
Parkinson’s Disease: Potential Actions of Lithium by Targeting the WNT/β-Catenin Pathway, Oxidative Stress, Inflammation and Glutamatergic Pathway


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Parkinson’s Disease and Oxidative Stress
3. Parkinson’s Disease and Inflammation
4. Parkinson’s Disease and Glutamatergic Pathway
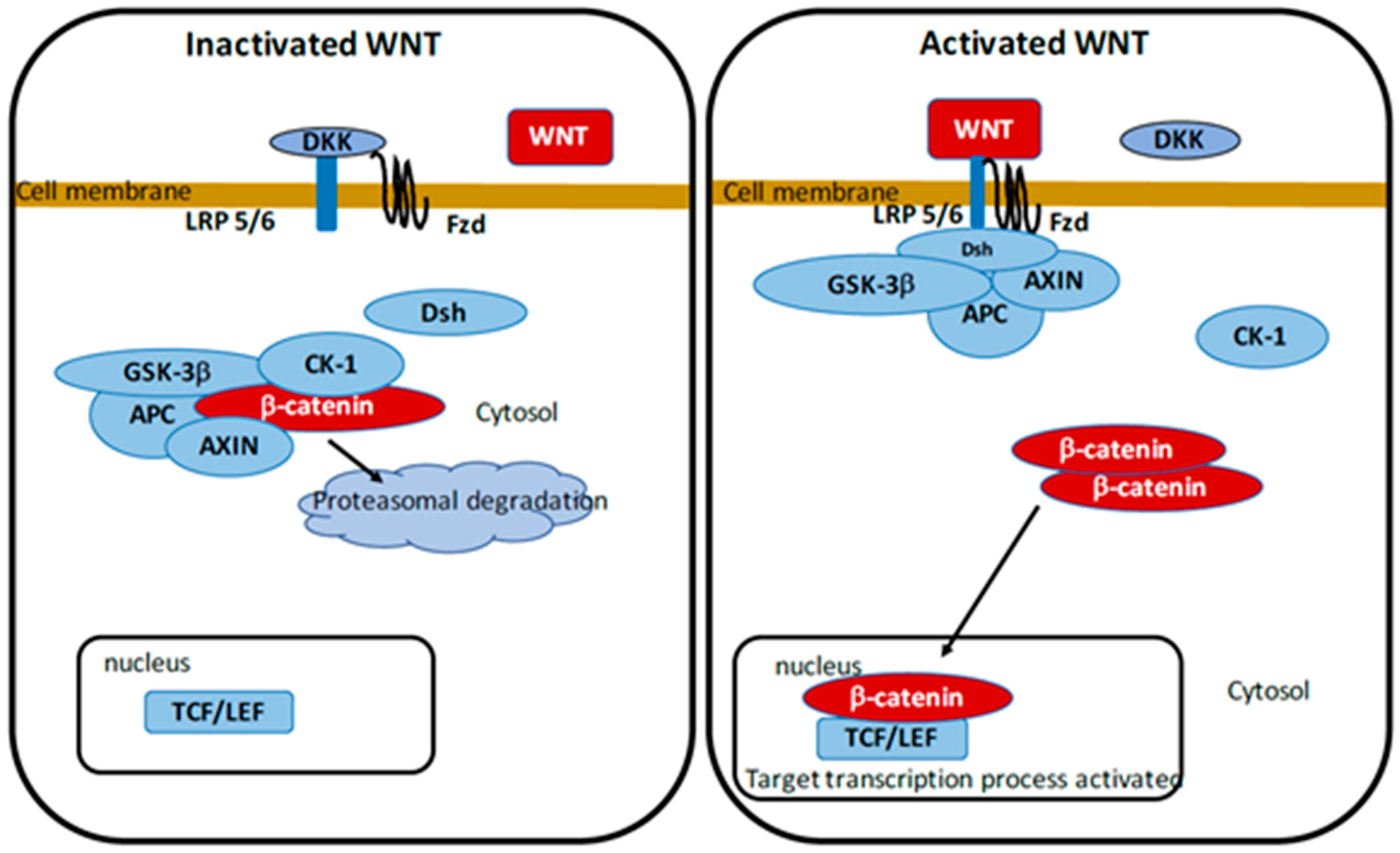
5. WNT/β-Catenin Pathway
5.1. Parkinson’s Disease and WNT/β-Catenin Pathway
5.2. WNT/β-Catenin Pathway and Oxidative Stress
5.3. WNT/β-Catenin Pathway and Inflammation
5.4. WNT/β-Catenin Pathway and Glutamatergic Pathway
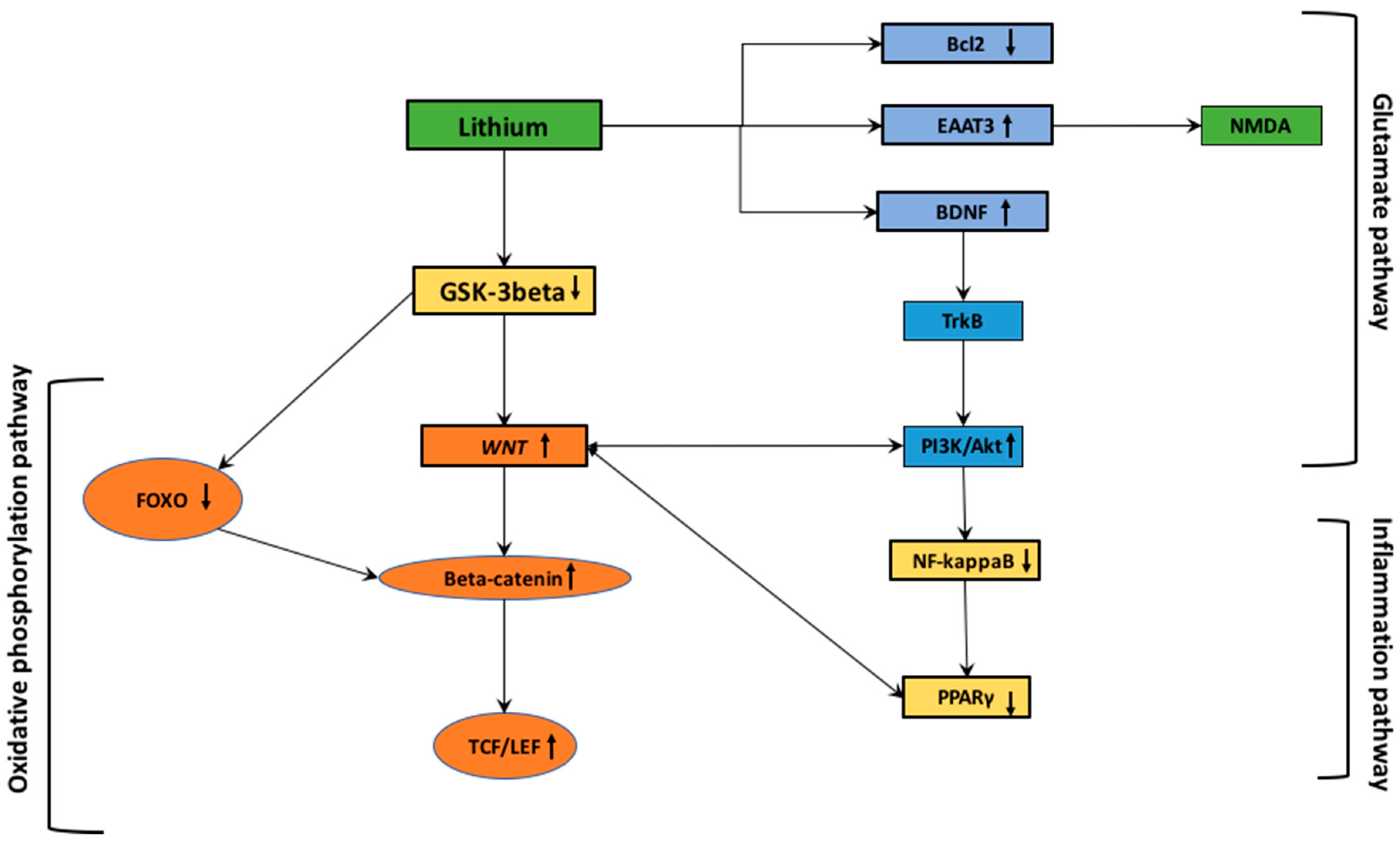
6. Parkinson’s Disease: Interactions between WNT/β-Catenin Pathway and Lithium
6.1. Lithium and Oxidative Stress
6.2. Lithium and Inflammation
6.3. Lithium and Glutamatergic Pathway
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Braak, H.; Ghebremedhin, E.; Rüb, U.; Bratzke, H.; Del Tredici, K. Stages in the Development of Parkinson’s Disease-Related Pathology. Cell Tissue Res. 2004, 318, 121–134. [Google Scholar] [CrossRef]
- Grinberg, L.T.; Rueb, U.; di Lorenzo Alho, A.T.; Heinsen, H. Brainstem Pathology and Non-Motor Symptoms in PD. J. Neurol. Sci. 2010, 289, 81–88. [Google Scholar] [CrossRef]
- Maguire-Zeiss, K.A.; Federoff, H.J. Future Directions for Immune Modulation in Neurodegenerative Disorders: Focus on Parkinson’s Disease. J. Neural Transm. Vienna Austria 1996 2010, 117, 1019–1025. [Google Scholar] [CrossRef]
- Yin, F.; Boveris, A.; Cadenas, E. Mitochondrial Energy Metabolism and Redox Signaling in Brain Aging and Neurodegeneration. Antioxid. Redox Signal. 2014, 20, 353–371. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.-N. Thermodynamics in Neurodegenerative Diseases: Interplay between Canonical WNT/Beta-Catenin Pathway-PPAR Gamma, Energy Metabolism and Circadian Rhythms. Neuromolecular Med. 2018, 20, 174–204. [Google Scholar] [CrossRef]
- Parish, C.L.; Castelo-Branco, G.; Rawal, N.; Tonnesen, J.; Sorensen, A.T.; Salto, C.; Kokaia, M.; Lindvall, O.; Arenas, E. Wnt5a-Treated Midbrain Neural Stem Cells Improve Dopamine Cell Replacement Therapy in Parkinsonian Mice. J. Clin. Invest. 2008, 118, 149–160. [Google Scholar] [CrossRef] [Green Version]
- Rawal, N.; Corti, O.; Sacchetti, P.; Ardilla-Osorio, H.; Sehat, B.; Brice, A.; Arenas, E. Parkin Protects Dopaminergic Neurons from Excessive Wnt/Beta-Catenin Signaling. Biochem. Biophys. Res. Commun. 2009, 388, 473–478. [Google Scholar] [CrossRef]
- Berwick, D.C.; Harvey, K. The Importance of Wnt Signalling for Neurodegeneration in Parkinson’s Disease. Biochem. Soc. Trans. 2012, 40, 1123–1128. [Google Scholar] [CrossRef]
- Smith, L.A.; Cornelius, V.; Warnock, A.; Bell, A.; Young, A.H. Effectiveness of Mood Stabilizers and Antipsychotics in the Maintenance Phase of Bipolar Disorder: A Systematic Review of Randomized Controlled Trials. Bipolar Disord. 2007, 9, 394–412. [Google Scholar] [CrossRef]
- Baldessarini, R.J.; Tondo, L.; Davis, P.; Pompili, M.; Goodwin, F.K.; Hennen, J. Decreased Risk of Suicides and Attempts during Long-Term Lithium Treatment: A Meta-Analytic Review. Bipolar Disord. 2006, 8, 625–639. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.-F.; Wang, Q.-G.; Han, B.-J.; William, C.P. Neuroprotective Effect and Cognitive Outcome of Chronic Lithium on Traumatic Brain Injury in Mice. Brain Res. Bull. 2010, 83, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, I.M.; Cuningham, J. Persisting Neurologic Sequelae of Lithium Carbonate Therapy. Arch. Neurol. 1983, 40, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Lazzara, C.A.; Kim, Y.-H. Potential Application of Lithium in Parkinson’s and Other Neurodegenerative Diseases. Front. Neurosci. 2015, 9, 403. [Google Scholar] [CrossRef] [Green Version]
- Aprahamian, I.; Santos, F.S.; dos Santos, B.; Talib, L.; Diniz, B.S.; Radanovic, M.; Gattaz, W.F.; Forlenza, O.V. Long-Term, Low-Dose Lithium Treatment Does Not Impair Renal Function in the Elderly: A 2-Year Randomized, Placebo-Controlled Trial Followed by Single-Blind Extension. J. Clin. Psychiatry 2014, 75, e672–678. [Google Scholar] [CrossRef] [PubMed]
- Baird-Gunning, J.; Lea-Henry, T.; Hoegberg, L.C.G.; Gosselin, S.; Roberts, D.M. Lithium Poisoning. J. Intensive Care Med. 2017, 32, 249–263. [Google Scholar] [CrossRef]
- Maddala, R.N.M.; Ashwal, A.J.; Rao, M.S.; Padmakumar, R. Chronic Lithium Intoxication: Varying Electrocardiogram Manifestations. Indian J. Pharmacol. 2017, 49, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Peet, M.; Pratt, J.P. Lithium. Current Status in Psychiatric Disorders. Drugs 1993, 46, 7–17. [Google Scholar] [CrossRef]
- Ferensztajn-Rochowiak, E.; Chłopocka-Woźniak, M.; Rybakowski, J.K. Ultra-Long-Term Lithium Therapy: All-Important Matters and a Case of Successful 50-Year Lithium Treatment. Rev. Bras. Psiquiatr. Sao Paulo Braz. 1999 2020. [Google Scholar] [CrossRef]
- Abou-Saleh, M.T.; Coppen, A. The Efficacy of Low-Dose Lithium: Clinical, Psychological and Biological Correlates. J. Psychiatr. Res. 1989, 23, 157–162. [Google Scholar] [CrossRef]
- Straten, G.; Saur, R.; Laske, C.; Gasser, T.; Annas, P.; Basun, H.; Leyhe, T. Influence of Lithium Treatment on GDNF Serum and CSF Concentrations in Patients with Early Alzheimer’s Disease. Curr. Alzheimer Res. 2011, 8, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Post, R.M. The New News about Lithium: An Underutilized Treatment in the United States. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2018, 43, 1174–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordano, S.; Darley-Usmar, V.; Zhang, J. Autophagy as an Essential Cellular Antioxidant Pathway in Neurodegenerative Disease. Redox Biol. 2014, 2, 82–90. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhou, Y.; Gao, Q.; Ping, D.; Wang, Y.; Wu, W.; Lin, X.; Fang, Y.; Zhang, J.; Shao, A. The Role of Exosomal MicroRNAs and Oxidative Stress in Neurodegenerative Diseases. Oxid. Med. Cell. Longev. 2020, 2020, 3232869. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Moreno, N.; Lane, J.D. Autophagy and Redox Homeostasis in Parkinson’s: A Crucial Balancing Act. Oxid. Med. Cell. Longev. 2020, 2020, 8865611. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Feng, Y.; Wang, X.-X.; Truong, D.; Wu, Y.-C. The Critical Role of SIRT1 in Parkinson’s Disease: Mechanism and Therapeutic Considerations. Aging Dis. 2020, 11, 1608–1622. [Google Scholar] [CrossRef] [PubMed]
- Dorszewska, J.; Kowalska, M.; Prendecki, M.; Piekut, T.; Kozłowska, J.; Kozubski, W. Oxidative Stress Factors in Parkinson’s Disease. Neural Regen. Res. 2021, 16, 1383–1391. [Google Scholar] [CrossRef]
- Cuevas, E.; Burks, S.; Raymick, J.; Robinson, B.; Gómez-Crisóstomo, N.P.; Escudero-Lourdes, C.; Lopez, A.G.G.; Chigurupati, S.; Hanig, J.; Ferguson, S.A.; et al. Tauroursodeoxycholic Acid (TUDCA) Is Neuroprotective in a Chronic Mouse Model of Parkinson’s Disease. Nutr. Neurosci. 2020, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Mat Taib, C.N.; Mustapha, M. MPTP-Induced Mouse Model of Parkinson’s Disease: A Promising Direction of Therapeutic Strategies. Bosn. J. Basic Med. Sci. 2020. [Google Scholar] [CrossRef]
- Franco-Iborra, S.; Vila, M.; Perier, C. The Parkinson Disease Mitochondrial Hypothesis: Where Are We At? Neurosci. Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2016, 22, 266–277. [Google Scholar] [CrossRef]
- Luque-Contreras, D.; Carvajal, K.; Toral-Rios, D.; Franco-Bocanegra, D.; Campos-Peña, V. Oxidative Stress and Metabolic Syndrome: Cause or Consequence of Alzheimer’s Disease? Oxid. Med. Cell. Longev. 2014, 2014, 497802. [Google Scholar] [CrossRef] [PubMed]
- Benilova, I.; Karran, E.; De Strooper, B. The Toxic Aβ Oligomer and Alzheimer’s Disease: An Emperor in Need of Clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Sochocka, M.; Koutsouraki, E.S.; Gasiorowski, K.; Leszek, J. Vascular Oxidative Stress and Mitochondrial Failure in the Pathobiology of Alzheimer’s Disease: A New Approach to Therapy. CNS Neurol. Disord. Drug Targets 2013, 12, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.T. Oxidative Stress and Mitochondrial Dysfunction-Linked Neurodegenerative Disorders. Neurol. Res. 2017, 39, 73–82. [Google Scholar] [CrossRef]
- Schapira, A.H.V. Mitochondria in the Aetiology and Pathogenesis of Parkinson’s Disease. Lancet Neurol. 2008, 7, 97–109. [Google Scholar] [CrossRef]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative Stress and Parkinson’s Disease. Front. Neuroanat. 2015, 9, 91. [Google Scholar] [CrossRef] [Green Version]
- Ames, B.N.; Shigenaga, M.K.; Hagen, T.M. Oxidants, Antioxidants, and the Degenerative Diseases of Aging. Proc. Natl. Acad. Sci. USA 1993, 90, 7915–7922. [Google Scholar] [CrossRef] [Green Version]
- Coyle, J.T.; Puttfarcken, P. Oxidative Stress, Glutamate, and Neurodegenerative Disorders. Science 1993, 262, 689–695. [Google Scholar] [CrossRef]
- Surace, M.J.; Block, M.L. Targeting Microglia-Mediated Neurotoxicity: The Potential of NOX2 Inhibitors. Cell. Mol. Life Sci. CMLS 2012, 69, 2409–2427. [Google Scholar] [CrossRef] [Green Version]
- Dias, V.; Junn, E.; Mouradian, M.M. The Role of Oxidative Stress in Parkinson’s Disease. J. Park. Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [Green Version]
- Puspita, L.; Chung, S.Y.; Shim, J.-W. Oxidative Stress and Cellular Pathologies in Parkinson’s Disease. Mol. Brain 2017, 10, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenner, P. Oxidative Stress in Parkinson’s Disease. Ann. Neurol. 2003, 53 (Suppl. 3). [Google Scholar] [CrossRef] [PubMed]
- Rahimmi, A.; Khosrobakhsh, F.; Izadpanah, E.; Moloudi, M.R.; Hassanzadeh, K. N-Acetylcysteine Prevents Rotenone-Induced Parkinson’s Disease in Rat: An Investigation into the Interaction of Parkin and Drp1 Proteins. Brain Res. Bull. 2015, 113, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Olanow, C.W.; Schapira, A.H.V.; LeWitt, P.A.; Kieburtz, K.; Sauer, D.; Olivieri, G.; Pohlmann, H.; Hubble, J. TCH346 as a Neuroprotective Drug in Parkinson’s Disease: A Double-Blind, Randomised, Controlled Trial. Lancet Neurol. 2006, 5, 1013–1020. [Google Scholar] [CrossRef]
- Lim, K.-L.; Tan, J.M.M. Role of the Ubiquitin Proteasome System in Parkinson’s Disease. BMC Biochem. 2007, 8 (Suppl. 1), S13. [Google Scholar] [CrossRef] [Green Version]
- Hassanzadeh, K.; Rahimmi, A. Oxidative Stress and Neuroinflammation in the Story of Parkinson’s Disease: Could Targeting These Pathways Write a Good Ending? J. Cell. Physiol. 2018, 234, 23–32. [Google Scholar] [CrossRef] [Green Version]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive Microglia Are Positive for HLA-DR in the Substantia Nigra of Parkinson’s and Alzheimer’s Disease Brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef]
- Anglade, P.; Vyas, S.; Javoy-Agid, F.; Herrero, M.T.; Michel, P.P.; Marquez, J.; Mouatt-Prigent, A.; Ruberg, M.; Hirsch, E.C.; Agid, Y. Apoptosis and Autophagy in Nigral Neurons of Patients with Parkinson’s Disease. Histol. Histopathol. 1997, 12, 25–31. [Google Scholar]
- Gupta, A.; Dawson, V.L.; Dawson, T.M. What Causes Cell Death in Parkinson’s Disease? Ann. Neurol. 2008, 64 (Suppl. 2), S3–S15. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms Underlying Inflammation in Neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [Green Version]
- Ramesh, G.; MacLean, A.G.; Philipp, M.T. Cytokines and Chemokines at the Crossroads of Neuroinflammation, Neurodegeneration, and Neuropathic Pain. Mediat. Inflamm. 2013, 2013, 480739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tufekci, K.U.; Meuwissen, R.; Genc, S.; Genc, K. Inflammation in Parkinson’s Disease. Adv. Protein Chem. Struct. Biol. 2012, 88, 69–132. [Google Scholar] [CrossRef] [PubMed]
- Rocha, N.P.; de Miranda, A.S.; Teixeira, A.L. Insights into Neuroinflammation in Parkinson’s Disease: From Biomarkers to Anti-Inflammatory Based Therapies. BioMed Res. Int. 2015, 2015, 628192. [Google Scholar] [CrossRef] [PubMed]
- Shih, R.-H.; Wang, C.-Y.; Yang, C.-M. NF-KappaB Signaling Pathways in Neurological Inflammation: A Mini Review. Front. Mol. Neurosci. 2015, 8, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mogi, M.; Togari, A.; Kondo, T.; Mizuno, Y.; Komure, O.; Kuno, S.; Ichinose, H.; Nagatsu, T. Caspase Activities and Tumor Necrosis Factor Receptor R1 (P55) Level Are Elevated in the Substantia Nigra from Parkinsonian Brain. J. Neural Transm. Vienna Austria 1996 2000, 107, 335–341. [Google Scholar] [CrossRef]
- Shimoji, M.; Pagan, F.; Healton, E.B.; Mocchetti, I. CXCR4 and CXCL12 Expression Is Increased in the Nigro-Striatal System of Parkinson’s Disease. Neurotox. Res. 2009, 16, 318–328. [Google Scholar] [CrossRef]
- Yacoubian, T.A.; Standaert, D.G. Targets for Neuroprotection in Parkinson’s Disease. Biochim. Biophys. Acta 2009, 1792, 676–687. [Google Scholar] [CrossRef]
- Gardet, A.; Benita, Y.; Li, C.; Sands, B.E.; Ballester, I.; Stevens, C.; Korzenik, J.R.; Rioux, J.D.; Daly, M.J.; Xavier, R.J.; et al. LRRK2 Is Involved in the IFN-Gamma Response and Host Response to Pathogens. J. Immunol. Baltim. Md 1950 2010, 185, 5577–5585. [Google Scholar] [CrossRef] [Green Version]
- Russo, I.; Berti, G.; Plotegher, N.; Bernardo, G.; Filograna, R.; Bubacco, L.; Greggio, E. Leucine-Rich Repeat Kinase 2 Positively Regulates Inflammation and down-Regulates NF-ΚB P50 Signaling in Cultured Microglia Cells. J. Neuroinflamm. 2015, 12, 230. [Google Scholar] [CrossRef] [Green Version]
- López de Maturana, R.; Lang, V.; Zubiarrain, A.; Sousa, A.; Vázquez, N.; Gorostidi, A.; Águila, J.; López de Munain, A.; Rodríguez, M.; Sánchez-Pernaute, R. Mutations in LRRK2 Impair NF-ΚB Pathway in IPSC-Derived Neurons. J. Neuroinflammation 2016, 13, 295. [Google Scholar] [CrossRef] [Green Version]
- Moussaud, S.; Jones, D.R.; Moussaud-Lamodière, E.L.; Delenclos, M.; Ross, O.A.; McLean, P.J. Alpha-Synuclein and Tau: Teammates in Neurodegeneration? Mol. Neurodegener. 2014, 9, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerreiro, P.S.; Gerhardt, E.; Lopes da Fonseca, T.; Bähr, M.; Outeiro, T.F.; Eckermann, K. LRRK2 Promotes Tau Accumulation, Aggregation and Release. Mol. Neurobiol. 2016, 53, 3124–3135. [Google Scholar] [CrossRef] [PubMed]
- Russo, I.; Bubacco, L.; Greggio, E. LRRK2 and Neuroinflammation: Partners in Crime in Parkinson’s Disease? J. Neuroinflammation 2014, 11, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guttuso, T.; Andrzejewski, K.L.; Lichter, D.G.; Andersen, J.K. Targeting Kinases in Parkinson’s Disease: A Mechanism Shared by LRRK2, Neurotrophins, Exenatide, Urate, Nilotinib and Lithium. J. Neurol. Sci. 2019, 402, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Helton, T.D.; Otsuka, T.; Lee, M.-C.; Mu, Y.; Ehlers, M.D. Pruning and Loss of Excitatory Synapses by the Parkin Ubiquitin Ligase. Proc. Natl. Acad. Sci. USA 2008, 105, 19492–19497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.W.; Chuang, D.M. Long Term Lithium Treatment Suppresses P53 and Bax Expression but Increases Bcl-2 Expression. A Prominent Role in Neuroprotection against Excitotoxicity. J. Biol. Chem. 1999, 274, 6039–6042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.-W.; Qin, Z.-H.; Ren, M.; Kanai, H.; Chalecka-Franaszek, E.; Leeds, P.; Chuang, D.-M. Regulation of C-Jun N-Terminal Kinase, P38 Kinase and AP-1 DNA Binding in Cultured Brain Neurons: Roles in Glutamate Excitotoxicity and Lithium Neuroprotection. J. Neurochem. 2003, 84, 566–575. [Google Scholar] [CrossRef]
- Loh, K.M.; van Amerongen, R.; Nusse, R. Generating Cellular Diversity and Spatial Form: Wnt Signaling and the Evolution of Multicellular Animals. Dev. Cell 2016, 38, 643–655. [Google Scholar] [CrossRef] [Green Version]
- Oren, O.; Smith, B.D. Eliminating Cancer Stem Cells by Targeting Embryonic Signaling Pathways. Stem Cell Rev. 2017, 13, 17–23. [Google Scholar] [CrossRef]
- Al-Harthi, L. Wnt/β-Catenin and Its Diverse Physiological Cell Signaling Pathways in Neurodegenerative and Neuropsychiatric Disorders. J. Neuroimmune Pharmacol. 2012, 7, 725–730. [Google Scholar] [CrossRef]
- Marchetti, B.; Pluchino, S. Wnt Your Brain Be Inflamed? Yes, It Wnt! Trends Mol. Med. 2013, 19, 144–156. [Google Scholar] [CrossRef] [Green Version]
- Lecarpentier, Y.; Claes, V.; Duthoit, G.; Hébert, J.-L. Circadian Rhythms, Wnt/Beta-Catenin Pathway and PPAR Alpha/Gamma Profiles in Diseases with Primary or Secondary Cardiac Dysfunction. Front. Physiol. 2014, 5, 429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecarpentier, Y.; Vallée, A. Opposite Interplay between PPAR Gamma and Canonical Wnt/Beta-Catenin Pathway in Amyotrophic Lateral Sclerosis. Front. Neurol. 2016, 7, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallée, A.; Lecarpentier, Y. Alzheimer Disease: Crosstalk between the Canonical Wnt/Beta-Catenin Pathway and PPARs Alpha and Gamma. Front. Neurosci. 2016, 10, 459. [Google Scholar] [CrossRef] [Green Version]
- He, T.C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of C-MYC as a Target of the APC Pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef]
- Shtutman, M.; Zhurinsky, J.; Simcha, I.; Albanese, C.; D’Amico, M.; Pestell, R.; Ben-Ze’ev, A. The Cyclin D1 Gene Is a Target of the Beta-Catenin/LEF-1 Pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 5522–5527. [Google Scholar] [CrossRef] [Green Version]
- Angers, S.; Moon, R.T. Proximal Events in Wnt Signal Transduction. Nat. Rev. Mol. Cell Biol. 2009. [Google Scholar] [CrossRef] [PubMed]
- Sharma, C.; Pradeep, A.; Wong, L.; Rana, A.; Rana, B. Peroxisome Proliferator-Activated Receptor Gamma Activation Can Regulate Beta-Catenin Levels via a Proteasome-Mediated and Adenomatous Polyposis Coli-Independent Pathway. J. Biol. Chem. 2004, 279, 35583–35594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosi, M.C.; Luccarini, I.; Grossi, C.; Fiorentini, A.; Spillantini, M.G.; Prisco, A.; Scali, C.; Gianfriddo, M.; Caricasole, A.; Terstappen, G.C.; et al. Increased Dickkopf-1 Expression in Transgenic Mouse Models of Neurodegenerative Disease. J. Neurochem. 2010, 112, 1539–1551. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Nusse, R. Wnt/β-Catenin Signaling and Disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inestrosa, N.C.; Montecinos-Oliva, C.; Fuenzalida, M. Wnt Signaling: Role in Alzheimer Disease and Schizophrenia. J. Neuroimmune Pharmacol. Off. J. Soc. NeuroImmune Pharmacol. 2012, 7, 788–807. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.-N. Interactions between TGF-Β1, Canonical WNT/β-Catenin Pathway and PPAR γ in Radiation-Induced Fibrosis. Oncotarget 2017, 8, 90579–90604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallée, A.; Lecarpentier, Y.; Vallée, J.-N. Hypothesis of Opposite Interplay Between the Canonical WNT/Beta-Catenin Pathway and PPAR Gamma in Primary Central Nervous System Lymphomas. Curr. Issues Mol. Biol. 2019, 31, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Aberle, H.; Bauer, A.; Stappert, J.; Kispert, A.; Kemler, R. β-Catenin Is a Target for the Ubiquitin–Proteasome Pathway. EMBO J. 1997, 16, 3797–3804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Pan, W. GSK3: A Multifaceted Kinase in Wnt Signaling. Trends Biochem. Sci. 2010, 35, 161–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hur, E.-M.; Zhou, F.-Q. GSK3 Signalling in Neural Development. Nat. Rev. Neurosci. 2010, 11, 539–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambacher, K.K.; Pitzul, K.B.; Karajgikar, M.; Hamilton, A.; Ferguson, S.S.; Cregan, S.P. The JNK- and AKT/GSK3β- Signaling Pathways Converge to Regulate Puma Induction and Neuronal Apoptosis Induced by Trophic Factor Deprivation. PLoS ONE 2012, 7, e46885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orellana, A.M.M.; Vasconcelos, A.R.; Leite, J.A.; de Sá Lima, L.; Andreotti, D.Z.; Munhoz, C.D.; Kawamoto, E.M.; Scavone, C. Age-Related Neuroinflammation and Changes in AKT-GSK-3β and WNT/ β-CATENIN Signaling in Rat Hippocampus. Aging 2015, 7, 1094–1111. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Arenas, E. Emerging Roles of Wnts in the Adult Nervous System. Nat. Rev. Neurosci. 2010, 11, 77–86. [Google Scholar] [CrossRef]
- Berwick, D.C.; Harvey, K. LRRK2 Signaling Pathways: The Key to Unlocking Neurodegeneration? Trends Cell Biol. 2011, 21, 257–265. [Google Scholar] [CrossRef]
- L’episcopo, F.; Serapide, M.F.; Tirolo, C.; Testa, N.; Caniglia, S.; Morale, M.C.; Pluchino, S.; Marchetti, B. A Wnt1 Regulated Frizzled-1/β-Catenin Signaling Pathway as a Candidate Regulatory Circuit Controlling Mesencephalic Dopaminergic Neuron-Astrocyte Crosstalk: Therapeutical Relevance for Neuron Survival and Neuroprotection. Mol. Neurodegener. 2011, 6, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Häbig, K.; Walter, M.; Poths, S.; Riess, O.; Bonin, M. RNA Interference of LRRK2-Microarray Expression Analysis of a Parkinson’s Disease Key Player. Neurogenetics 2008, 9, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Libro, R.; Bramanti, P.; Mazzon, E. The Role of the Wnt Canonical Signaling in Neurodegenerative Diseases. Life Sci. 2016, 158, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Zu, G.; Zhang, X.; Wang, X.; Li, S.; Gong, X.; Liang, Z.; Zhao, J. Neuroprotective Effects of Ginsenoside Rg1 through the Wnt/β-Catenin Signaling Pathway in Both in Vivo and in Vitro Models of Parkinson’s Disease. Neuropharmacology 2016, 101, 480–489. [Google Scholar] [CrossRef]
- L’Episcopo, F.; Tirolo, C.; Testa, N.; Caniglia, S.; Morale, M.C.; Deleidi, M.; Serapide, M.F.; Pluchino, S.; Marchetti, B. Plasticity of Subventricular Zone Neuroprogenitors in MPTP (1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine) Mouse Model of Parkinson’s Disease Involves Cross Talk between Inflammatory and Wnt/β-Catenin Signaling Pathways: Functional Consequences for Neuroprotection and Repair. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 2062–2085. [Google Scholar] [CrossRef] [Green Version]
- Barthel, A.; Schmoll, D.; Unterman, T.G. FoxO Proteins in Insulin Action and Metabolism. Trends Endocrinol. Metab. TEM 2005, 16, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.; Ambrogini, E.; Han, L.; Manolagas, S.C.; Jilka, R.L. Increased Lipid Oxidation Causes Oxidative Stress, Increased Peroxisome Proliferator-Activated Receptor-Gamma Expression, and Diminished pro-Osteogenic Wnt Signaling in the Skeleton. J. Biol. Chem. 2009, 284, 27438–27448. [Google Scholar] [CrossRef] [Green Version]
- Essers, M.A.G.; de Vries-Smits, L.M.M.; Barker, N.; Polderman, P.E.; Burgering, B.M.T.; Korswagen, H.C. Functional Interaction between Beta-Catenin and FOXO in Oxidative Stress Signaling. Science 2005, 308, 1181–1184. [Google Scholar] [CrossRef]
- Hoogeboom, D.; Essers, M.A.G.; Polderman, P.E.; Voets, E.; Smits, L.M.M.; Burgering, B.M.T. Interaction of FOXO with Beta-Catenin Inhibits Beta-Catenin/T Cell Factor Activity. J. Biol. Chem. 2008, 283, 9224–9230. [Google Scholar] [CrossRef] [Green Version]
- Reif, K.; Burgering, B.M.; Cantrell, D.A. Phosphatidylinositol 3-Kinase Links the Interleukin-2 Receptor to Protein Kinase B and P70 S6 Kinase. J. Biol. Chem. 1997, 272, 14426–14433. [Google Scholar] [CrossRef] [Green Version]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt Promotes Cell Survival by Phosphorylating and Inhibiting a Forkhead Transcription Factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef] [Green Version]
- Stahl, M.; Dijkers, P.F.; Kops, G.J.P.L.; Lens, S.M.A.; Coffer, P.J.; Burgering, B.M.T.; Medema, R.H. The Forkhead Transcription Factor FoxO Regulates Transcription of P27Kip1 and Bim in Response to IL-2. J. Immunol. Baltim. Md 1950 2002, 168, 5024–5031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, M.; Fernandez de Mattos, S.; van der Horst, A.; Klompmaker, R.; Kops, G.J.P.L.; Lam, E.W.-F.; Burgering, B.M.T.; Medema, R.H. Cell Cycle Inhibition by FoxO Forkhead Transcription Factors Involves Downregulation of Cyclin D. Mol. Cell. Biol. 2002, 22, 7842–7852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández de Mattos, S.; Essafi, A.; Soeiro, I.; Pietersen, A.M.; Birkenkamp, K.U.; Edwards, C.S.; Martino, A.; Nelson, B.H.; Francis, J.M.; Jones, M.C.; et al. FoxO3a and BCR-ABL Regulate Cyclin D2 Transcription through a STAT5/BCL6-Dependent Mechanism. Mol. Cell. Biol. 2004, 24, 10058–10071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manolopoulos, K.N.; Klotz, L.-O.; Korsten, P.; Bornstein, S.R.; Barthel, A. Linking Alzheimer’s Disease to Insulin Resistance: The FoxO Response to Oxidative Stress. Mol. Psychiatry 2010, 15, 1046–1052. [Google Scholar] [CrossRef] [Green Version]
- Shang, Y.C.; Chong, Z.Z.; Hou, J.; Maiese, K. Wnt1, FoxO3a, and NF-KappaB Oversee Microglial Integrity and Activation during Oxidant Stress. Cell. Signal. 2010, 22, 1317–1329. [Google Scholar] [CrossRef] [Green Version]
- Halleskog, C.; Mulder, J.; Dahlström, J.; Mackie, K.; Hortobágyi, T.; Tanila, H.; Kumar Puli, L.; Färber, K.; Harkany, T.; Schulte, G. WNT Signaling in Activated Microglia Is Proinflammatory. Glia 2011, 59, 119–131. [Google Scholar] [CrossRef] [Green Version]
- Ma, B.; Hottiger, M.O. Crosstalk between Wnt/β-Catenin and NF-ΚB Signaling Pathway during Inflammation. Front. Immunol. 2016, 7, 378. [Google Scholar] [CrossRef]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFκB System. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 227–241. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Miller, S.A.; Wang, H.-Y.; Xia, W.; Wen, Y.; Zhou, B.P.; Li, Y.; Lin, S.-Y.; Hung, M.-C. Beta-Catenin Interacts with and Inhibits NF-Kappa B in Human Colon and Breast Cancer. Cancer Cell 2002, 2, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Liao, Y.; Ma, K.; Wang, Y.; Zhang, G.; Yang, R.; Deng, J. PI3K Is Required for the Physical Interaction and Functional Inhibition of NF-ΚB by β-Catenin in Colorectal Cancer Cells. Biochem. Biophys. Res. Commun. 2013, 434, 760–766. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Rehani, K.; Jope, R.S.; Michalek, S.M. Toll-like Receptor-Mediated Cytokine Production Is Differentially Regulated by Glycogen Synthase Kinase 3. Nat. Immunol. 2005, 6, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Manicassamy, S.; Reizis, B.; Ravindran, R.; Nakaya, H.; Salazar-Gonzalez, R.M.; Wang, Y.-C.; Pulendran, B. Activation of Beta-Catenin in Dendritic Cells Regulates Immunity versus Tolerance in the Intestine. Science 2010, 329, 849–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.H.; Song, J.S.; Yu, J.M.; Yu, S.S.; Choi, S.J.; Kim, D.H.; Jung, J.S. Differential Effect of NF-KappaB Activity on Beta-Catenin/Tcf Pathway in Various Cancer Cells. FEBS Lett. 2008, 582, 616–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fliniaux, I.; Mikkola, M.L.; Lefebvre, S.; Thesleff, I. Identification of Dkk4 as a Target of Eda-A1/Edar Pathway Reveals an Unexpected Role of Ectodysplasin as Inhibitor of Wnt Signalling in Ectodermal Placodes. Dev. Biol. 2008, 320, 60–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoeflich, K.P.; Luo, J.; Rubie, E.A.; Tsao, M.S.; Jin, O.; Woodgett, J.R. Requirement for Glycogen Synthase Kinase-3beta in Cell Survival and NF-KappaB Activation. Nature 2000, 406, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Michalek, S.M.; Jope, R.S. Innate and Adaptive Immune Responses Regulated by Glycogen Synthase Kinase-3 (GSK3). Trends Immunol. 2010, 31, 24–31. [Google Scholar] [CrossRef] [Green Version]
- Lutgen, V.; Narasipura, S.D.; Sharma, A.; Min, S.; Al-Harthi, L. β-Catenin Signaling Positively Regulates Glutamate Uptake and Metabolism in Astrocytes. J. Neuroinflammation 2016, 13, 242. [Google Scholar] [CrossRef] [Green Version]
- Narasipura, S.D.; Henderson, L.J.; Fu, S.W.; Chen, L.; Kashanchi, F.; Al-Harthi, L. Role of β-Catenin and TCF/LEF Family Members in Transcriptional Activity of HIV in Astrocytes. J. Virol. 2012, 86, 1911–1921. [Google Scholar] [CrossRef] [Green Version]
- Lecarpentier, Y.; Schussler, O.; Hébert, J.-L.; Vallée, A. Molecular Mechanisms Underlying the Circadian Rhythm of Blood Pressure in Normotensive Subjects. Curr. Hypertens. Rep. 2020, 22, 50. [Google Scholar] [CrossRef]
- Giese, K.P. GSK-3: A Key Player in Neurodegeneration and Memory. IUBMB Life 2009, 61, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Alda, M.; Priller, J.; Young, L.T.; International Group For The Study Of Lithium Treated Patients (IGSLI). Implications of the Neuroprotective Effects of Lithium for the Treatment of Bipolar and Neurodegenerative Disorders. Pharmacopsychiatry 2003, 36 (Suppl. 3), S250–S254. [Google Scholar] [CrossRef] [PubMed]
- Rowe, M.K.; Chuang, D.-M. Lithium Neuroprotection: Molecular Mechanisms and Clinical Implications. Expert Rev. Mol. Med. 2004, 6, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Rowe, M.K.; Wiest, C.; Chuang, D.-M. GSK-3 Is a Viable Potential Target for Therapeutic Intervention in Bipolar Disorder. Neurosci. Biobehav. Rev. 2007, 31, 920–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alural, B.; Ozerdem, A.; Allmer, J.; Genc, K.; Genc, S. Lithium Protects against Paraquat Neurotoxicity by NRF2 Activation and MiR-34a Inhibition in SH-SY5Y Cells. Front. Cell. Neurosci. 2015, 9, 209. [Google Scholar] [CrossRef] [Green Version]
- Beaulieu, J.-M.; Gainetdinov, R.R.; Caron, M.G. The Akt-GSK-3 Signaling Cascade in the Actions of Dopamine. Trends Pharmacol. Sci. 2007, 28, 166–172. [Google Scholar] [CrossRef]
- Gould, T.D.; Chen, G.; Manji, H.K. In Vivo Evidence in the Brain for Lithium Inhibition of Glycogen Synthase Kinase-3. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2004, 29, 32–38. [Google Scholar] [CrossRef]
- O’Brien, W.T.; Harper, A.D.; Jové, F.; Woodgett, J.R.; Maretto, S.; Piccolo, S.; Klein, P.S. Glycogen Synthase Kinase-3beta Haploinsufficiency Mimics the Behavioral and Molecular Effects of Lithium. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 6791–6798. [Google Scholar] [CrossRef] [Green Version]
- Jope, R.S.; Johnson, G.V.W. The Glamour and Gloom of Glycogen Synthase Kinase-3. Trends Biochem. Sci. 2004, 29, 95–102. [Google Scholar] [CrossRef]
- Marmol, F. Lithium: Bipolar Disorder and Neurodegenerative Diseases Possible Cellular Mechanisms of the Therapeutic Effects of Lithium. Prog. Neuropsychopharmacol. Biol. Psychiatry 2008, 32, 1761–1771. [Google Scholar] [CrossRef]
- Gould, T.D.; Einat, H.; O’Donnell, K.C.; Picchini, A.M.; Schloesser, R.J.; Manji, H.K. Beta-Catenin Overexpression in the Mouse Brain Phenocopies Lithium-Sensitive Behaviors. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2007, 32, 2173–2183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gould, T.D.; O’Donnell, K.C.; Picchini, A.M.; Dow, E.R.; Chen, G.; Manji, H.K. Generation and Behavioral Characterization of Beta-Catenin Forebrain-Specific Conditional Knock-out Mice. Behav. Brain Res. 2008, 189, 117–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gould, T.D.; Quiroz, J.A.; Singh, J.; Zarate, C.A.; Manji, H.K. Emerging Experimental Therapeutics for Bipolar Disorder: Insights from the Molecular and Cellular Actions of Current Mood Stabilizers. Mol. Psychiatry 2004, 9, 734–755. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-H.; Rane, A.; Lussier, S.; Andersen, J.K. Lithium Protects against Oxidative Stress-Mediated Cell Death in α-Synuclein-Overexpressing in Vitro and in Vivo Models of Parkinson’s Disease. J. Neurosci. Res. 2011, 89, 1666–1675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieu, C.A.; Dewey, C.M.; Chinta, S.J.; Rane, A.; Rajagopalan, S.; Batir, S.; Kim, Y.-H.; Andersen, J.K. Lithium Prevents Parkinsonian Behavioral and Striatal Phenotypes in an Aged Parkin Mutant Transgenic Mouse Model. Brain Res. 2014, 1591, 111–117. [Google Scholar] [CrossRef] [Green Version]
- Hou, L.; Xiong, N.; Liu, L.; Huang, J.; Han, C.; Zhang, G.; Li, J.; Xu, X.; Lin, Z.; Wang, T. Lithium Protects Dopaminergic Cells from Rotenone Toxicity via Autophagy Enhancement. BMC Neurosci. 2015, 16, 82. [Google Scholar] [CrossRef] [Green Version]
- Qi, L.; Tang, Y.; He, W.; Pan, H.; Jiang, W.; Wang, L.; Deng, W. Lithium Chloride Promotes Neuronal Differentiation of Rat Neural Stem Cells and Enhances Neural Regeneration in Parkinson’s Disease Model. Cytotechnology 2017, 69, 277–287. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Liu, H.; Cheng, J.; Zhu, Y.; Xiao, Q.; Bai, Y.; Tao, J. Neuroprotective Effects of Lithium on a Chronic MPTP Mouse Model of Parkinson’s Disease via Regulation of A-synuclein Methylation. Mol. Med. Rep. 2019, 19, 4989–4997. [Google Scholar] [CrossRef]
- Wen, J.; Sawmiller, D.; Wheeldon, B.; Tan, J. A Review for Lithium: Pharmacokinetics, Drug Design, and Toxicity. CNS Neurol. Disord. Drug Targets 2019, 18, 769–778. [Google Scholar] [CrossRef]
- Freland, L.; Beaulieu, J.-M. Inhibition of GSK3 by Lithium, from Single Molecules to Signaling Networks. Front. Mol. Neurosci. 2012, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Noble, W.; Planel, E.; Zehr, C.; Olm, V.; Meyerson, J.; Suleman, F.; Gaynor, K.; Wang, L.; LaFrancois, J.; Feinstein, B.; et al. Inhibition of Glycogen Synthase Kinase-3 by Lithium Correlates with Reduced Tauopathy and Degeneration in Vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 6990–6995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/Beta-Catenin Signaling: Components, Mechanisms, and Diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Cen, L.; Qu, S.; Wei, L.; Mo, M.; Feng, J.; Sun, C.; Xiao, Y.; Luo, Q.; Li, S.; et al. Enhancing Beta-Catenin Activity via GSK3beta Inhibition Protects PC12 Cells against Rotenone Toxicity through Nurr1 Induction. PLoS ONE 2016, 11, e0152931. [Google Scholar] [CrossRef] [PubMed]
- Aarsland, D.; Creese, B.; Politis, M.; Chaudhuri, K.R.; Ffytche, D.H.; Weintraub, D.; Ballard, C. Cognitive Decline in Parkinson Disease. Nat. Rev. Neurol. 2017, 13, 217–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.; Ryder, J.; Li, B.; Wu, X.; Fox, N.; Solenberg, P.; Brune, K.; Paul, S.; Zhou, Y.; Liu, F.; et al. Lithium, a Common Drug for Bipolar Disorder Treatment, Regulates Amyloid-Beta Precursor Protein Processing. Biochemistry 2004, 43, 6899–6908. [Google Scholar] [CrossRef]
- Xilouri, M.; Brekk, O.R.; Stefanis, L. Autophagy and Alpha-Synuclein: Relevance to Parkinson’s Disease and Related Synucleopathies. Mov. Disord. Off. J. Mov. Disord. Soc. 2016, 31, 178–192. [Google Scholar] [CrossRef]
- Lipton, J.O.; Sahin, M. The Neurology of MTOR. Neuron 2014, 84, 275–291. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired Degradation of Mutant Alpha-Synuclein by Chaperone-Mediated Autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef]
- Motoi, Y.; Shimada, K.; Ishiguro, K.; Hattori, N. Lithium and Autophagy. ACS Chem. Neurosci. 2014, 5, 434–442. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Krishna, G.; Imarisio, S.; Saiki, S.; O’Kane, C.J.; Rubinsztein, D.C. A Rational Mechanism for Combination Treatment of Huntington’s Disease Using Lithium and Rapamycin. Hum. Mol. Genet. 2008, 17, 170–178. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Wang, H.; Wang, S.; Xu, M.; Liu, M.; Liao, M.; Frank, J.A.; Adhikari, S.; Bower, K.A.; Shi, X.; et al. GSK3β Signaling Is Involved in Ultraviolet B-Induced Activation of Autophagy in Epidermal Cells. Int. J. Oncol. 2012, 41, 1782–1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, T.; Eisenberg, F. Purification, Structure, and Catalytic Properties of L-Myo-Inositol-1-Phosphate Synthase from Rat Testis. J. Biol. Chem. 1980, 255, 8458–8464. [Google Scholar] [CrossRef]
- Sarkar, S.; Floto, R.A.; Berger, Z.; Imarisio, S.; Cordenier, A.; Pasco, M.; Cook, L.J.; Rubinsztein, D.C. Lithium Induces Autophagy by Inhibiting Inositol Monophosphatase. J. Cell Biol. 2005, 170, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Criollo, A.; Vicencio, J.M.; Tasdemir, E.; Maiuri, M.C.; Lavandero, S.; Kroemer, G. The Inositol Trisphosphate Receptor in the Control of Autophagy. Autophagy 2007, 3, 350–353. [Google Scholar] [CrossRef] [Green Version]
- Cannon, J.R.; Tapias, V.; Na, H.M.; Honick, A.S.; Drolet, R.E.; Greenamyre, J.T. A Highly Reproducible Rotenone Model of Parkinson’s Disease. Neurobiol. Dis. 2009, 34, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Lazzara, C.A.; Riley, R.R.; Rane, A.; Andersen, J.K.; Kim, Y.-H. The Combination of Lithium and L-Dopa/Carbidopa Reduces MPTP-Induced Abnormal Involuntary Movements (AIMs) via Calpain-1 Inhibition in a Mouse Model: Relevance for Parkinson׳s Disease Therapy. Brain Res. 2015, 1622, 127–136. [Google Scholar] [CrossRef] [Green Version]
- Xiong, N.; Jia, M.; Chen, C.; Xiong, J.; Zhang, Z.; Huang, J.; Hou, L.; Yang, H.; Cao, X.; Liang, Z.; et al. Potential Autophagy Enhancers Attenuate Rotenone-Induced Toxicity in SH-SY5Y. Neuroscience 2011, 199, 292–302. [Google Scholar] [CrossRef]
- Li, X.; Chen, X.; Zhao, K.; Bai, L.; Zhang, H.; Zhou, X. Therapeutic Effects of Valproate Combined with Lithium Carbonate on MPTP-Induced Parkinsonism in Mice: Possible Mediation through Enhanced Autophagy. Int. J. Neurosci. 2013, 123, 73–79. [Google Scholar] [CrossRef]
- Ferrucci, M.; Pasquali, L.; Ruggieri, S.; Paparelli, A.; Fornai, F. Alpha-Synuclein and Autophagy as Common Steps in Neurodegeneration. Parkinsonism Relat. Disord. 2008, 14 (Suppl. 2), S180–S184. [Google Scholar] [CrossRef]
- Mao, Z.; Liu, L.; Zhang, R.; Li, X. Lithium Reduces FoxO3a Transcriptional Activity by Decreasing Its Intracellular Content. Biol. Psychiatry 2007, 62, 1423–1430. [Google Scholar] [CrossRef]
- Shao, L.; Young, L.T.; Wang, J.-F. Chronic Treatment with Mood Stabilizers Lithium and Valproate Prevents Excitotoxicity by Inhibiting Oxidative Stress in Rat Cerebral Cortical Cells. Biol. Psychiatry 2005, 58, 879–884. [Google Scholar] [CrossRef] [PubMed]
- De Vasconcellos, A.P.S.; Nieto, F.B.; Crema, L.M.; Diehl, L.A.; de Almeida, L.M.; Prediger, M.E.; da Rocha, E.R.; Dalmaz, C. Chronic Lithium Treatment Has Antioxidant Properties but Does Not Prevent Oxidative Damage Induced by Chronic Variate Stress. Neurochem. Res. 2006, 31, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Shao, L.; Young, L.T.; Wang, J.-F. Role of Glutathione in Neuroprotective Effects of Mood Stabilizing Drugs Lithium and Valproate. Neuroscience 2007, 144, 1447–1453. [Google Scholar] [CrossRef] [PubMed]
- Frey, B.N.; Andreazza, A.C.; Kunz, M.; Gomes, F.A.; Quevedo, J.; Salvador, M.; Gonçalves, C.A.; Kapczinski, F. Increased Oxidative Stress and DNA Damage in Bipolar Disorder: A Twin-Case Report. Prog. Neuropsychopharmacol. Biol. Psychiatry 2007, 31, 283–285. [Google Scholar] [CrossRef] [PubMed]
- Machado-Vieira, R.; Andreazza, A.C.; Viale, C.I.; Zanatto, V.; Cereser, V.; da Silva Vargas, R.; Kapczinski, F.; Portela, L.V.; Souza, D.O.; Salvador, M.; et al. Oxidative Stress Parameters in Unmedicated and Treated Bipolar Subjects during Initial Manic Episode: A Possible Role for Lithium Antioxidant Effects. Neurosci. Lett. 2007, 421, 33–36. [Google Scholar] [CrossRef]
- Rodríguez de la Concepción, M.L.; Yubero, P.; Iglesias, R.; Giralt, M.; Villarroya, F. Lithium Inhibits Brown Adipocyte Differentiation. FEBS Lett. 2005, 579, 1670–1674. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-H.; Olson, P.; Evans, R.M. Minireview: Lipid Metabolism, Metabolic Diseases, and Peroxisome Proliferator-Activated Receptors. Endocrinology 2003, 144, 2201–2207. [Google Scholar] [CrossRef] [Green Version]
- Marx, N.; Duez, H.; Fruchart, J.-C.; Staels, B. Peroxisome Proliferator-Activated Receptors and Atherogenesis: Regulators of Gene Expression in Vascular Cells. Circ. Res. 2004, 94, 1168–1178. [Google Scholar] [CrossRef]
- Cunard, R.; Ricote, M.; DiCampli, D.; Archer, D.C.; Kahn, D.A.; Glass, C.K.; Kelly, C.J. Regulation of Cytokine Expression by Ligands of Peroxisome Proliferator Activated Receptors. J. Immunol. Baltim. Md 1950 2002, 168, 2795–2802. [Google Scholar] [CrossRef] [Green Version]
- Ricote, M.; Li, A.C.; Willson, T.M.; Kelly, C.J.; Glass, C.K. The Peroxisome Proliferator-Activated Receptor-Gamma Is a Negative Regulator of Macrophage Activation. Nature 1998, 391, 79–82. [Google Scholar] [CrossRef]
- Giannini, S.; Serio, M.; Galli, A. Pleiotropic Effects of Thiazolidinediones: Taking a Look beyond Antidiabetic Activity. J. Endocrinol. Invest. 2004, 27, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y. Crosstalk Between Peroxisome Proliferator-Activated Receptor Gamma and the Canonical WNT/β-Catenin Pathway in Chronic Inflammation and Oxidative Stress During Carcinogenesis. Front. Immunol. 2018, 9, 745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallée, A.; Vallée, J.-N.; Lecarpentier, Y. PPARγ Agonists: Potential Treatment for Autism Spectrum Disorder by Inhibiting the Canonical WNT/β-Catenin Pathway. Mol. Psychiatry 2018. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.-N. Thermodynamics in Gliomas: Interactions between the Canonical WNT/Beta-Catenin Pathway and PPAR Gamma. Front. Physiol. 2017, 8, 352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.-N. Demyelination in Multiple Sclerosis: Reprogramming Energy Metabolism and Potential PPARγ Agonist Treatment Approaches. Int. J. Mol. Sci. 2018, 19, 1212. [Google Scholar] [CrossRef] [Green Version]
- Park, K.S.; Lee, R.D.; Kang, S.-K.; Han, S.Y.; Park, K.L.; Yang, K.H.; Song, Y.S.; Park, H.J.; Lee, Y.M.; Yun, Y.P.; et al. Neuronal Differentiation of Embryonic Midbrain Cells by Upregulation of Peroxisome Proliferator-Activated Receptor-Gamma via the JNK-Dependent Pathway. Exp. Cell Res. 2004, 297, 424–433. [Google Scholar] [CrossRef]
- Vallée, A.; Vallée, J.-N. Warburg Effect Hypothesis in Autism Spectrum Disorders. Mol. Brain 2018, 11, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimes, C.A.; Jope, R.S. The Multifaceted Roles of Glycogen Synthase Kinase 3beta in Cellular Signaling. Prog. Neurobiol. 2001, 65, 391–426. [Google Scholar] [CrossRef]
- Jeon, M.; Rahman, N.; Kim, Y.-S. Wnt/β-Catenin Signaling Plays a Distinct Role in Methyl Gallate-Mediated Inhibition of Adipogenesis. Biochem. Biophys. Res. Commun. 2016, 479, 22–27. [Google Scholar] [CrossRef]
- Gustafson, B.; Eliasson, B.; Smith, U. Thiazolidinediones Increase the Wingless-Type MMTV Integration Site Family (WNT) Inhibitor Dickkopf-1 in Adipocytes: A Link with Osteogenesis. Diabetologia 2010, 53, 536–540. [Google Scholar] [CrossRef] [Green Version]
- Basu, A.; Haldar, S. The Relationship between BcI2, Bax and P53: Consequences for Cell Cycle Progression and Cell Death. Mol. Hum. Reprod. 1998, 4, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Rajkowska, G.; Du, F.; Seraji-Bozorgzad, N.; Manji, H.K. Enhancement of Hippocampal Neurogenesis by Lithium. J. Neurochem. 2000, 75, 1729–1734. [Google Scholar] [CrossRef] [PubMed]
- Youdim, M.B.H.; Arraf, Z. Prevention of MPTP (N-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine) Dopaminergic Neurotoxicity in Mice by Chronic Lithium: Involvements of Bcl-2 and Bax. Neuropharmacology 2004, 46, 1130–1140. [Google Scholar] [CrossRef] [PubMed]
- Ruvolo, P.P.; Deng, X.; May, W.S. Phosphorylation of Bcl2 and Regulation of Apoptosis. Leukemia 2001, 15, 515–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-L.; Lin, C.-F.; Chiang, C.-W.; Jan, M.-S.; Lin, Y.-S. Lithium Inhibits Ceramide- and Etoposide-Induced Protein Phosphatase 2A Methylation, Bcl-2 Dephosphorylation, Caspase-2 Activation, and Apoptosis. Mol. Pharmacol. 2006, 70, 510–517. [Google Scholar] [CrossRef]
- Abousaab, A.; Lang, F. Up-Regulation of Excitatory Amino Acid Transporters EAAT3 and EAAT4 by Lithium Sensitive Glycogen Synthase Kinase GSK3ß. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2016, 40, 1252–1260. [Google Scholar] [CrossRef]
- Yoshii, A.; Constantine-Paton, M. Postsynaptic BDNF-TrkB Signaling in Synapse Maturation, Plasticity, and Disease. Dev. Neurobiol. 2010, 70, 304–322. [Google Scholar] [CrossRef] [Green Version]
- Scheuing, L.; Chiu, C.-T.; Liao, H.-M.; Chuang, D.-M. Antidepressant Mechanism of Ketamine: Perspective from Preclinical Studies. Front. Neurosci. 2015, 9, 249. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.-T.; Scheuing, L.; Liu, G.; Liao, H.-M.; Linares, G.R.; Lin, D.; Chuang, D.-M. The Mood Stabilizer Lithium Potentiates the Antidepressant-like Effects and Ameliorates Oxidative Stress Induced by Acute Ketamine in a Mouse Model of Stress. Int. J. Neuropsychopharmacol. 2014, 18. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.-T.; Chuang, D.-M. Molecular Actions and Therapeutic Potential of Lithium in Preclinical and Clinical Studies of CNS Disorders. Pharmacol. Ther. 2010, 128, 281–304. [Google Scholar] [CrossRef] [Green Version]
- Verhoef, L.G.G.C.; Lindsten, K.; Masucci, M.G.; Dantuma, N.P. Aggregate Formation Inhibits Proteasomal Degradation of Polyglutamine Proteins. Hum. Mol. Genet. 2002, 11, 2689–2700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, H.; Rubinsztein, D.C. Control of Autophagy as a Therapy for Neurodegenerative Disease. Nat. Rev. Neurol. 2011, 8, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Lorzadeh, S.; Kohan, L.; Ghavami, S.; Azarpira, N. Autophagy and the Wnt Signaling Pathway: A Focus on Wnt/β-Catenin Signaling. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1868, 118926. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vallée, A.; Vallée, J.-N.; Lecarpentier, Y. Parkinson’s Disease: Potential Actions of Lithium by Targeting the WNT/β-Catenin Pathway, Oxidative Stress, Inflammation and Glutamatergic Pathway. Cells 2021, 10, 230. https://doi.org/10.3390/cells10020230
Vallée A, Vallée J-N, Lecarpentier Y. Parkinson’s Disease: Potential Actions of Lithium by Targeting the WNT/β-Catenin Pathway, Oxidative Stress, Inflammation and Glutamatergic Pathway. Cells. 2021; 10(2):230. https://doi.org/10.3390/cells10020230
Chicago/Turabian StyleVallée, Alexandre, Jean-Noël Vallée, and Yves Lecarpentier. 2021. "Parkinson’s Disease: Potential Actions of Lithium by Targeting the WNT/β-Catenin Pathway, Oxidative Stress, Inflammation and Glutamatergic Pathway" Cells 10, no. 2: 230. https://doi.org/10.3390/cells10020230