
Deregulated Immune Pathway Associated with Palbociclib Resistance in Preclinical Breast Cancer Models: Integrative Genomics and Transcriptomics

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
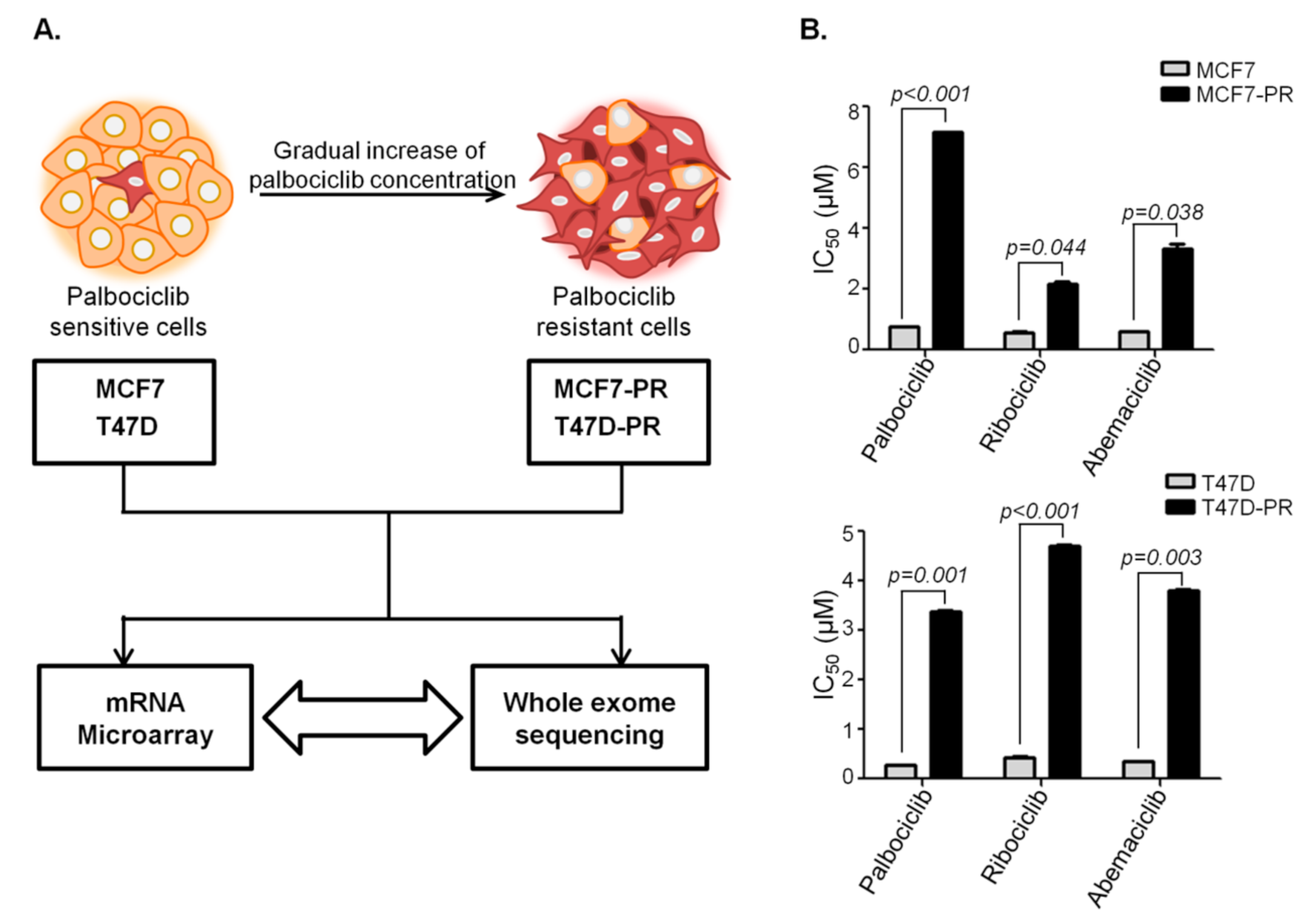
2.1. Cell Culture and Resistant Cell Line Establishment
2.2. Gene Expression via Microarray Analyses
2.3. Whole Exome Sequencing (WES)
2.4. Pathway Analysis of Differentially Expressed Genes (DEGs)
2.5. Quantitative Real-Time PCR (qRT-PCR)
2.6. Cell-Mediated Cytotoxicity Asssay
2.7. Identification of Driver or Pathogenic Mutations
2.8. Pathway Analysis for Genes with Driver Mutations by Visualizing Gene Ontology (GO)
3. Results
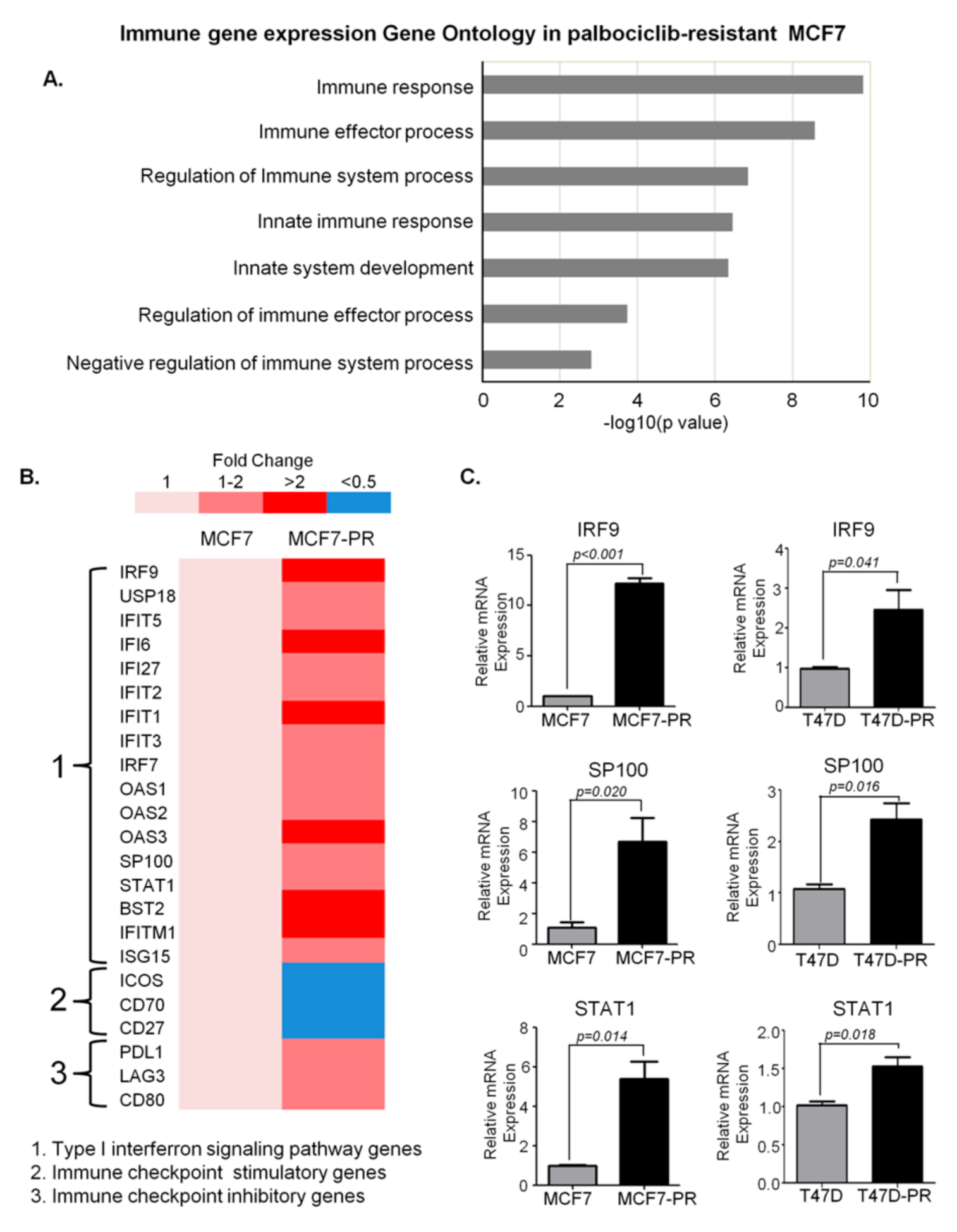
3.1. DEG Analysis Revealing Deregulation of Immune Pathway in Palbociclib-Resistant Cells
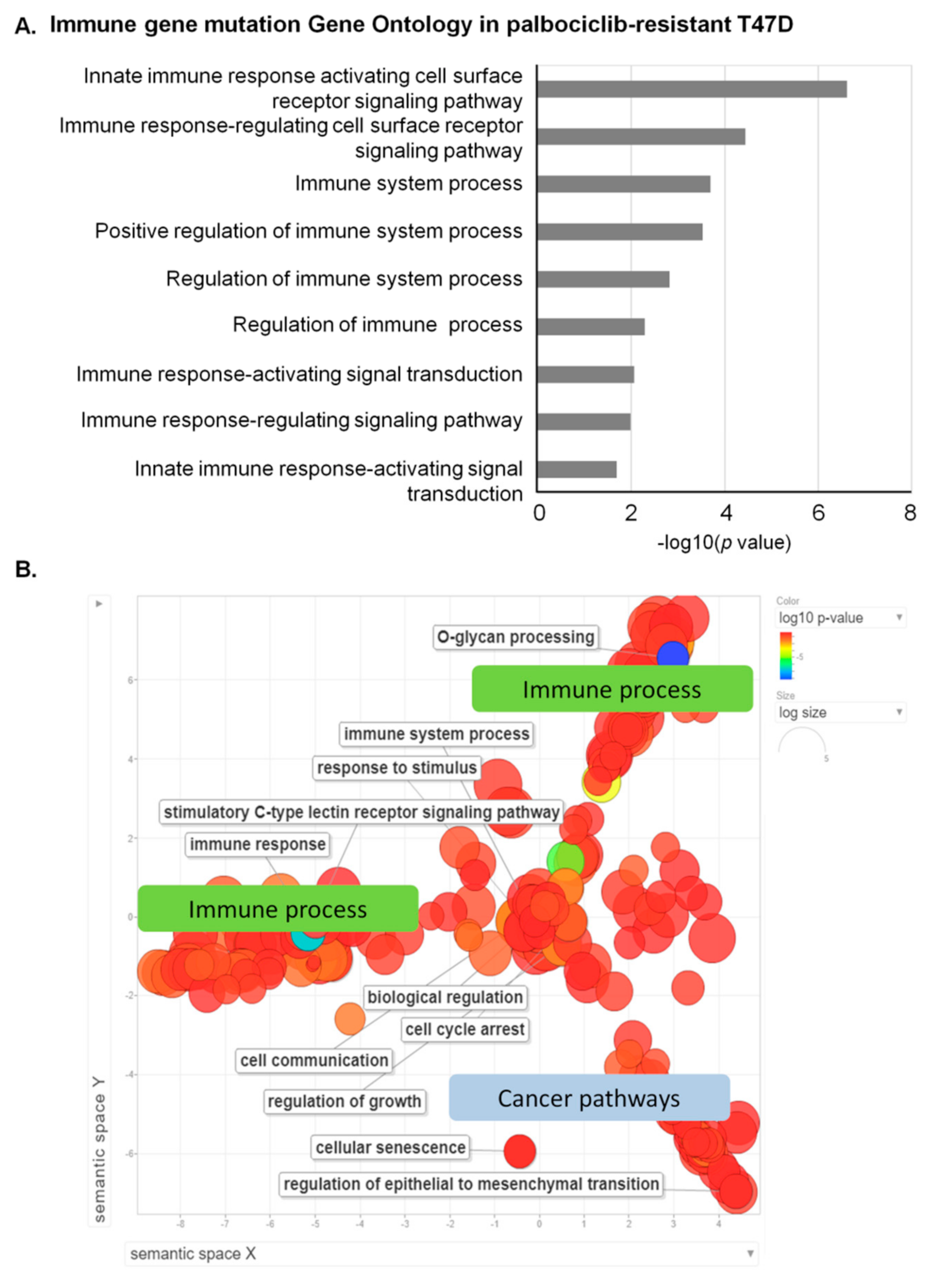
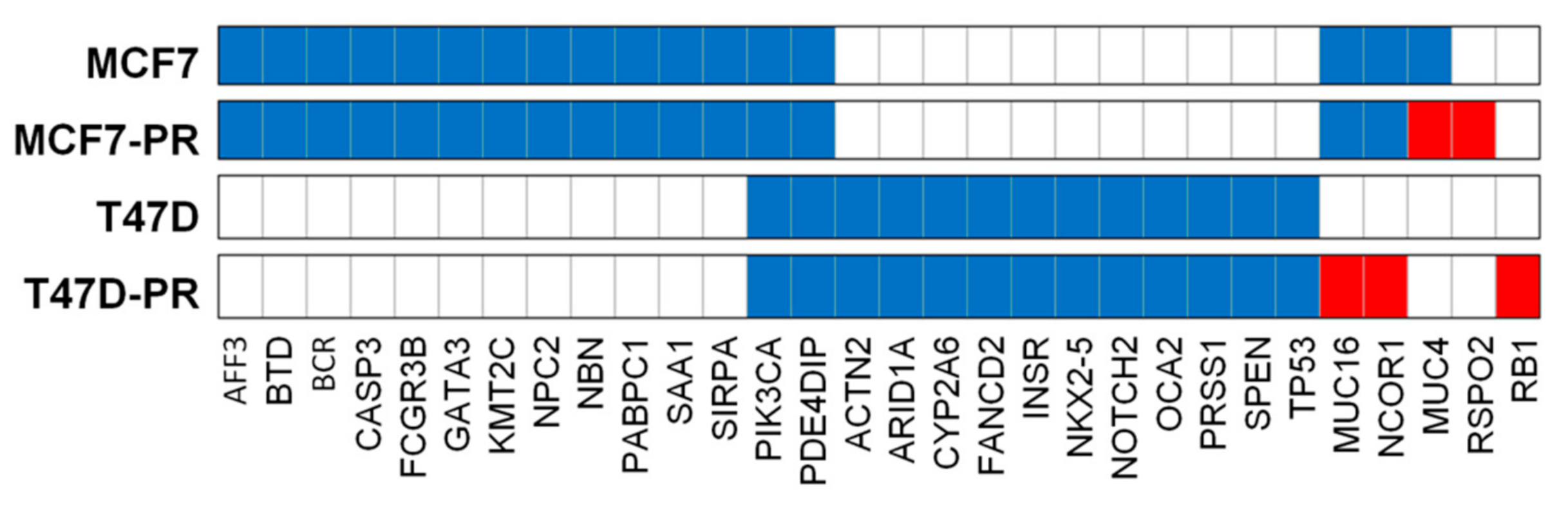
3.2. Mutation Profiling Revealing Deregulation of Immune Pathway in Palbociclib-Resistant Cells
3.3. Visualization of GO Revealing Prominent Immune Process Involvement in Palbociclib-Resistant Cells
3.4. Mutation Dot Plots
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Momenimovahed, Z.; Salehiniya, H. Epidemiological characteristics of and risk factors for breast cancer in the world. Breast Cancer Targets Ther. 2019, 11, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, C.D.; Migliaccio, I.; Malorni, L.; Guarducci, C.; Biganzoli, L.; Di Leo, A. Challenges in the management of advanced, ER-positive, HER2-negative breast cancer. Nat. Rev. Clin. Oncol. 2015, 12, 541. [Google Scholar] [CrossRef] [PubMed]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.-S.; Sonke, G.S.; Paluch-Shimon, S.; Campone, M.; Blackwell, K.L.; André, F.; Winer, E.P. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N. Engl. J. Med. 2016, 375, 1738–1748. [Google Scholar] [CrossRef] [PubMed]
- Sobhani, N.; D’Angelo, A.; Pittacolo, M.; Roviello, G.; Miccoli, A.; Corona, S.P.; Bernocchi, O.; Generali, D.; Otto, T. Updates on the CDK4/6 inhibitory strategy and combinations in breast cancer. Cells 2019, 8, 321. [Google Scholar] [CrossRef] [Green Version]
- De Leeuw, R.; McNair, C.; Schiewer, M.J.; Neupane, N.P.; Brand, L.J.; Augello, M.A.; Li, Z.; Cheng, L.C.; Yoshida, A.; Courtney, S.M. MAPK reliance via acquired CDK4/6 inhibitor resistance in cancer. Clin. Cancer Res. 2018, 24, 4201–4214. [Google Scholar] [CrossRef] [Green Version]
- Pandey, K.; An, H.J.; Kim, S.K.; Lee, S.A.; Kim, S.; Lim, S.M.; Kim, G.M.; Sohn, J.; Moon, Y.W. Molecular mechanisms of resistance to CDK4/6 inhibitors in breast cancer: A review. Int. J. Cancer 2019, 145, 1179–1188. [Google Scholar] [CrossRef] [Green Version]
- Portman, N.; Alexandrou, S.; Carson, E.; Wang, S.; Lim, E.; Caldon, C.E. Overcoming CDK4/6 inhibitor resistance in ER-positive breast cancer. Endocr. Relat. Cancer 2019, 26, R15–R30. [Google Scholar] [CrossRef] [Green Version]
- Herrera-Abreu, M.T.; Palafox, M.; Asghar, U.; Rivas, M.A.; Cutts, R.J.; Garcia-Murillas, I.; Pearson, A.; Guzman, M.; Rodriguez, O.; Grueso, J. Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor–positive breast cancer. Cancer Res. 2016, 76, 2301–2313. [Google Scholar] [CrossRef] [Green Version]
- Guarducci, C.; Bonechi, M.; Benelli, M.; Biagioni, C.; Boccalini, G.; Romagnoli, D.; Verardo, R.; Schiff, R.; Osborne, C.K.; De Angelis, C. Cyclin E1 and Rb modulation as common events at time of resistance to palbociclib in hormone receptor-positive breast cancer. NPJ Breast Cancer 2018, 4, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Mao, P.; Kusiel, J.; Cohen, O.; Wagle, N. Abstract PD4-01: The role of FGF/FGFR axis in resistance to SERDs and CDK4/6 inhibitors in ER+ breast cancer. Cancer Res. 2018, 78. [Google Scholar] [CrossRef]
- Costa, C.; Wang, Y.; Ly, A.; Hosono, Y.; Murchie, E.; Walmsley, C.S.; Huynh, T.; Healy, C.; Peterson, R.; Yanase, S. PTEN loss mediates clinical cross-resistance to CDK4/6 and PI3Kα inhibitors in breast cancer. Cancer Discov. 2020, 10, 72–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilgelm, A.E.; Saleh, N.; Shattuck-Brandt, R.; Riemenschneider, K.; Slesur, L.; Chen, S.-C.; Johnson, C.A.; Yang, J.; Blevins, A.; Yan, C. MDM2 antagonists overcome intrinsic resistance to CDK4/6 inhibition by inducing p21. Sci. Transl. Med. 2019, 11, eaav7171. [Google Scholar] [CrossRef] [PubMed]
- Demuth, C.; Andersen, M.N.; Jakobsen, K.R.; Madsen, A.T.; Sørensen, B.S. Increased PD-L1 expression in erlotinib-resistant NSCLC cells with MET gene amplification is reversed upon MET-TKI treatment. Oncotarget 2017, 8, 68221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinshaw, D.C.; Shevde, L.A. The tumor microenvironment innately modulates cancer progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [Green Version]
- Zamarron, B.F.; Chen, W. Dual roles of immune cells and their factors in cancer development and progression. Int. J. Biol. Sci. 2011, 7, 651. [Google Scholar] [CrossRef]
- Wei, S.; Duffy, C.; Allison, J. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [Green Version]
- Seliger, B. Combinatorial approaches with checkpoint inhibitors to enhance anti-tumor immunity. Front. Immunol. 2019, 10, 999. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R. CDK4/6 inhibition augments antitumor immunity by enhancing T-cell activation. Cancer Discov. 2018, 8, 216–233. [Google Scholar] [CrossRef] [Green Version]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef]
- Pandey, K.; Park, N.; Park, K.S.; Hur, J.; Cho, Y.B.; Kang, M.; An, H.J.; Kim, S.; Hwang, S.; Moon, Y.W. Combined CDK2 and CDK4/6 Inhibition Overcomes Palbociclib Resistance in Breast Cancer by Enhancing Senescence. Cancers 2020, 12, 3566. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, L.; Fairley, S.; Zheng-Bradley, X.; Streeter, I.; Perry, E.; Lowy, E.; Tassé, A.-M.; Flicek, P. The international Genome sample resource (IGSR): A worldwide collection of genome variation incorporating the 1000 Genomes Project data. Nucleic Acids Res. 2017, 45, D854–D859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC Cancer Gene Census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [Green Version]
- Post, A.E.; Smid, M.; Nagelkerke, A.; Martens, J.W.; Bussink, J.; Sweep, F.C.; Span, P.N. Interferon-stimulated genes are involved in cross-resistance to radiotherapy in tamoxifen-resistant breast cancer. Clin. Cancer Res. 2018, 24, 3397–3408. [Google Scholar] [CrossRef] [Green Version]
- De Angelis, C.; Fu, X.; Cataldo, M.L.; Nardone, A.; Jansen, V.M.; Veeraraghavan, J.; Nanda, S.; Qin, L.; Sethunath, V.; Pereira, R. Abstract GS2-01: High levels of interferon-response gene signatures are associated with de novo and acquired resistance to CDK4/6 inhibitors in ER+ breast cancer. Cancer Res. 2020, 80. [Google Scholar] [CrossRef]
- Vijayaraghavan, S.; Doostan, I.; Carey, J.P.; Keyomarsi, K. Characterizing acquired resistance to palbociclib in breast cancer. Cancer Res. 2017, 77. [Google Scholar] [CrossRef]
- Kufe, D.W. Mucins in cancer: Function, prognosis and therapy. Nat. Rev. Cancer 2009, 9, 874–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrawal, B.; Gupta, N.; Konowalchuk, J.D. MUC1 mucin: A putative regulatory (checkpoint) molecule of T cells. Front. Immunol. 2018, 9, 2391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanson, R.L.; Hollingsworth, M.A. Functional consequences of differential O-glycosylation of MUC1, MUC4, and MUC16 (downstream effects on signaling). Biomolecules 2016, 6, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; Kaur, K.; Liu, C. ClinVar: Improvements to accessing data. Nucleic Acids Res. 2020, 48, D835–D844. [Google Scholar] [CrossRef]
- Gautam, S.K.; Kumar, S.; Cannon, A.; Hall, B.; Bhatia, R.; Nasser, M.W.; Mahapatra, S.; Batra, S.K.; Jain, M. MUC4 mucin-a therapeutic target for pancreatic ductal adenocarcinoma. Expert Opin. Ther. Targets 2017, 21, 657–669. [Google Scholar] [CrossRef]
- Aithal, A.; Rauth, S.; Kshirsagar, P.; Shah, A.; Lakshmanan, I.; Junker, W.M.; Jain, M.; Ponnusamy, M.P.; Batra, S.K. MUC16 as a novel target for cancer therapy. Expert Opin. Ther. Targets 2018, 22, 675–686. [Google Scholar] [CrossRef]
- Ahad, A.; Stevanin, M.; Smita, S.; Mishra, G.P.; Gupta, D.; Waszak, S.; Sarkar, U.A.; Basak, S.; Gupta, B.; Acha-Orbea, H. NCoR1: Putting the brakes on the dendritic cell immune tolerance. iScience 2019, 19, 996–1011. [Google Scholar] [CrossRef] [Green Version]
- Han, X.H.; Jin, Y.-R.; Seto, M.; Yoon, J.K. A WNT/β-catenin signaling activator, R-spondin, plays positive regulatory roles during skeletal myogenesis. J. Biol. Chem. 2011, 286, 10649–10659. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Xiang, Y.; Li, F.; Yi, Q.; Li, B.; Ke, X. WNT/β-catenin signaling pathway regulating T cell-inflammation in tumor microenvironment. Front. Immunol. 2019, 10, 2293. [Google Scholar] [CrossRef] [Green Version]
- Schneck, H.; Blassl, C.; Meier-Stiegen, F.; Neves, R.P.; Janni, W.; Fehm, T.; Neubauer, H. Analysing the mutational status of PIK3CA in circulating tumor cells from metastatic breast cancer patients. Mol. Oncol. 2013, 7, 976–986. [Google Scholar] [CrossRef]
- Condorelli, R.; Spring, L.; O’shaughnessy, J.; Lacroix, L.; Bailleux, C.; Scott, V.; Dubois, J.; Nagy, R.; Lanman, R.; Iafrate, A. Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Ann. Oncol. 2018, 29, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Liu, Y.; Zhu, Z.; Loi, S.; Colleoni, M.; Loibl, S.; DeMichele, A.; Harbeck, N.; André, F.; Bayar, M.A. Cyclin E1 expression and palbociclib efficacy in previously treated hormone receptor–positive metastatic breast cancer. J. Clin. Oncol. 2019, 37, 1169. [Google Scholar] [CrossRef] [PubMed]
- Wander, S.A.; Cohen, O.; Gong, X.; Johnson, G.N.; Buendia-Buendia, J.E.; Lloyd, M.R.; Kim, D.; Luo, F.; Mao, P.; Helvie, K. The genomic landscape of intrinsic and acquired resistance to cyclin-dependent kinase 4/6 inhibitors in patients with hormone receptor positive metastatic breast cancer. Cancer Discov. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, E.P.; Jansen, V.M.; Nash Smyth, E.N.; Schlauch, D.; Cuyun Carter, G.; Willard, M.D.; Misch, A.; Bowman, L.; Zhu, Y.E.; McNeely, S. Next-generation sequencing (NGS) results among hormone receptor-positive (HR+), human epidermal growth factor receptor 2-negative (HER2-) metastatic breast cancer (MBC) patients treated with a CDK4 & 6 inhibitor: A retrospective observational study based on real-world data. J. Clin. Oncol. 2019, 37, 1042. [Google Scholar]
- Nan, Y.; Wu, C.; Zhang, Y.-J. Interplay between Janus kinase/signal transducer and activator of transcription signaling activated by type I interferons and viral antagonism. Front. Immunol. 2017, 8, 1758. [Google Scholar] [CrossRef]
- Huang, R.; Faratian, D.; Sims, A.H.; Wilson, D.; Thomas, J.S.; Harrison, D.J.; Langdon, S.P. Increased STAT1 signaling in endocrine-resistant breast cancer. PLoS ONE 2014, 9, e94226. [Google Scholar] [CrossRef] [Green Version]
- Kettner, N.M.; Vijayaraghavan, S.; Durak, M.G.; Bui, T.; Kohansal, M.; Ha, M.J.; Liu, B.; Rao, X.; Wang, J.; Yi, M. Combined inhibition of STAT3 and DNA repair in palbociclib-resistant ER-positive breast cancer. Clin. Cancer Res. 2019, 25, 3996–4013. [Google Scholar] [CrossRef] [Green Version]
- Tolaney, S.M.; Kabos, P.; Dickler, M.N.; Gianni, L.; Jansen, V.; Lu, Y.; Young, S.; Rugo, H.S. Updated efficacy, safety, & PD-L1 status of patients with HR+, HER2-metastatic breast cancer administered abemaciclib plus pembrolizumab. J. Clin. Oncol 2018, 36, 1059. [Google Scholar]
- Masuda, J.; Tsurutani, J.; Masuda, N.; Futamura, M.; Matsumoto, K.; Aogi, K.; Takahashi, M.; Iwata, H.; Iwasa, T.; Mukohara, T. Abstract OT2-04-07: Phase II study of nivolumab in combination with abemaciclib plus endocrine therapy in patients with hormone receptor-positive, human epidermal growth factor receptor-2 negative metastatic breast cancer (WJOG11418B, NEWFLAME trial). Am. Assoc. Cancer Res. 2020, 80. [Google Scholar] [CrossRef]
- King, R.J.; Yu, F.; Singh, P.K. Genomic alterations in mucins across cancers. Oncotarget 2017, 8, 67152. [Google Scholar] [CrossRef] [Green Version]
- del Mar Noblejas-Lopez, M.; Morcillo-García, S.; Nieto-Jimenez, C.; Nuncia-Cantarero, M.; Győrffy, B.; Galan-Moya, E.M.; Pandiella, A.; Ocaña, A. Evaluation of transcriptionally regulated genes identifies NCOR1 in hormone receptor negative breast tumors and lung adenocarcinomas as a potential tumor suppressor gene. PLoS ONE 2018, 13, e0207776. [Google Scholar]
- Bhatia, R.; Gautam, S.K.; Cannon, A.; Thompson, C.; Hall, B.R.; Aithal, A.; Banerjee, K.; Jain, M.; Solheim, J.C.; Kumar, S. Cancer-associated mucins: Role in immune modulation and metastasis. Cancer Metastasis Rev. 2019, 38, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Müller, L.; Hainberger, D.; Stolz, V.; Ellmeier, W. NCOR1—A new player on the field of T cell development. J. Leukoc. Biol. 2018, 104, 1061–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GO Biological Process | Count | Genes | p-Value | Adjusted p-Value |
|---|---|---|---|---|
| immune response | 54 | ADCY5, ANG, ANXA3, BLNK, BST2, CD22, CEBPG, CLEC2D, CLU, CTSC, CTSH, CTSK, DDX58, DDX60, EGR1, EPRS, FFAR3, FRK, FTH1, GBP2, HERC5, HMOX1, IFI6, IFIH1, IFIT1, IFITM1, IGHD, IL20, IRF9, ISG15, KIR2DS2, KIR2DS4, KIR3DL2, KYNU, LYN, MYB, NFIL3, OAS1, OAS2, OAS3, PTGER4, RAET1G, RIPK2, S100A8, S100A9, SEMA3C, STAT1, SUSD2, TAB1, TXNIP, ULBP1, UNC13D, USP18, VIPR1 | 1.5 × 10−10 | 4.3 × 10−9 |
| immune effector process | 33 | ACKR3, ANXA3, BST2, CEBPG, CLU, CTSC, CTSH, DDX58, DDX60, FFAR3, HERC5, HMOX1, IFIH1, IFIT1, IFITM1, IGHD, IRF9, ISG15, LYN, MTSS1, MYB, OAS1, OAS2, OAS3, PDK4, PTGER4, RAET1G, RIPK2, STAT1, TNIK, ULBP1, UNC13D, ZNF189 | 2.7 × 10−9 | 6.8 × 10−8 |
| regulation of immune system process | 45 | ACKR3, ANXA1, BMP4, BST2, C5AR2, CDK6, CLEC2D, CLU, CTSH, CTSK, DDX58, DDX60, FFAR3, FLT3, HERC5, HMOX1, IFIH1, IFIT1, IFITM1, IGHD, IL20, ISG15, KIR2DS2, KIR3DL2, LYN, MITF, MTSS1, MYB, MYC, PDE5A, PDK4, PLCB1, PTGER4, RIPK2, S100A7, SHPK, STAT1, TAB1, TGFBR2, TNIK, TRIB1, ULBP1, UNC13D, USP18, ZNF189 | 1.4 × 10−7 | 2.8 × 10−6 |
| innate immune response | 36 | ADCY5, ANG, BST2, CEBPG, CLU, CTSK, DDX58, DDX60, EGR1, EPRS, FRK, GBP2, HERC5, IFI6, IFIH1, IFIT1, IFITM1, IRF9, ISG15, KIR2DS2, KIR2DS4, KYNU, LYN, OAS1, OAS2, OAS3, RAET1G, RIPK2, S100A8, S100A9, STAT1, TAB1, TXNIP, ULBP1, UNC13D, USP18 | 3.5 × 10−7 | 6.5 × 10−6 |
| immune system development | 30 | ANXA1, BMP4, CALCR, CDK6, CEBPG, DHRS2, EGR1, FLT3, G6PD, HERC6, IGHD, IL20, ISG15, KRT75, L3MBTL3, LYN, MITF, MPZL2, MYB, MYC, ONECUT1, PTGER4, RIPK2, RUNX2, SIX1, TGFBR2, TMOD2, TRIB1, ZFP36L2, ZNF385A | 4.5 × 10−7 | 8.1 × 10−6 |
| regulation of immune effector process | 16 | ACKR3, BST2, DDX58, DDX60, FFAR3, HERC5, HMOX1, IFIT1, LYN, MTSS1, MYB, PDK4, RIPK2, TNIK, UNC13D, ZNF189 | 0.00018 | 0.00182 |
| negative regulation of immune system process | 13 | BMP4, BST2, C5AR2, CDK6, FLT3, HMOX1, IFIT1, LYN, MYC, PDE5A, PLCB1, PTGER4, TRIB1 | 0.00155 | 0.01199 |
| GO Biological Process | Count | Genes | p-Value | Adjusted p-Value |
|---|---|---|---|---|
| innate immune response activating cell surface receptor signaling pathway | 7 | MUC3A, MUC4, MUC5B, MUC6, MUC12, MUC16, MUC17 | 2.41 × 10−7 | 7.24 × 10−5 |
| immune response-regulating cell surface receptor signaling pathway | 8 | HSP90AB1, MUC3A, MUC4, MUC5B, MUC6, MUC12, MUC17, SOS1 | 3.62 × 10−5 | 0.0048 |
| innate immune response-activating signal transduction | 6 | MUC3A, MUC4, MUC5B, MUC6, MUC12, MUC17 | 0.0002 | 0.0238 |
| immune response-regulating signaling pathway | 8 | HSP90AB1, MUC3A, MUC4, MUC5B, MUC6, MUC12, MUC17, SOS1 | 0.0003 | 0.0238 |
| immune response-activating signal transduction | 7 | HSP90AB1, MUC3A, MUC4, MUC5B, MUC6, MUC17, MUC12 | 0.0015 | 0.0895 |
| regulation of immune response | 8 | HSP90AB1, MUC3A, MUC4, MUC5B, MUC6, MUC17, MUC12, SOS1 | 0.0052 | 0.1476 |
| regulation of immune system process | 11 | HSP90AB1, KMT2C, MUC3A, MUC4, MUC5B, MUC6, MUC12, MUC17, POU4F1, RB1, SOS1 | 0.0086 | 0.1476 |
| positive regulation of immune system process | 9 | HSP90AB1, MUC3A, MUC4, MUC5B, MUC6, MUC12, MUC17, POU4F1, RB1 | 0.0103 | 0.1476 |
| immune system process | 12 | PRSS3, KMT2C, HSP90AB1, TGFBR1, SOS1, MUC5B, MUC4, VCP, MUC17, MUC3A, MUC6, MUC12 | 0.0203 | 0.1476 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pandey, K.; Lee, E.; Park, N.; Hur, J.; Cho, Y.B.; Katuwal, N.B.; Kim, S.K.; Lee, S.A.; Kim, I.; An, H.J.; et al. Deregulated Immune Pathway Associated with Palbociclib Resistance in Preclinical Breast Cancer Models: Integrative Genomics and Transcriptomics. Genes 2021, 12, 159. https://doi.org/10.3390/genes12020159
Pandey K, Lee E, Park N, Hur J, Cho YB, Katuwal NB, Kim SK, Lee SA, Kim I, An HJ, et al. Deregulated Immune Pathway Associated with Palbociclib Resistance in Preclinical Breast Cancer Models: Integrative Genomics and Transcriptomics. Genes. 2021; 12(2):159. https://doi.org/10.3390/genes12020159
Chicago/Turabian StylePandey, Kamal, Eunbyeol Lee, Nahee Park, Jin Hur, Young Bin Cho, Nar Bahadur Katuwal, Seung Ki Kim, Seung Ah Lee, Isaac Kim, Hee Jung An, and et al. 2021. "Deregulated Immune Pathway Associated with Palbociclib Resistance in Preclinical Breast Cancer Models: Integrative Genomics and Transcriptomics" Genes 12, no. 2: 159. https://doi.org/10.3390/genes12020159