui, S.T., Cochran, H.D., Cummings, P.T.: Rheology of lubricant basestocks: a molecular dynamics study of C30 isomers. J. Chem. Phys. 113, 8833–8840 (2000). https://doi.org/10.1063/1.1318768
Spikes, H.A.: Stress-augmented thermal activation: tribology feels the force. Friction 6, 1–31 (2018). https://doi.org/10.1007/s40544-018-0201-2
Plimpton, S.: Fast parallel algorithms for short-range molecular-dynamics. J. Comput. Phys. 117, 1–19 (1995). https://doi.org/10.1006/jcph.1995.1039
Thompson, M.W., Gilmer, J.B., Matsumoto, R.A., Quach, C.D., Shamaprasad, P., Yang, A.H., Iacovella, C.R., McCabe, M., Cummings, P.T.: Towards molecular simulations that are transparent, reproducible, usable by others, and extensible (TRUE). Mol. Phys. 118, e1742938 (2020). https://doi.org/10.1080/00268976.2020.1742938
Jadhao, V., Robbins, M.O.: Rheological properties of liquids under conditions of elastohydrodynamic lubrication. Tribol. Lett. 67, 66 (2019). https://doi.org/10.1007/s11249-019-1178-3
Cui, S.T., Gupta, S.A., Cummings, P.T., Cochran, H.D.: Molecular dynamics simulations of the rheology of normal decane, hexadecane, and tetracosane. J. Chem. Phys. 105, 1214–1220 (1996). https://doi.org/10.1063/1.471971
Kob, W., Andersen, H.C.: Testing mode-coupling theory for a supercooled binary Lennard-Jones mixture: the van Hove correlation function. Phys. Rev. E 51, 4626 (1995). https://doi.org/10.1103/PhysRevE.51.4626
Lin, L., Kedzierski, M.A.: Density and viscosity of a polyol ester lubricant: measurement and molecular dynamics simulation. Int. J. Refrig. 118, 188–201 (2020). https://doi.org/10.1016/j.ijrefrig.2020.07.004
Siu, S.W.I., Pluhackova, K., Bockmann, R.A.: Optimization of the OPLS-AA force field for long hydrocarbons. J. Chem. Theory Comput. 8, 1459–1470 (2012). https://doi.org/10.1021/ct200908r
Prentice, I.J., Liu, X., Nerushev, O.A., Balakrishnan, S., Pulham, C.R., Camp, P.J.: Experimental and simulation study of the high-pressure behavior of squalane and poly-α-olefins. J. Chem. Phys. 152, 074504 (2020). https://doi.org/10.1063/1.5139723
Rottler, J., Robbins, M.O.: Shear yielding of amorphous glassy solids: effect of temperature and strain rate. Phys. Rev. E. 68, 011507 (2003). https://doi.org/10.1103/PhysRevE.68.011507
Baljon, A.R.C., Robbins, M.O.: Energy dissipation during rupture of adhesive bonds. Science 271, 482–484 (1996). https://doi.org/10.1126/science.271.5248.482
Kremer, K., Grest, G.S.: Dynamics of entangled linear polymer melts: a molecular dynamics simulation. J. Chem. Phys. 92, 5057 (1990). https://doi.org/10.1063/1.458541
Thompson, P.A., Grest, G.S., Robbins, M.O.: Phase transitions and universal dynamics in confined films. Phys. Rev. Lett. 68, 3448–3451 (1992). https://doi.org/10.1103/PhysRevLett.68.3448
Daivis, P.J., Evans, D.J.: Comparison of constant pressure and constant volume nonequilibrium simulations of sheared model decane. J. Chem. Phys. 100, 541–547 (1994). https://doi.org/10.1063/1.466970
Lue, L., Jepps, O.G., Delhommelle, J., Evans, D.J.: Configurational thermostats for molecular systems. Mol. Phys. 100, 2387–2395 (2002). https://doi.org/10.1080/00268970210122145
Braga, C., Travis, K.P.: A configurational temperature Nosé-Hoover thermostat. J. Chem. Phys. 123, 134101 (2005). https://doi.org/10.1063/1.2013227
Edberg, R., Morriss, G.P., Evans, D.J.: Rheology of n-alkanes by nonequilibrium molecular dynamics. J. Chem. Phys. 86, 4555–4570 (1987). https://doi.org/10.1063/1.452731
Travis, K.P., Daivis, P.J., Evans, D.J.: Computer simulation algorithms for molecules undergoing planar Couette flow: a nonequilibrium molecular dynamics study. J. Chem. Phys. 103, 1109–1118 (1995). https://doi.org/10.1063/1.469821
Todd, B.D., Daivis, P.J.: Nonequilibrium Molecular Dynamics: Theory Algorithms and Applications. Cambridge University Press, Cambridge (2017)
Bair, S.: Viscous heating in compressed liquid films. Tribol. Lett. 68, 7 (2020). https://doi.org/10.1007/s11249-019-1252-x
Hoover, W.Q., Ladd, A.J.C., Moran, B.: High-strain-rate plastic flow studied via nonequilibrium molecular dynamics. Phys. Rev. Lett. 48, 1818 (1982). https://doi.org/10.1103/PhysRevLett.48.1818
Ewen, J.P., Gattinoni, C., Zhang, J., Heyes, D.M., Spikes, H.A., Dini, D.: On the effect of confined fluid molecular structure on nonequilibrium phase behaviour and friction. Phys. Chem. Chem. Phys. 19, 17883–17894 (2017). https://doi.org/10.1039/C7CP01895A
Porras-Vazquez, A., Martinie, L., Vergne, P., Fillot, N.: Independence between friction and velocity distribution in fluids. Phys. Chem. Chem. Phys. 20, 27280–27293 (2018). https://doi.org/10.1039/C8CP04620D
McCabe, C., Cui, S.T., Cummings, P.T., Gordon, P.A., Saeger, R.B.: Examining the rheology of 9-octylheptadecane to giga-pascal pressures. J. Chem. Phys. 114, 1887–1891 (2001). https://doi.org/10.1063/1.1334676
Barus, C.: Isothermals, isopiestics and isometrics relative to viscosity. Am. J. Sci. 45, 87–96 (1893). https://doi.org/10.2475/ajs.s3-45.266.87
Liu, P.Z., Yu, H.L., Ren, N., Lockwood, F.E., Wang, Q.J.: Pressure-viscosity coefficient of hydrocarbon base oil through molecular dynamics simulations. Tribol. Lett. 60, 34 (2015). https://doi.org/10.1007/s11249-015-0610-6
McEwen, E.: The effect of variation of viscosity with pressure on the load-carrying capacity of the oil film between gear-teeth. J. Inst. Pet. 38, 646–672 (1952)
Case, F., Chaka, A., Friend, D.G., Frurip, D., Golab, J., Johnson, R., Moore, J., Mountain, R.D., Olson, J., Schiller, M., Storer, J.: The first industrial fluid properties simulation challenge. Fluid Phase Equilib. 217, 1–10 (2004). https://doi.org/10.1016/s0378-3812(03)00208-5
Bair, S.: The pressure dependence of viscosity for 2,2,4 trimethylhexane to 1 GPa along the 20 °C isotherm. Fluid Phase Equilib. 488, 9–12 (2019). https://doi.org/10.1016/j.fluid.2019.01.021
Galvani Cunha, M.A., Robbins, M.O.: Determination of pressure-viscosity relation of 2,2,4-trimethylhexane by all-atom molecular dynamics simulation. Fluid Phase Equilib. 495, 28–32 (2019). https://doi.org/10.1016/j.fluid.2019.05.008
O’Connor, T.C., Andzelm, J., Robbins, M.O.: AIREBO-M: a reactive model for hydrocarbons at extreme pressures. J. Chem. Phys. 142, 024903 (2015). https://doi.org/10.1063/1.4905549
Ewen, J.P., Gattinoni, C., Thakkar, F.M., Morgan, N., Spikes, H., Dini, D.: A comparison of classical force-fields for molecular dynamics simulations of lubricants. Materials. 9, 651 (2016). https://doi.org/10.3390/ma9080651
Stuart, S.J., Tutein, A.B., Harrison, J.A.: A reactive potential for hydrocarbons with intermolecular interactions. J. Chem. Phys. 112, 6472–6486 (2000). https://doi.org/10.1063/1.481208
Casalini, R., Ransom, T.C.: On the experimental determination of the repulsive component of the potential from high pressure measurements: what is special about twelve? J. Chem. Phys. 151, 194504 (2019). https://doi.org/10.1063/1.5123614
Kondratyuk, N.D., Pisarev, V.: V: Calculation of viscosities of branched alkanes from 0.1 to 1000 MPa by molecular dynamics methods using COMPASS force field. Fluid Phase Equilib. 498, 151–159 (2019). https://doi.org/10.1016/j.fluid.2019.06.023
Bair, S.: Choosing pressure-viscosity relations. High Temp. High Press. 44, 415–428 (2015)
Dymond, J.H., Malhotra, R.: The Tait equation: 100 years on. Int. J. Thermophys. 9, 941–951 (1988). https://doi.org/10.1007/BF01133262
Paluch, M., Dendzik, Z., Rzoska, S.J.: Scaling of high-pressure viscosity data in low-molecular-weight glass-forming liquids. Phys. Rev. B. 60, 2979–2982 (1999). https://doi.org/10.1103/PhysRevB.60.2979
Debenedetti, P.G., Stillinger, F.H.: Supercooled liquids and the glass transition. Nature 410, 259–267 (2001). https://doi.org/10.1038/35065704
Sun, H.: COMPASS: An ab initio force-field optimized for condensed-phase applications—overview with details on alkane and benzene compounds. J. Phys. Chem. B. 102, 7338–7364 (1998). https://doi.org/10.1021/jp980939v
Green, M.S.: Markoff random processes and the statistical mechanics of time-dependent phenomena. II. Irreversible processes in fluids. J. Chem. Phys. 22, 398–413 (1954). https://doi.org/10.1063/1.1740082
Kubo, R.: Statistical-mechanical theory of irreversible processes. I. General theory and simple applications to magnetic and conduction problems. J. Phys. Soc. Jpn. 12, 570–586 (1957). https://doi.org/10.1143/JPSJ.12.570
Maginn, E.J., Messerly, R.A., Carlson, D.J., Roe, D.R., Elliott, J.R.: Best practices for computing transport properties 1. Self-diffusivity and viscosity from equilibrium molecular dynamics [Article v1.0]. Living J. Comput. Mol. Sci. 1, 6324 (2018). https://doi.org/10.33011/livecoms.1.1.6324
Zhang, Y., Otani, A., Maginn, E.J.: Reliable viscosity calculation from equilibrium molecular dynamics simulations: a time decomposition method. J. Chem. Theory Comput. 11, 3537–3546 (2015). https://doi.org/10.1021/acs.jctc.5b00351
Messerly, R.A., Anderson, M.C., Razavi, S.M., Elliott, J.R., Anderson, M.C., Elliott, J.R.: Mie 16–6 force field predicts viscosity with faster-than-exponential pressure dependence for 2,2,4-trimethylhexane. Fluid Phase Equilib. 495, 76–85 (2019). https://doi.org/10.1016/j.fluid.2019.05.013
Martin, M.G., Siepmann, J.I.: Novel configurational-bias Monte Carlo method for branched molecules. Transferable potentials for phase equilibria. 2. United-atom description of branched alkanes. J. Phys. Chem. B. 103, 4508–4517 (1999). https://doi.org/10.1021/jp984742e
Mick, J.R., Soroush Barhaghi, M., Jackman, B., Schwiebert, L., Potoff, J.J.: Optimized mie potentials for phase equilibria: application to branched alkanes. J. Chem. Eng. Data. 62, 1806–1818 (2017). https://doi.org/10.1021/acs.jced.6b01036
Heyes, D.M.: Transport coefficients of simple fluids with steeply repulsive potentials. J. Phys. Condens. Matter. 6, 6409–6421 (1994). https://doi.org/10.1088/0953-8984/6/32/004
Gordon, P.A.: Development of intermolecular potentials for predicting transport properties of hydrocarbons. J. Chem. Phys. 125, 014504 (2006). https://doi.org/10.1063/1.2208359
Manheim, J., Wehde, K., Zhang, W.T.J., Vozka, P., Romanczyk, M., Kilaz, G., Kenttämaa, H.I.: Identification and quantitation of linear alkanes in lubricant base oils by using GC×GC/EI TOF mass spectrometry. J. Am. Soc. Mass Spectrom. 30, 2670–2677 (2019). https://doi.org/10.1007/s13361-019-02336-x
Hermann, J., Distasio, R.A., Tkatchenko, A.: First-principles models for van der Waals interactions in molecules and materials: concepts, theory, and applications. Chem. Rev. 117, 4714–4758 (2017). https://doi.org/10.1021/acs.chemrev.6b00446
Jadhao, V., Robbins, M.O.: Crossover to Arrhenius behavior at high viscosities in squalane. Proc. Natl. Acad. Sci. 114, E8807–E8808 (2017). https://doi.org/10.1073/pnas.1715298114
Lu, J., Wang, Q.J., Ren, N., Lockwood, F.E.: Correlation between pressure-viscosity coefficient and traction coefficient of the base stocks in traction lubricants: a molecular dynamic approach. Tribol. Int. 134, 328–334 (2019). https://doi.org/10.1016/j.triboint.2019.02.013
Keasler, S.J., Charan, S.M., Wick, C.D., Economou, I.G., Siepmann, J.I.: Transferable potentials for phase equilibria-united atom description of five- and six-membered cyclic alkanes and ethers. J. Phys. Chem. B. 116, 11234–11246 (2012). https://doi.org/10.1021/jp302975c
Kondratyuk, N.D., Pisarev, V.V., Ewen, J.P.: Probing the high-pressure viscosity of hydrocarbon mixtures using molecular dynamics simulations. J. Chem. Phys. 153, 154502 (2020). https://doi.org/10.1063/5.0028393
Grunberg, L., Nissan, A.H.: Mixture law for viscosity. Nature 164, 799–800 (1949). https://doi.org/10.1038/164799b0
Kajita, S., Kinjo, T., Nishi, T.: Autonomous molecular design by Monte-Carlo tree search and rapid evaluations using molecular dynamics simulations. Commun. Phys. 3, 77 (2020). https://doi.org/10.1038/s42005-020-0338-y
Dyre, J.C.: Colloquium: the glass transition and elastic models of glass-forming liquids. Rev. Mod. Phys. 78, 953–972 (2006). https://doi.org/10.1103/RevModPhys.78.953
Hansen, H.W., Frick, B., Hecksher, T., Dyre, J.C., Niss, K.: Connection between fragility, mean-squared displacement, and shear modulus in two van der Waals bonded glass-forming liquids. Phys. Rev. B. 95, 104202 (2017). https://doi.org/10.1103/PhysRevB.95.104202
Evans, C.R., Johnson, K.L.: The rheological properties of elastohydrodynamic lubricants. Proc. Inst. Mech. Eng. Part C. 200, 303–312 (1986). https://doi.org/10.1243/PIME_PROC_1986_200_134_02
Alsaad, M., Bair, S., Sanborn, D.M., Winer, W.O.: Glass transitions in lubricants: its relation to elastohydrodynamic lubrication (EHD). J. Lubr. Technol. 100, 404–416 (1978). https://doi.org/10.1627/jpi1959.19.1
Ehret, P., Dowson, D., Taylor, C.M.: On lubricant transport conditions in elastohydrodynamic conjunctions. Proc. R. Soc. London Ser. A. 454, 763–787 (1998). https://doi.org/10.1098/rspa.1998.0185
Ponjavic, A., di Mare, L., Wong, J.S.S.: Effect of pressure on the flow behavior of polybutene. J. Polym. Sci. B Polym. Phys. 52, 708–715 (2014). https://doi.org/10.1002/polb.23472
Jeffreys, S., di Mare, L., Liu, X., Morgan, N., Wong, J.S.S.: Elastohydrodynamic lubricant flow with nanoparticle tracking. RSC Adv. 9, 1441–1450 (2019). https://doi.org/10.1039/c8ra09396b
Bair, S., Qureshi, F., Winer, W.O.: Observations of shear localization in liquid lubricants under pressure. J. Tribol. 115, 507–514 (1993). https://doi.org/10.1115/1.2921667
Bair, S., Qureshi, F., Khonsari, M.: Adiabatic shear localization in a liquid lubricant under pressure. J. Tribol. 116, 705 (1994). https://doi.org/10.1115/1.2927321
Galmiche, B., Ponjavic, A., Wong, J.S.S.: Flow measurements of a polyphenyl ether oil in an elastohydrodynamic contact. J. Phys. Condens. Matter. 28, 134005 (2016). https://doi.org/10.1088/0953-8984/28/13/134005
Yong, X., Zhang, L.T.: Thermostats and thermostat strategies for molecular dynamics simulations of nanofluidics. J. Chem. Phys. 138, 084503 (2013). https://doi.org/10.1063/1.4792202
Khare, R., de Pablo, J., Yethiraj, A.: Molecular simulation and continuum mechanics study of simple fluids in non-isothermal planar Couette flows. J. Chem. Phys. 107, 2589 (1997). https://doi.org/10.1063/1.474570
Martini, A., Hsu, H.Y., Patankar, N.A., Lichter, S.: Slip at high shear rates. Phys. Rev. Lett. 100, 206001 (2008). https://doi.org/10.1103/PhysRevLett.100.206001
Cao, J., Likhtman, A.E.: Shear banding in molecular dynamics of polymer melts. Phys. Rev. Lett. 108, 028302 (2012). https://doi.org/10.1103/PhysRevLett.108.028302
Ewen, J.P., Gao, H., Müser, M.H., Dini, D.: Shear heating, flow, and friction of confined molecular fluids at high pressure. Phys. Chem. Chem. Phys. 21, 5813 (2019). https://doi.org/10.1039/C8CP07436D
Thompson, P.A., Robbins, M.O.: Shear flow near solids: epitaxial order and flow boundary conditions. Phys. Rev. A. 41, 6830–6837 (1990). https://doi.org/10.1103/PhysRevA.41.6830
Butler, S., Harrowell, P.: Factors determining crystal-liquid coexistence under shear. Nature 415, 2000–2003 (2002). https://doi.org/10.1038/4151008a
Varnik, F., Bocquet, L., Barrat, J.-L., Berthier, L.: Shear localization in a model glass. Phys. Rev. Lett. 90, 095702 (2003). https://doi.org/10.1103/PhysRevLett.90.095702
Thompson, P.A., Robbins, M.O., Grest, G.S.: Structure and shear response in nanometer thick films. Isr. J. Chem. 35, 30 (1995). https://doi.org/10.1002/ijch.199500015
Washizu, H., Ohmori, T.: Molecular dynamics simulations of elastohydrodynamic lubrication oil film. Lubr. Sci. 22, 323–340 (2010). https://doi.org/10.1002/ls.126
Heyes, D.M., Smith, E.R., Dini, D., Spikes, H.A., Zaki, T.A.: Pressure dependence of confined liquid behavior subjected to boundary-driven shear. J. Chem. Phys. 136, 134705 (2012). https://doi.org/10.1063/1.3698601
Gattinoni, C., Heyes, D.M., Lorenz, C.D., Dini, D.: Traction and nonequilibrium phase behavior of confined sheared liquids at high pressure. Phys. Rev. E. 88, 052406 (2013). https://doi.org/10.1103/PhysRevE.88.052406
Maćkowiak, S., Heyes, D.M., Dini, D., Brańka, A.C.: Non-equilibrium phase behavior and friction of confined molecular films under shear: a non-equilibrium molecular dynamics study. J. Chem. Phys. 145, 164704 (2016). https://doi.org/10.1063/1.4965829
Martinie, L., Vergne, P.: Lubrication at extreme conditions: a discussion about the limiting shear stress concept. Tribol. Lett. 63, 21 (2016). https://doi.org/10.1007/s11249-016-0709-4
Washizu, H., Ohmori, T., Suzuki, A.: Molecular origin of limiting shear stress of elastohydrodynamic lubrication oil film studied by molecular dynamics. Chem. Phys. Lett. 678, 1–4 (2017). https://doi.org/10.1016/j.cplett.2017.04.020
Kobayashi, H., Fujita, Y.: Mechanisms for three kinds of limiting shear stresses appearing in the traction modes of viscous, viscoelastic, and glassy states of lubricants. J. Appl. Phys. 115, 223509 (2014). https://doi.org/10.1063/1.4881276
Echeverri Restrepo, S., van Eijk, M.C.P., Ewen, J.P.: Behaviour of n-alkanes confined between iron oxide surfaces at high pressure and shear rate: a nonequilibrium molecular dynamics study. Tribol. Int. 137, 420–432 (2019). https://doi.org/10.1016/j.triboint.2019.05.008
Gao, H., Müser, M.H.: Why liquids can appear to solidify during squeeze-out—even when they don’t. J. Colloid Interface Sci. 562, 273–278 (2020). https://doi.org/10.1016/j.jcis.2019.10.097
Robbins, M.O., Smith, E.D.: Connecting molecular-scale and macroscopic tribology. Langmuir 12, 4543–4547 (1996). https://doi.org/10.1021/la9505576
Lu, J., Reddyhoff, T., Dini, D.: A study of thermal effects in EHL rheology and friction using infrared microscopy. Tribol. Int. 146, 106179 (2020). https://doi.org/10.1016/j.triboint.2020.106179
Savio, D., Pastewka, L., Gumbsch, P.: Boundary lubrication of heterogeneous surfaces and the onset of cavitation in frictional contacts. Sci. Adv. 2, e1501585 (2016). https://doi.org/10.1126/sciadv.1501585
Thompson, P.A., Troian, S.M.: A general boundary condition for liquid flow at solid surfaces. Nature 389, 360–362 (1997). https://doi.org/10.1038/38686
Polajnar, M., Kalin, M.: The effect of wetting and surface energy on the friction and slip in oil-lubricated contacts. Tribol. Lett. 52, 185–194 (2013). https://doi.org/10.1007/s11249-013-0194-y
Gao, J.P., Luedtke, W.D., Landman, U.: Structures, solvation forces and shear of molecular films in a rough nano-confinement. Tribol. Lett. 9, 3–13 (2000). https://doi.org/10.1023/a:1018840023845
Washizu, H., Hyodo, S., Ohmori, T., Nishino, N., Suzuki, A.: Macroscopic no-slip boundary condition confirmed in full atomistic simulation of oil film. Tribol. Online. 9, 45–50 (2014). https://doi.org/10.2474/trol.9.45
Ponjavic, A., Wong, J.S.S.: The effect of boundary slip on elastohydrodynamic lubrication. RSC Adv. 4, 20821–20829 (2014). https://doi.org/10.1039/c4ra01714e
Bigelow, W.C., Pickett, D.L., Zisman, W.A.: Oleophobic monolayers: I. Films adsorbed from solution in non-polar liquids. J. Colloid Sci. 1, 513–538 (1946)
Ewen, J.P., Gattinoni, C., Morgan, N., Spikes, H., Dini, D.: Nonequilibrium molecular dynamics simulations of organic friction modifiers adsorbed on iron oxide surfaces. Langmuir 32, 4450–4463 (2016). https://doi.org/10.1021/acs.langmuir.6b00586
Choo, J.H., Forrest, A.K., Spikes, H.A.: Influence of organic friction modifier on liquid slip: a new mechanism of organic friction modifier action. Tribol. Lett. 27, 239–244 (2007). https://doi.org/10.1007/s11249-007-9231-z
Ewen, J.P., Kannam, S.K., Todd, B.D., Dini, D.: Slip of alkanes confined between surfactant monolayers adsorbed on solid surfaces. Langmuir 34, 3864–3873 (2018). https://doi.org/10.1021/acs.langmuir.8b00189
Kalin, M., Kus, M.: New strategy for reducing the EHL friction in steel contacts using additive-formed oleophobic boundary films. Friction. (2020). https://doi.org/10.1007/s40544-020-0403-2
Vakis, A.I., Yastrebov, V.A., Scheibert, J., Minfray, C., Nicola, L., Dini, D., Almqvist, A., Paggi, M., Lee, S., Limbert, G., Molinari, J.F., Anciaux, G., Aghababaei, R., Echeverri Restrepo, S., Papangelo, A., Cammarata, A., Nicolini, P., Putignano, C., Carbone, G., Stupkiewicz, S., Lengiewicz, J., Costagliola, G., Bosia, F., Guarino, R., Pugno, N.M., Müser, M.H., Ciavarella, M.: Modeling and simulation in tribology across scales: an overview. Tribol. Int. 125, 169–199 (2018). https://doi.org/10.1016/j.triboint.2018.02.005
Nielsen, S.O., Lopez, C.F., Srinivas, G., Klein, M.L.: Coarse grain models and the computer simulation of soft materials. J. Phys. Condens. Matter. 16, R481 (2004). https://doi.org/10.1088/0953-8984/16/15/R03
Zheng, L., Trusler, J.P.M., Bresme, F., Müller, E.A.: Predicting the pressure dependence of the viscosity of 2,2,4-trimethylhexane using the SAFT coarse-grained force field. Fluid Phase Equilib. 496, 1–6 (2019). https://doi.org/10.1016/j.fluid.2019.05.017
Hartinger, M., Dumont, M.-L., Ioannides, S., Gosman, D., Spikes, H.: CFD modeling of a thermal and shear-thinning elastohydrodynamic line contact. J. Tribol. 130, 041503 (2008). https://doi.org/10.1115/1.2958077
Hajishafiee, A., Dini, D., Kadiric, A., Ioannides, S.: A fully-coupled finite volume solver for elasto-hydrodynamic lubrication problems with particular application to rolling element bearings. Tribol. Int. 109, 258–273 (2017). https://doi.org/10.1016/j.triboint.2016.12.046
Hartinger, M., Reddyhoff, T.: CFD modeling compared to temperature and friction measurements of an EHL line contact. Tribol. Int. 126, 144–152 (2018). https://doi.org/10.1016/j.triboint.2018.05.012
Tošic, M., Larsson, R., Jovanovic, J., Lohner, T., Bjorling, M., Stahl, K.: A computational fluid dynamics study on shearing mechanisms in thermal elastohydrodynamic. Lubricants 7, 69 (2019). https://doi.org/10.3390/lubricants7080069
Martini, A., Liu, Y., Snurr, R.Q., Wang, Q.J.: Molecular dynamics characterization of thin film viscosity for EHL simulation. Tribol. Lett. 21, 217–225 (2006). https://doi.org/10.1007/s11249-006-9023-x
Holland, D.M., Lockerby, D.A., Borg, M.K., Nicholls, W.D., Reese, J.M.: Molecular dynamics pre-simulations for nanoscale computational fluid dynamics. Microfluid. Nanofluidics. 18, 461–474 (2015). https://doi.org/10.1007/s10404-014-1443-6
Fillot, N., Berro, H., Vergne, P.: From continuous to molecular scale in modelling elastohydrodynamic lubrication: nanoscale surface slip effects on film thickness and friction. Tribol. Lett. 43, 257–266 (2011). https://doi.org/10.1007/s11249-011-9804-8
Savio, D., Fillot, N., Vergne, P., Hetzler, H., Seemann, W., Morales-Espejel, G.E.: A multiscale study on the wall slip effect in a ceramic-steel contact with nanometer-thick lubricant film by a nano-to-elastohydrodynamic lubrication approach. J. Tribol. 137, 031502 (2015). https://doi.org/10.1115/1.4029937
O’Connell, S.T., Thompson, P.A.: Molecular dynamics-continuum hybrid computations: a tool for studying complex fluid flows. Phys. Rev. E. 52, 5792–5795 (1995). https://doi.org/10.1103/PhysRevE.52.R5792
Mohamed, K.M., Mohamad, A.A.: A review of the development of hybrid atomistic-continuum methods for dense fluids. Microfluid. Nanofluidics. 8, 283–302 (2010). https://doi.org/10.1007/s10404-009-0529-z
Nie, X.B., Chen, S.Y., Robbins, M.O.: A continuum and molecular dynamics hybrid method for micro- and nano-fluid flow. J. Fluid Mech. 500, 55–64 (2004). https://doi.org/10.1017/S0022112003007225
Nie, X., Robbins, M.O., Chen, S.: Resolving singular forces in cavity flow: multiscale modeling from atomic to millimeter scales. Phys. Rev. Lett. 96, 134501 (2006). https://doi.org/10.1103/PhysRevLett.96.134501
Liu, J., Chen, S., Nie, X., Robbins, M.O.: A continuum-atomistic simulation of heat transfer in micro- and nano-flows. J. Comput. Phys. 227, 279–291 (2007). https://doi.org/10.1016/j.jcp.2007.07.014
Luan, B.Q., Hyun, S., Molinari, J.F., Bernstein, N., Robbins, M.O.: Multiscale modeling of two-dimensional contacts. Phys. Rev. E 74, 046710 (2006). https://doi.org/10.1103/PhysRevE.74.046710
Luan, B., Robbins, M.O.: Hybrid atomistic/continuum study of contact and friction between rough solids. Tribol. Lett. 36, 1–16 (2009). https://doi.org/10.1007/s11249-009-9453-3
Miller, R.E., Tadmor, E.B.: A unified framework and performance benchmark of fourteen multiscale atomistic/continuum coupling methods. Model. Simul. Mater. Sci. Eng. 17, 053001 (2009). https://doi.org/10.1088/0965-0393/17/5/053001
Liu, J., Chen, S., Nie, X., Robbins, M.O.: A continuum-atomistic multi-timescale algorithm for micro/nano flows. Commun. Comput. Phys. 4, 1279–1291 (2008)
Weller, H.G., Tabor, G., Jasak, H., Fureby, C.: A tensorial approach to computational continuum mechanics using object-oriented techniques. Comput. Phys. 12, 620 (1998). https://doi.org/10.1063/1.168744
Cosden, I.A., Lukes, J.R.: A hybrid atomistic-continuum model for fluid flow using LAMMPS and OpenFOAM. Comput. Phys. Commun. 184, 1958–1965 (2013). https://doi.org/10.1016/j.cpc.2013.03.009
Smith, E.R., Trevelyan, D.J., Ramos-Fernandez, E., Sufian, A., O’Sullivan, C., Dini, D.: CPL library—a minimal framework for coupled particle and continuum simulation. Comput. Phys. Commun. 250, 107068 (2020). https://doi.org/10.1016/j.cpc.2019.107068
Tong, Z.X., He, Y.L., Tao, W.Q.: A review of current progress in multiscale simulations for fluid flow and heat transfer problems: the frameworks, coupling techniques and future perspectives. Int. J. Heat Mass Transf. 137, 1263–1289 (2019). https://doi.org/10.1016/j.ijheatmasstransfer.2019.04.004
Acknowledgements
We are indebted to Mark Robbins for numerous stimulating discussions, the field of tribology will not be the same without him.
Funding
J.P.E. thanks the Royal Academy of Engineering for funding through a Research Fellowship. The support of the Engineering and Physical Research Council (EPSRC) is acknowledged through grant EP/P030211/1. D.D. thanks the EPSRC for an Established Career Fellowship EP/N025954/1.
Author information
Authors and Affiliations
Corresponding author
Additional information
Special Issue Dedicated to the Memory of Mark Robbins.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ewen, J.P., Spikes, H.A. & Dini, D. Contributions of Molecular Dynamics Simulations to Elastohydrodynamic Lubrication. Tribol Lett 69, 24 (2021). https://doi.org/10.1007/s11249-021-01399-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11249-021-01399-w