JAK-STAT Pathway Inhibition Partially Restores Intestinal Homeostasis in Hdac1- and Hdac2-Intestinal Epithelial Cell-Deficient Mice

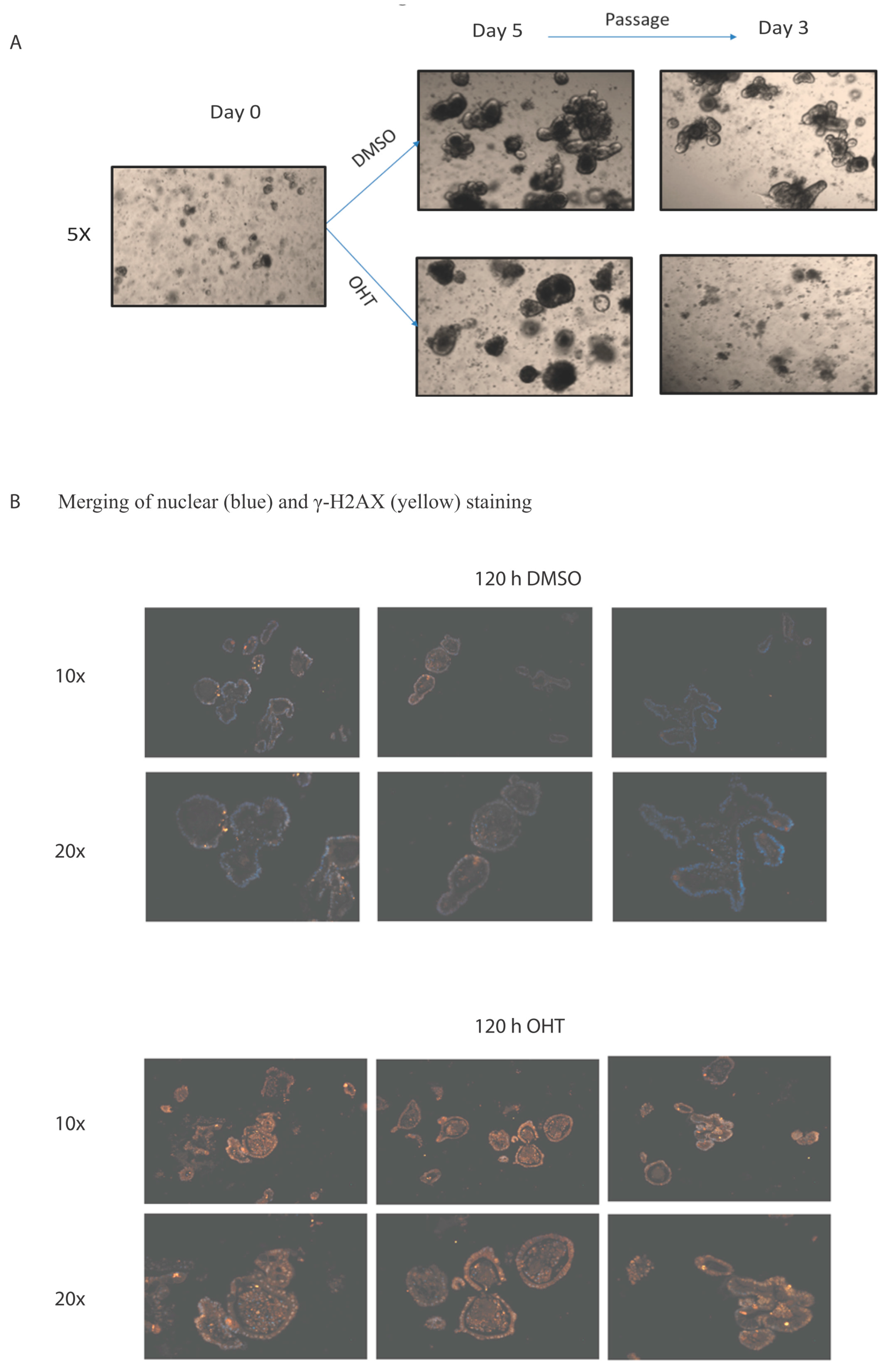{kind=link}
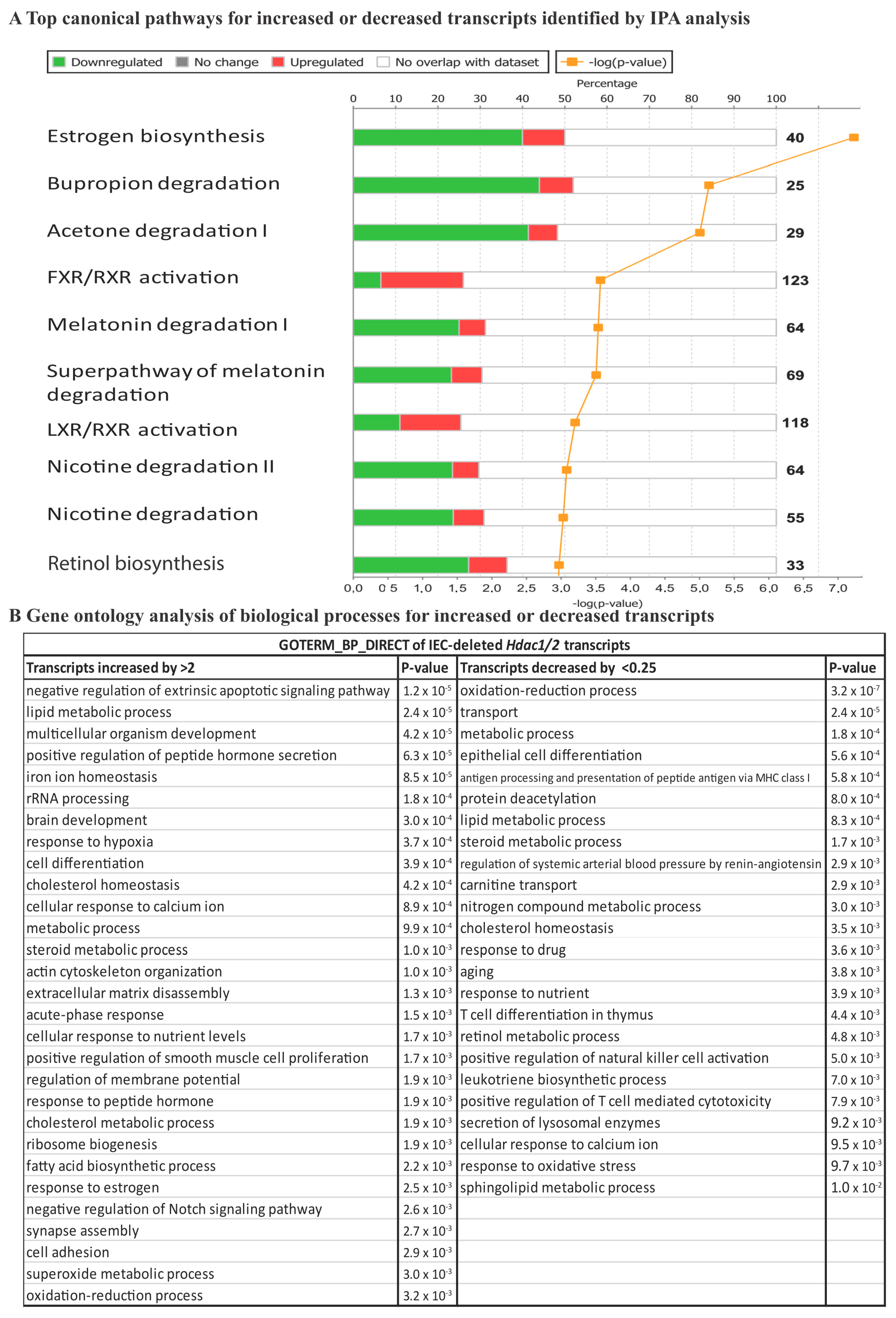{kind=link}
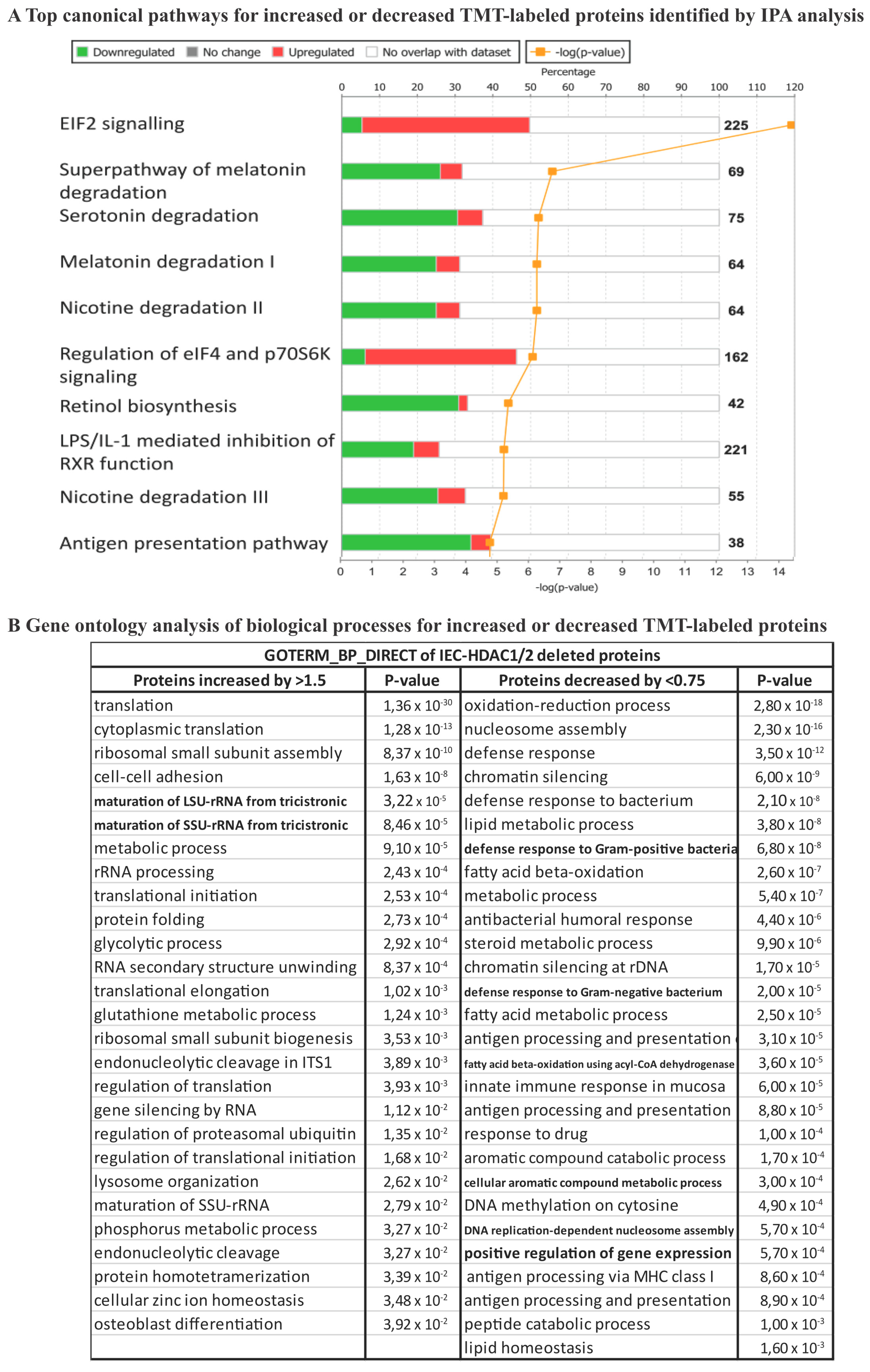{kind=link}
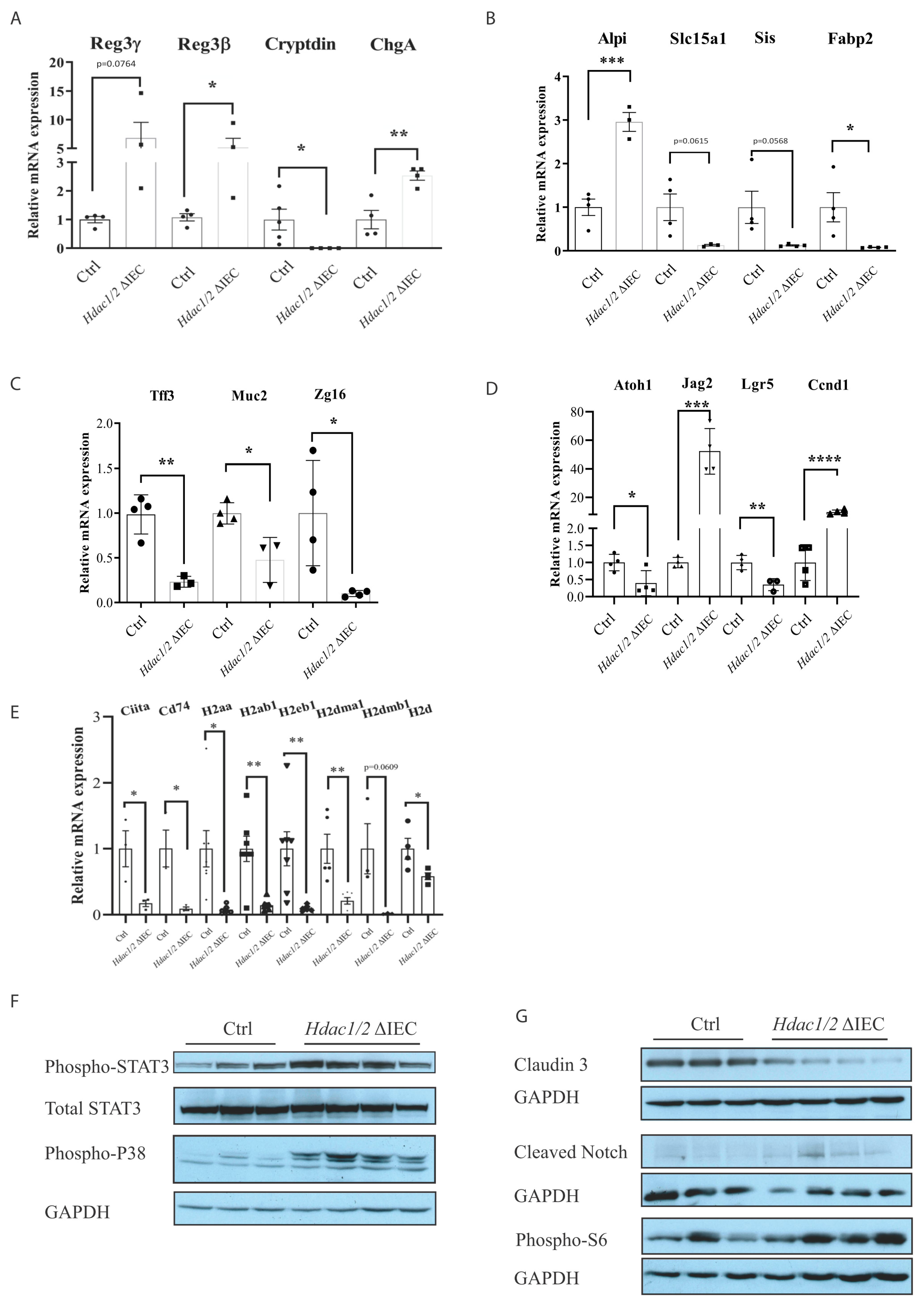{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Mice
2.2. Enteroid Cultures
2.3. RNA Isolation and Quantitative Real-Time PCR (qPCR)
2.4. RNA-Seq Analysis
2.5. Proteome Analysis
2.6. Bioinformatics Analysis
2.7. Histological Analysis, Immunofluorescence and Immunocytochemistry
2.8. Western Blot Analysis
2.9. Statistical Analysis
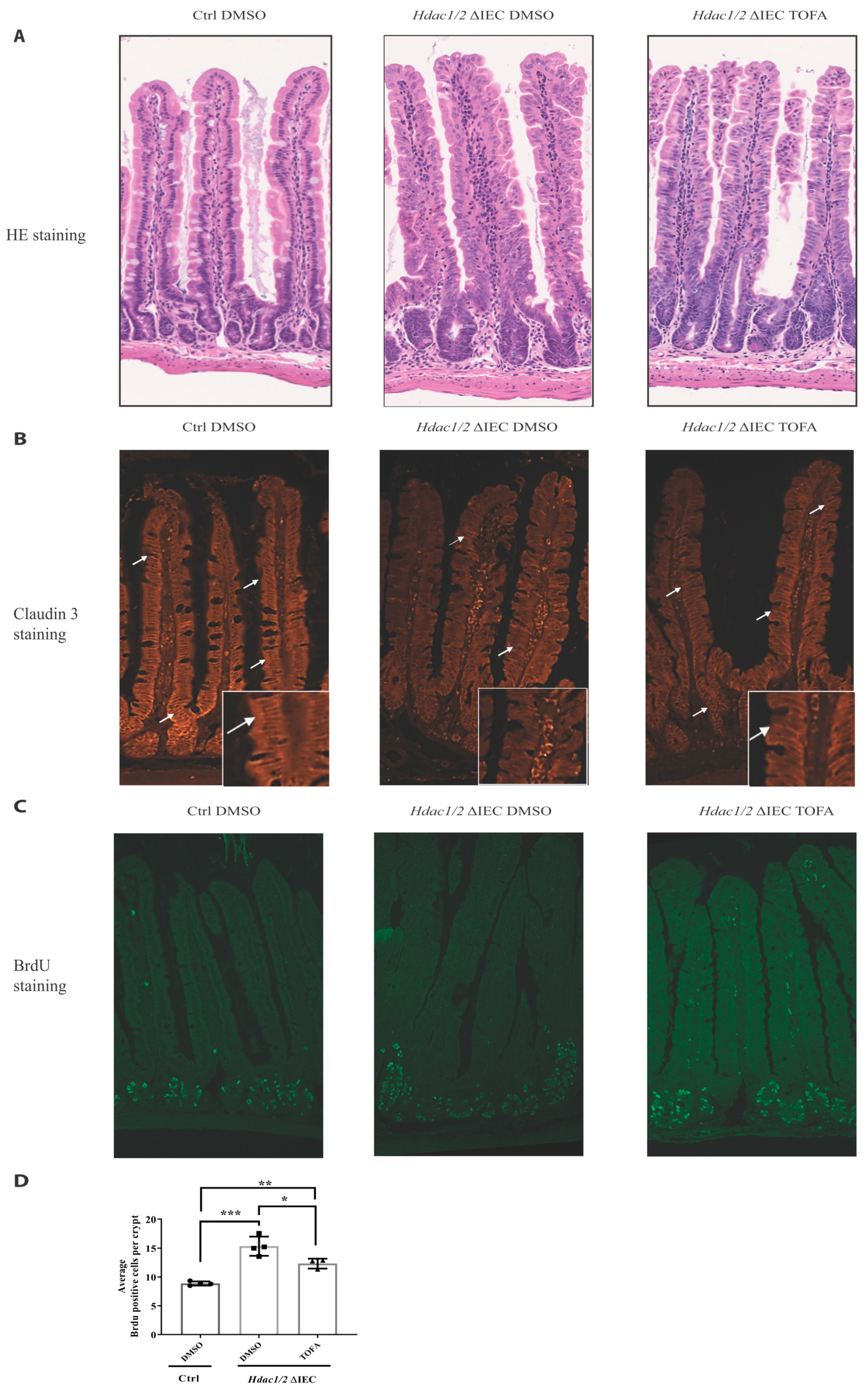
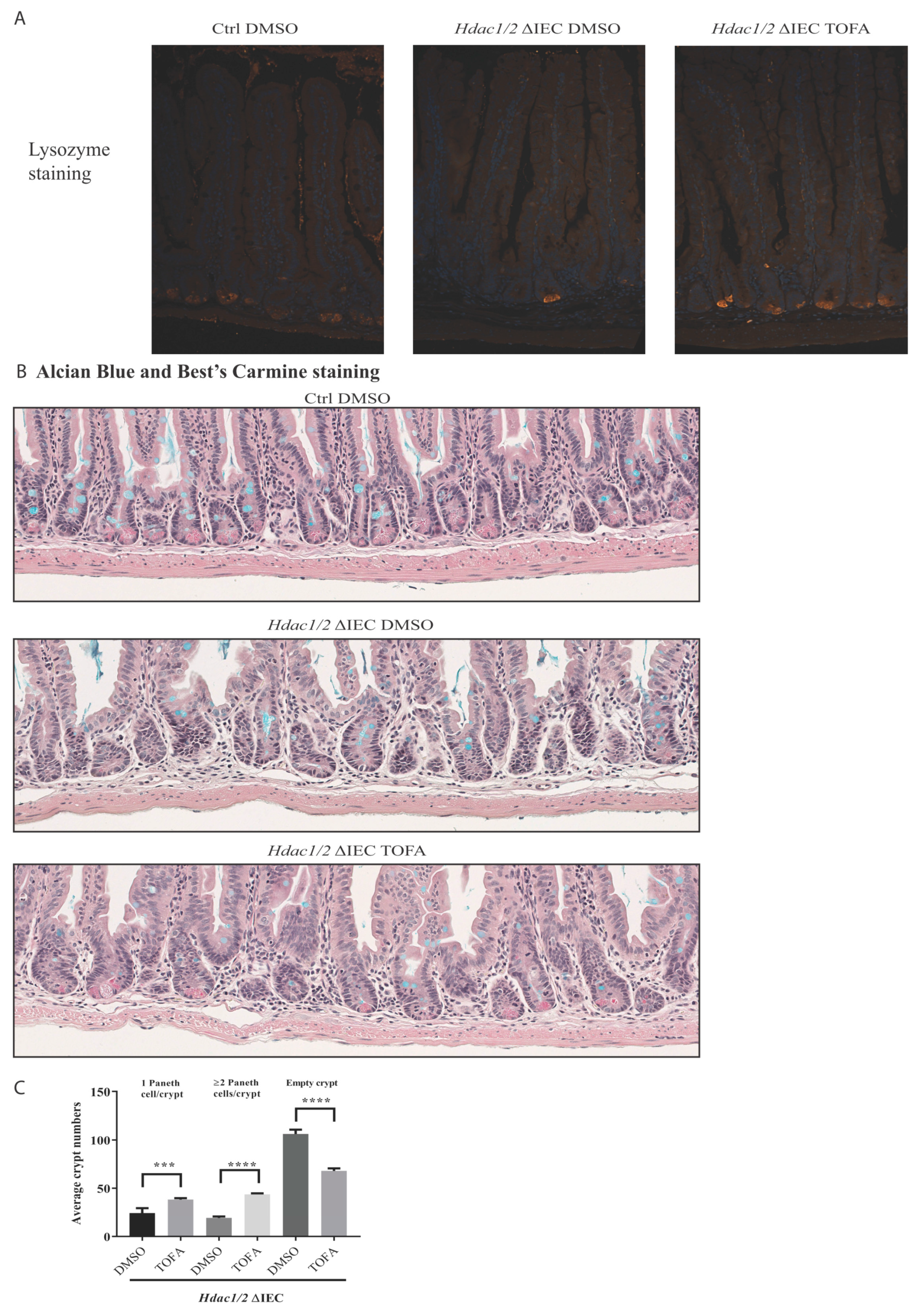
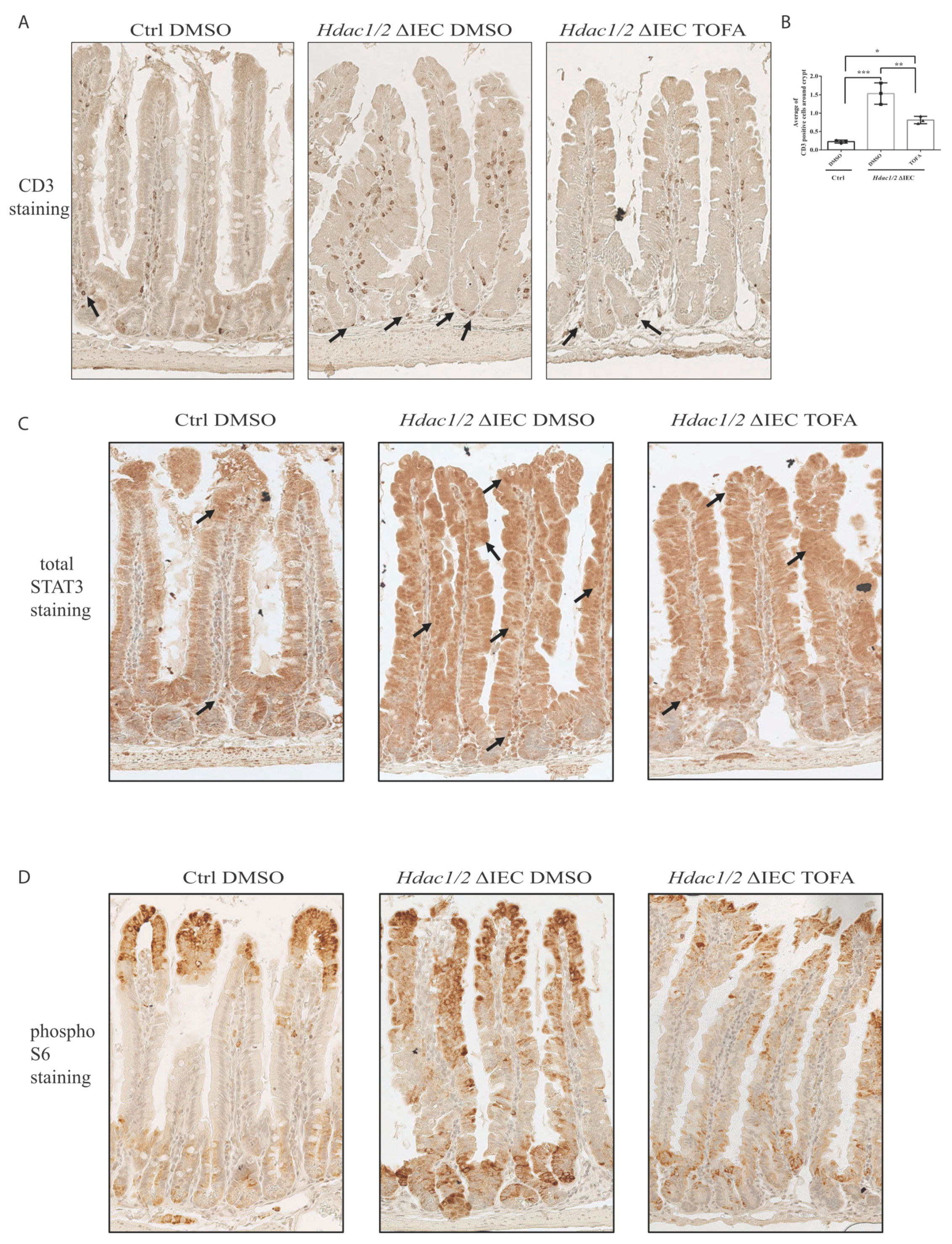
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Asselin, C.; Gendron, F.P. Shuttling of information between the mucosal and luminal environment drives intestinal homeostasis. FEBS Lett. 2014, 588, 4148–4157. [Google Scholar] [CrossRef] [PubMed]
- Allaire, J.M.; Crowley, S.M.; Law, H.T.; Chang, S.Y.; Ko, H.J.; Vallance, B.A. The Intestinal Epithelium: Central Coordinator of Mucosal Immunity: (Trends in Immunology 39, 677–696, 2018). Trends Immunol. 2019, 40, 174. [Google Scholar] [CrossRef] [PubMed]
- Gehart, H.; Clevers, H. Tales from the crypt: New insights into intestinal stem cells. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Moor, A.E.; Harnik, Y.; Ben-Moshe, S.; Massasa, E.E.; Rozenberg, M.; Eilam, R.; Bahar Halpern, K.; Itzkovitz, S. Spatial Reconstruction of Single Enterocytes Uncovers Broad Zonation along the Intestinal Villus Axis. Cell 2018, 175, 1156–1167.e15. [Google Scholar] [CrossRef] [Green Version]
- Knoop, K.A.; Newberry, R.D. Goblet cells: Multifaceted players in immunity at mucosal surfaces. Mucosal Immunol. 2018, 11, 1551–1557. [Google Scholar] [CrossRef]
- Gassler, N. Paneth cells in intestinal physiology and pathophysiology. World J. Gastrointest. Pathophysiol. 2017, 8, 150–160. [Google Scholar] [CrossRef]
- Beumer, J.; Artegiani, B.; Post, Y.; Reimann, F.; Gribble, F.; Nguyen, T.N.; Zeng, H.; Van den Born, M.; Van Es, J.H.; Clevers, H. Enteroendocrine cells switch hormone expression along the crypt-to-villus BMP signalling gradient. Nat. Cell Biol. 2018, 20, 909–916. [Google Scholar] [CrossRef]
- Beumer, J.; Gehart, H.; Clevers, H. Enteroendocrine Dynamics—New Tools Reveal Hormonal Plasticity in the Gut. Endocrine Rev. 2020, 41. [Google Scholar] [CrossRef]
- Graham, D.B.; Xavier, R.J. Pathway paradigms revealed from the genetics of inflammatory bowel disease. Nature 2020, 578, 527–539. [Google Scholar] [CrossRef]
- Ray, G.; Longworth, M.S. Epigenetics, DNA Organization, and Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2019, 25, 235–247. [Google Scholar] [CrossRef]
- Mokry, M.; Middendorp, S.; Wiegerinck, C.L.; Witte, M.; Teunissen, H.; Meddens, C.A.; Cuppen, E.; Clevers, H.; Nieuwenhuis, E.E. Many inflammatory bowel disease risk loci include regions that regulate gene expression in immune cells and the intestinal epithelium. Gastroenterology 2014, 146, 1040–1047. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.; Kotliar, M.; Woo, V.; Jagannathan, S.; Whitt, J.; Moncivaiz, J.; Aronow, B.J.; Dubinsky, M.C.; Hyams, J.S.; Markowitz, J.F.; et al. Microbiota-sensitive epigenetic signature predicts inflammation in Crohn’s disease. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Pietrocola, F.; Galluzzi, L.; Bravo-San Pedro, J.M.; Madeo, F.; Kroemer, G. Acetyl coenzyme A: A central metabolite and second messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, J.C.; Verdin, E. Ketone bodies as signaling metabolites. Trends Endocrinol. Metab. 2014, 25, 42–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Wang, J.; He, T.; Becker, S.; Zhang, G.; Li, D.; Ma, X. Butyrate: A Double-Edged Sword for Health? Adv. Nutr. 2018, 9, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Kelly, R.D.; Cowley, S.M. The physiological roles of histone deacetylase (HDAC) 1 and 2: Complex co-stars with multiple leading parts. Biochem. Soc. Trans. 2013, 41, 741–749. [Google Scholar] [CrossRef] [Green Version]
- Moser, M.A.; Hagelkruys, A.; Seiser, C. Transcription and beyond: The role of mammalian class I lysine deacetylases. Chromosoma 2014, 123, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Turgeon, N.; Blais, M.; Gagne, J.M.; Tardif, V.; Boudreau, F.; Perreault, N.; Asselin, C. HDAC1 and HDAC2 restrain the intestinal inflammatory response by regulating intestinal epithelial cell differentiation. PLoS ONE 2013, 8, e73785. [Google Scholar] [CrossRef] [Green Version]
- Leppkes, M.; Neurath, M.F.; Herrmann, M.; Becker, C. Immune deficiency vs. immune excess in inflammatory bowel diseases-STAT3 as a rheo-STAT of intestinal homeostasis. J. Leukoc. Biol. 2016, 99, 57–66. [Google Scholar] [CrossRef]
- Fritsch, S.D.; Weichhart, T. Metabolic and immunologic control of intestinal cell function by mTOR. Int. Immunol. 2020, 32, 455–465. [Google Scholar] [CrossRef]
- Montgomery, R.L.; Davis, C.A.; Potthoff, M.J.; Haberland, M.; Fielitz, J.; Qi, X.; Hill, J.A.; Richardson, J.A.; Olson, E.N. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007, 21, 1790–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madison, B.B.; Dunbar, L.; Qiao, X.T.; Braunstein, K.; Braunstein, E.; Gumucio, D.L. Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J. Biol. Chem. 2002, 277, 33275–33283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- el Marjou, F.; Janssen, K.P.; Chang, B.H.; Li, M.; Hindie, V.; Chan, L.; Louvard, D.; Chambon, P.; Metzger, D.; Robine, S. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis 2004, 39, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Turgeon, N.; Gagne, J.M.; Blais, M.; Gendron, F.P.; Boudreau, F.; Asselin, C. The acetylome regulators Hdac1 and Hdac2 differently modulate intestinal epithelial cell dependent homeostatic responses in experimental colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G594–G605. [Google Scholar] [CrossRef] [PubMed]
- Gonneaud, A.; Turgeon, N.; Boudreau, F.; Perreault, N.; Rivard, N.; Asselin, C. Distinct Roles for Intestinal Epithelial Cell-Specific Hdac1 and Hdac2 in the Regulation of Murine Intestinal Homeostasis. J. Cell. Physiol. 2016, 231, 436–448. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Janus kinase (JAK) inhibitors in the treatment of inflammatory and neoplastic diseases. Pharmacol. Res. 2016, 111, 784–803. [Google Scholar] [CrossRef]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef]
- Sato, T.; Clevers, H. Growing self-organizing mini-guts from a single intestinal stem cell: Mechanism and applications. Science 2013, 340, 1190–1194. [Google Scholar] [CrossRef] [Green Version]
- Gonneaud, A.; Jones, C.; Turgeon, N.; Levesque, D.; Asselin, C.; Boudreau, F.; Boisvert, F.M. A SILAC-Based Method for Quantitative Proteomic Analysis of Intestinal Organoids. Sci. Rep. 2016, 6, 38195. [Google Scholar] [CrossRef] [Green Version]
- Gonneaud, A.; Turgeon, N.; Jones, C.; Couture, C.; Levesque, D.; Boisvert, F.M.; Boudreau, F.; Asselin, C. HDAC1 and HDAC2 independently regulate common and specific intrinsic responses in murine enteroids. Sci. Rep. 2019, 9, 5363. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Dennis, G., Jr.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, P3. [Google Scholar] [CrossRef] [PubMed]
- Kramer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, A.E.; Jones, T.R.; Lamprecht, M.R.; Clarke, C.; Kang, I.H.; Friman, O.; Guertin, D.A.; Chang, J.H.; Lindquist, R.A.; Moffat, J.; et al. CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006, 7, R100. [Google Scholar] [CrossRef] [Green Version]
- Roos, W.P.; Krumm, A. The multifaceted influence of histone deacetylases on DNA damage signalling and DNA repair. Nucleic Acids Res. 2016, 44, 10017–10030. [Google Scholar] [CrossRef] [Green Version]
- Zimberlin, C.D.; Lancini, C.; Sno, R.; Rosekrans, S.L.; McLean, C.M.; Vlaming, H.; van den Brink, G.R.; Bots, M.; Medema, J.P.; Dannenberg, J.H. HDAC1 and HDAC2 collectively regulate intestinal stem cell homeostasis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 2070–2080. [Google Scholar] [CrossRef]
- Zhang, Y.; Edwards, P.A. FXR signaling in metabolic disease. Febs Lett. 2008, 582, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Iyer, M.; McEachin, R.; Zhao, M.; Wu, Y.M.; Cao, X.; Oravecz-Wilson, K.; Zajac, C.; Mathewson, N.; Wu, S.J.; et al. Genome-Wide STAT3 Binding Analysis after Histone Deacetylase Inhibition Reveals Novel Target Genes in Dendritic Cells. J. Innate Immun. 2017, 9, 126–144. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.H.; Seeley, R.J. Reg3 Proteins as Gut Hormones? Endocrinology 2019, 160, 1506–1514. [Google Scholar] [CrossRef]
- Haber, A.L.; Biton, M.; Rogel, N.; Herbst, R.H.; Shekhar, K.; Smillie, C.; Burgin, G.; Delorey, T.M.; Howitt, M.R.; Katz, Y.; et al. A single-cell survey of the small intestinal epithelium. Nature 2017, 551, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Spanier, B.; Rohm, F. Proton Coupled Oligopeptide Transporter 1 (PepT1) Function, Regulation, and Influence on the Intestinal Homeostasis. Compr. Physiol. 2018, 8, 843–869. [Google Scholar] [CrossRef] [PubMed]
- Lalles, J.P. Recent advances in intestinal alkaline phosphatase, inflammation, and nutrition. Nutr. Rev. 2019, 77, 710–724. [Google Scholar] [CrossRef] [PubMed]
- Bilski, J.; Mazur-Bialy, A.; Wojcik, D.; Zahradnik-Bilska, J.; Brzozowski, B.; Magierowski, M.; Mach, T.; Magierowska, K.; Brzozowski, T. The Role of Intestinal Alkaline Phosphatase in Inflammatory Disorders of Gastrointestinal Tract. Mediat. Inflamm. 2017, 2017, 9074601. [Google Scholar] [CrossRef] [PubMed]
- Braga Emidio, N.; Hoffmann, W.; Brierley, S.M.; Muttenthaler, M. Trefoil Factor Family: Unresolved Questions and Clinical Perspectives. Trends Biochem. Sci. 2019, 44, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Bergstrom, J.H.; Birchenough, G.M.; Katona, G.; Schroeder, B.O.; Schutte, A.; Ermund, A.; Johansson, M.E.; Hansson, G.C. Gram-positive bacteria are held at a distance in the colon mucus by the lectin-like protein ZG16. Proc. Natl. Acad. Sci. USA 2016, 113, 13833–13838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wosen, J.E.; Mukhopadhyay, D.; Macaubas, C.; Mellins, E.D. Epithelial MHC Class II Expression and Its Role in Antigen Presentation in the Gastrointestinal and Respiratory Tracts. Front. Immunol. 2018, 9, 2144. [Google Scholar] [CrossRef]
- Biton, M.; Haber, A.L.; Rogel, N.; Burgin, G.; Beyaz, S.; Schnell, A.; Ashenberg, O.; Su, C.W.; Smillie, C.; Shekhar, K.; et al. T Helper Cell Cytokines Modulate Intestinal Stem Cell Renewal and Differentiation. Cell 2018, 175, 1307–1320.e22. [Google Scholar] [CrossRef] [Green Version]
- Demitrack, E.S.; Samuelson, L.C. Notch regulation of gastrointestinal stem cells. J. Physiol. 2016, 594, 4791–4803. [Google Scholar] [CrossRef] [Green Version]
- Meng, D.; Frank, A.R.; Jewell, J.L. mTOR signaling in stem and progenitor cells. Development 2018, 145. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Hernandez, V.; Quiros, M.; Nusrat, A. Intestinal epithelial claudins: Expression and regulation in homeostasis and inflammation. Ann. New York Acad. Sci. 2017, 1397, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. Regulation of mTORC1 and its impact on gene expression at a glance. J. Cell Sci. 2013, 126, 1713–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Meng, Y.; Kwiatkowski, D.J.; Chen, X.; Peng, H.; Sun, Q.; Zha, X.; Wang, F.; Wang, Y.; Jing, Y.; et al. Mammalian target of rapamycin regulates murine and human cell differentiation through STAT3/p63/Jagged/Notch cascade. J. Clin. Investig. 2010, 120, 103–114. [Google Scholar] [CrossRef] [Green Version]
- Poser, S.W.; Park, D.M.; Androutsellis-Theotokis, A. The STAT3-Ser/Hes3 signaling axis: An emerging regulator of endogenous regeneration and cancer growth. Front. Physiol. 2013, 4, 273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajvani, U.B.; Qiang, L.; Kangsamaksin, T.; Kitajewski, J.; Ginsberg, H.N.; Accili, D. Inhibition of Notch uncouples Akt activation from hepatic lipid accumulation by decreasing mTorc1 stability. Nat. Med. 2013, 19, 1054–1060. [Google Scholar] [CrossRef] [Green Version]
- Shimobayashi, M.; Hall, M.N. Making new contacts: The mTOR network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 2014, 15, 155–162. [Google Scholar] [CrossRef]
- van Es, J.H.; van Gijn, M.E.; Riccio, O.; van den Born, M.; Vooijs, M.; Begthel, H.; Cozijnsen, M.; Robine, S.; Winton, D.J.; Radtke, F.; et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature 2005, 435, 959–963. [Google Scholar] [CrossRef]
- Salas, A.; Hernandez-Rocha, C.; Duijvestein, M.; Faubion, W.; McGovern, D.; Vermeire, S.; Vetrano, S.; Vande Casteele, N. JAK-STAT pathway targeting for the treatment of inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 323–337. [Google Scholar] [CrossRef]
- Rahner, C.; Mitic, L.L.; Anderson, J.M. Heterogeneity in expression and subcellular localization of claudins 2, 3, 4, and 5 in the rat liver, pancreas, and gut. Gastroenterology 2001, 120, 411–422. [Google Scholar] [CrossRef]
- Beumer, J.; Clevers, H. Cell fate specification and differentiation in the adult mammalian intestine. Nat. Rev. Mol. Cell Biol. 2021, 22, 39–53. [Google Scholar] [CrossRef]
- Danese, S.; Grisham, M.; Hodge, J.; Telliez, J.B. JAK inhibition using tofacitinib for inflammatory bowel disease treatment: A hub for multiple inflammatory cytokines. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G155–G162. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonneaud, A.; Turgeon, N.; Boisvert, F.-M.; Boudreau, F.; Asselin, C. JAK-STAT Pathway Inhibition Partially Restores Intestinal Homeostasis in Hdac1- and Hdac2-Intestinal Epithelial Cell-Deficient Mice. Cells 2021, 10, 224. https://doi.org/10.3390/cells10020224
Gonneaud A, Turgeon N, Boisvert F-M, Boudreau F, Asselin C. JAK-STAT Pathway Inhibition Partially Restores Intestinal Homeostasis in Hdac1- and Hdac2-Intestinal Epithelial Cell-Deficient Mice. Cells. 2021; 10(2):224. https://doi.org/10.3390/cells10020224
Chicago/Turabian StyleGonneaud, Alexis, Naomie Turgeon, Francois-Michel Boisvert, Francois Boudreau, and Claude Asselin. 2021. "JAK-STAT Pathway Inhibition Partially Restores Intestinal Homeostasis in Hdac1- and Hdac2-Intestinal Epithelial Cell-Deficient Mice" Cells 10, no. 2: 224. https://doi.org/10.3390/cells10020224