
Evaluation of DNA Extraction Methods Developed for Forensic and Ancient DNA Applications Using Bone Samples of Different Age

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples and Extraction Methods
2.1.1. Adapted Loreille Method
2.1.2. Adapted Dabney Method
2.2. Real-Time Quantitative PCR
2.3. mtDNA Massively Parallel Sequencing
2.4. Nuclear DNA STR and SNP Typing
3. Results and Discussion
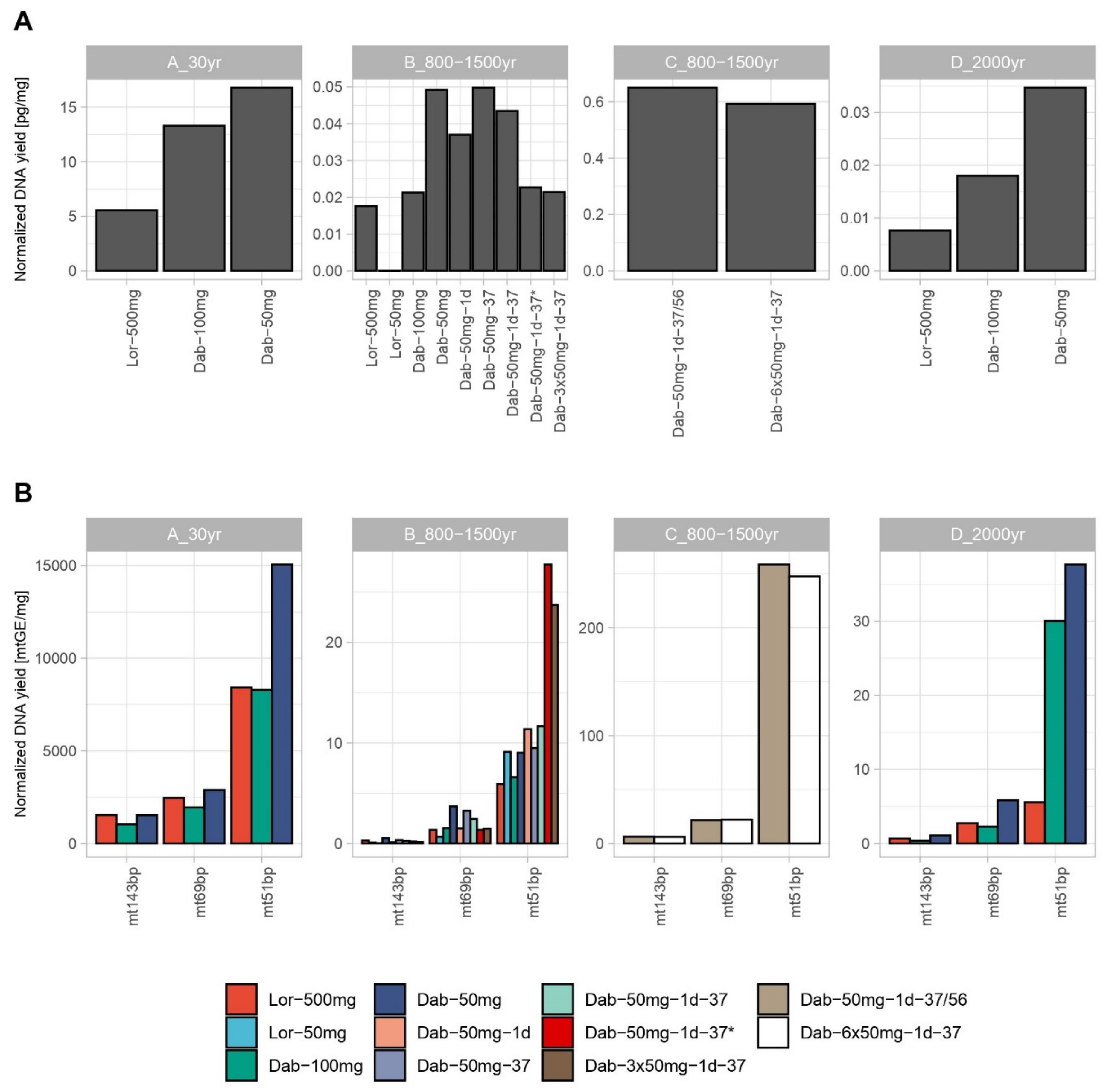
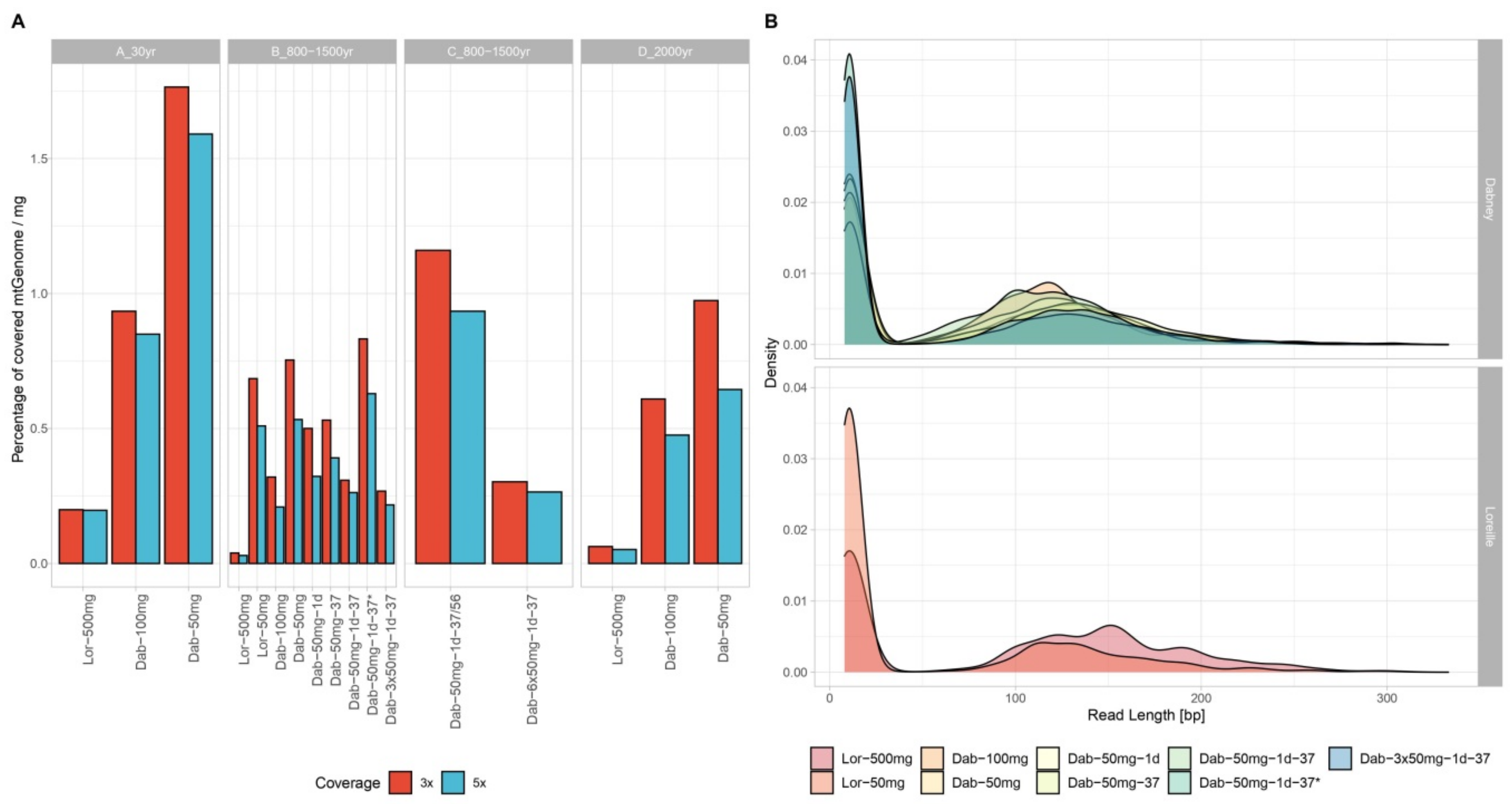
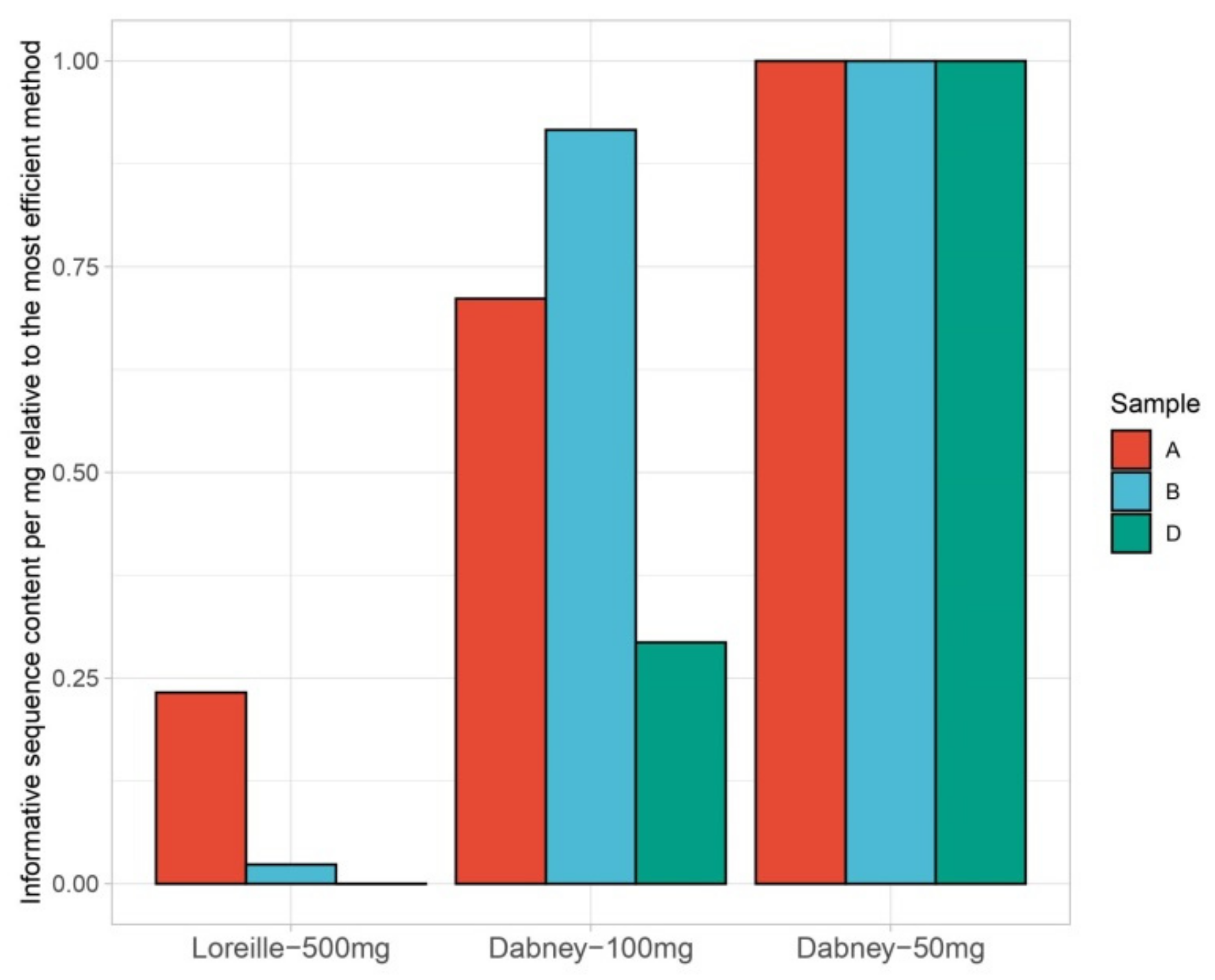
3.1. Nuclear and mtDNA Quantification
3.2. MtDNA Sequencing
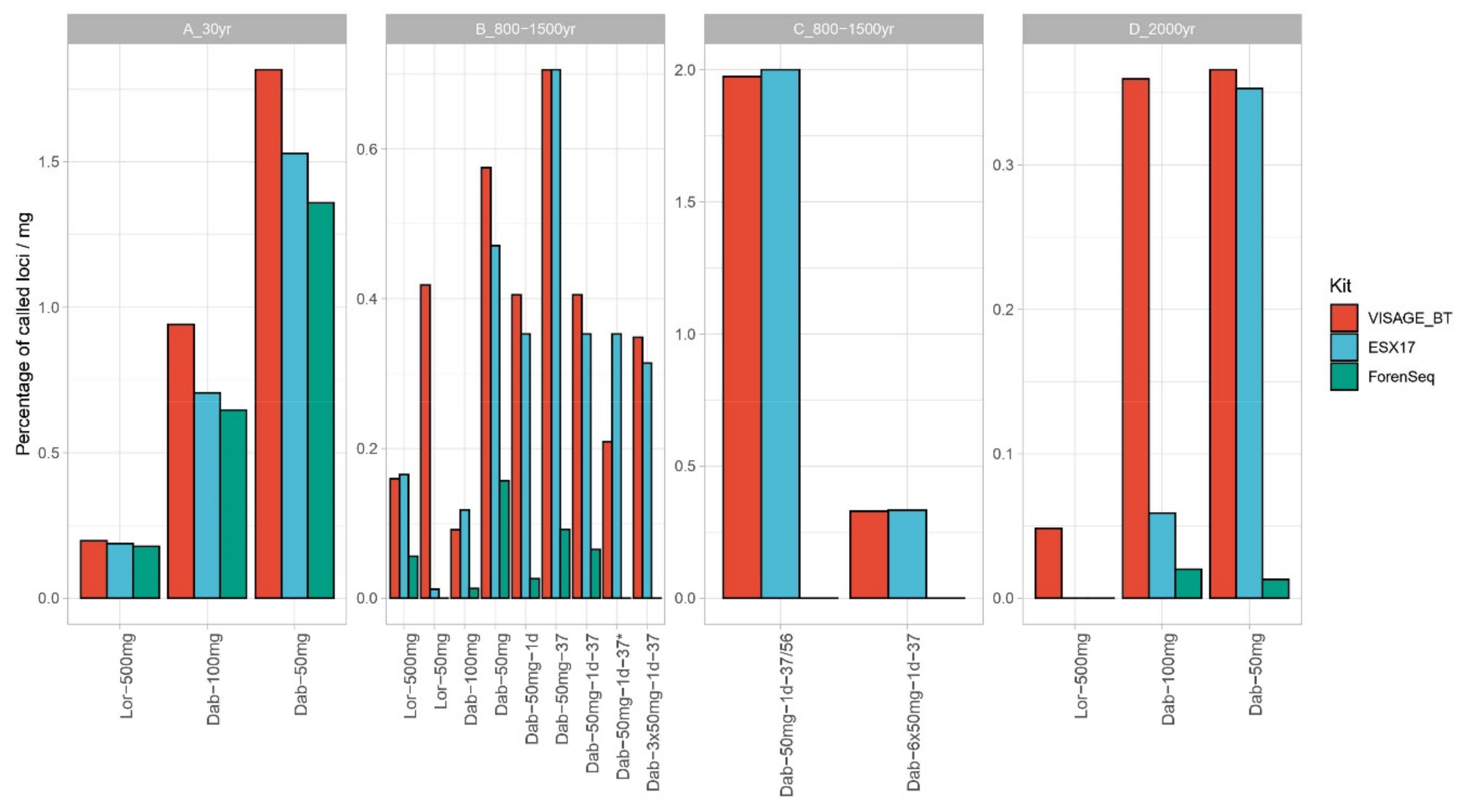
3.3. Nuclear DNA–STR Typing and SNP Sequencing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meyer, M.; Fu, Q.; Aximu-Petri, A.; Glocke, I.; Nickel, B.; Arsuaga, J.-L.; Martínez, I.; Gracia, A.; de Castro, J.M.B.; Carbonell, E.; et al. A mitochondrial genome sequence of a hominin from Sima de los Huesos. Nature 2014, 505, 403–406. [Google Scholar] [CrossRef]
- Orlando, L.; Ginolhac, A.; Zhang, G.; Froese, D.; Albrechtsen, A.; Stiller, M.; Schubert, M.; Cappellini, E.; Petersen, B.; Moltke, I.; et al. Recalibrating Equus evolution using the genome sequence of an early Middle Pleistocene horse. Nature 2013, 499, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Willerslev, E.; Hansen, A.J.; Binladen, J.; Brand, T.B.; Gilbert, M.T.P.; Shapiro, B.; Bunce, M.; Wiuf, C.; Gilichinsky, D.A.; Cooper, A. Diverse Plant and Animal Genetic Records from Holocene and Pleistocene Sediments. Science 2003, 300, 791–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slon, V.; Hopfe, C.; Weiß, C.L.; Mafessoni, F.; de la Rasilla, M.; Lalueza-Fox, C.; Rosas, A.; Soressi, M.; Knul, M.V.; Miller, R.; et al. Neandertal and Denisovan DNA from Pleistocene sediments. Science 2017, 356, 605–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Höss, M.; Pääbo, S. DNA extraction from Pleistocene bones by a silica-based purification method. Nucleic Acids Res. 1993, 21, 3913–3914. [Google Scholar] [CrossRef] [Green Version]
- Hofreiter, M.; Rabeder, G.; Jaenicke-Després, V.; Withalm, G.; Nagel, D.; Paunovic, M.; Jambrĕsić, G.; Pääbo, S. Evidence for Reproductive Isolation between Cave Bear Populations. Curr. Biol. 2004, 14, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Noonan, J.P.; Hofreiter, M.; Smith, D.; Priest, J.R.; Rohland, N.; Rabeder, G.; Krause, J.; Detter, J.C.; Pääbo, S.; Rubin, E.M. Genomic sequencing of Pleistocene cave bears. Science 2005, 309, 597–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohland, N.; Hofreiter, M. Ancient DNA extraction from bones and teeth. Nat. Protoc. 2007, 2, 1756–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohland, N.; Hofreiter, M. Comparison and optimization of ancient DNA extraction. Biotechniques 2007, 42, 343–352. [Google Scholar] [CrossRef] [Green Version]
- Poinar, H.N.; Schwarz, C.; Qi, J.; Shapiro, B.; Macphee, R.D.; Buigues, B.; Tikhonov, A.; Huson, D.H.; Tomsho, L.P.; Auch, A.; et al. Metagenomics to paleogenomics: Large-scale sequencing of mammoth DNA. Science 2006, 311, 392–394. [Google Scholar] [CrossRef] [Green Version]
- Dabney, J.; Knapp, M.; Glocke, I.; Gansauge, M.-T.; Weihmann, A.; Nickel, B.; Valdiosera, C.; García, N.; Pääbo, S.; Arsuaga, J.-L.; et al. Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proc. Natl. Acad. Sci. USA 2013, 110, 15758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieves-Colón, M.A.; Ozga, A.T.; Pestle, W.J.; Cucina, A.; Tiesler, V.; Stanton, T.W.; Stone, A.C. Comparison of two ancient DNA extraction protocols for skeletal remains from tropical environments. Am. J. Phys. Anthropol. 2018, 166, 824–836. [Google Scholar] [CrossRef] [PubMed]
- Glocke, I.; Meyer, M. Extending the spectrum of DNA sequences retrieved from ancient bones and teeth. Genome Res. 2017, 27, 1230–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohland, N.; Glocke, I.; Aximu-Petri, A.; Meyer, M. Extraction of highly degraded DNA from ancient bones, teeth and sediments for high-throughput sequencing. Nat. Protoc. 2018, 13, 2447–2461. [Google Scholar] [CrossRef]
- Epp, L.S.; Zimmermann, H.H.; Stoof-Leichsenring, K.R. Sampling and Extraction of Ancient DNA from Sediments. Methods Mol. Biol. Clifton N.J. 2019, 1963, 31–44. [Google Scholar] [CrossRef]
- Adler, C.J.; Haak, W.; Donlon, D.; Cooper, A. Survival and recovery of DNA from ancient teeth and bones. J. Archaeol. Sci. 2011, 38, 956–964. [Google Scholar] [CrossRef]
- Allentoft, M.E.; Collins, M.; Harker, D.; Haile, J.; Oskam, C.L.; Hale, M.L.; Campos, P.F.; Samaniego, J.A.; Gilbert, M.T.; Willerslev, E.; et al. The half-life of DNA in bone: Measuring decay kinetics in 158 dated fossils. Proc. Biol. Sci. 2012, 279, 4724–4733. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, M.; Vissing, J. Paternal Inheritance of Mitochondrial DNA. N. Engl. J. Med. 2002, 347, 576–580. [Google Scholar] [CrossRef]
- Jobling, M.A.; Gill, P. Encoded evidence: DNA in forensic analysis. Nat. Rev. Genet. 2004, 5, 739–751. [Google Scholar] [CrossRef]
- Berger, C.; Parson, W. Mini-midi-mito: Adapting the amplification and sequencing strategy of mtDNA to the degradation state of crime scene samples. Forensic Sci. Int. Genet. 2009, 3, 149–153. [Google Scholar] [CrossRef]
- Senge, T.; Madea, B.; Junge, A.; Rothschild, M.A.; Schneider, P.M. STRs, mini STRs and SNPs—A comparative study for typing degraded DNA. Leg. Med. 2011, 13, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Churchill, J.D.; Schmedes, S.E.; King, J.L.; Budowle, B. Evaluation of the Illumina® Beta Version ForenSeq™ DNA Signature Prep Kit for use in genetic profiling. Forensic Sci. Int. Genet. 2016, 20, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Jäger, A.C.; Alvarez, M.L.; Davis, C.P.; Guzmán, E.; Han, Y.; Way, L.; Walichiewicz, P.; Silva, D.; Pham, N.; Caves, G.; et al. Developmental validation of the MiSeq FGx Forensic Genomics System for Targeted Next Generation Sequencing in Forensic DNA Casework and Database Laboratories. Forensic Sci. Int. Genet. 2017, 28, 52–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eduardoff, M.; Santos, C.; de la Puente, M.; Gross, T.E.; Fondevila, M.; Strobl, C.; Sobrino, B.; Ballard, D.; Schneider, P.M.; Carracedo, Á.; et al. Inter-laboratory evaluation of SNP-based forensic identification by massively parallel sequencing using the Ion PGM™. Forensic Sci. Int. Genet. 2015, 17, 110–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loreille, O.M.; Diegoli, T.M.; Irwin, J.A.; Coble, M.D.; Parsons, T.J. High efficiency DNA extraction from bone by total demineralization. Forensic Sci. Int. Genet. 2007, 1, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Amory, S.; Huel, R.; Bilić, A.; Loreille, O.; Parsons, T.J. Automatable full demineralization DNA extraction procedure from degraded skeletal remains. Forensic Sci. Int. Genet. 2012, 6, 398–406. [Google Scholar] [CrossRef]
- Eduardoff, M.; Xavier, C.; Strobl, C.; Casas-Vargas, A.; Parson, W. Optimized mtDNA Control Region Primer Extension Capture Analysis for Forensically Relevant Samples and Highly Compromised mtDNA of Different Age and Origin. Genes 2017, 8, 237. [Google Scholar] [CrossRef] [Green Version]
- Parson, W.; Eduardoff, M.; Xavier, C.; Bertoglio, B.; Teschler-Nicola, M. Resolving the matrilineal relationship of seven Late Bronze Age individuals from Stillfried, Austria. Forensic Sci. Int. Genet. 2018, 36, 148–151. [Google Scholar] [CrossRef]
- Marshall, C.; Sturk-Andreaggi, K.; Daniels-Higginbotham, J.; Oliver, R.S.; Barritt-Ross, S.; McMahon, T.P. Performance evaluation of a mitogenome capture and Illumina sequencing protocol using non-probative, case-type skeletal samples: Implications for the use of a positive control in a next-generation sequencing procedure. Forensic Sci. Int. Genet. 2017, 31, 198–206. [Google Scholar] [CrossRef] [Green Version]
- Emery, M.V.; Bolhofner, K.; Winingear, S.; Oldt, R.; Montes, M.; Kanthaswamy, S.; Buikstra, J.E.; Fulginiti, L.C.; Stone, A.C. Reconstructing full and partial STR profiles from severely burned human remains using comparative ancient and forensic DNA extraction techniques. Forensic Sci. Int. Genet. 2020, 46, 102272. [Google Scholar] [CrossRef]
- Bauer, C.M.; Niederstätter, H.; McGlynn, G.; Stadler, H.; Parson, W. Comparison of morphological and molecular genetic sex-typing on mediaeval human skeletal remains. Forensic Sci. Int. Genet. 2013, 7, 581–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casas-Vargas, A.; Romero, L.M.; Usaquén, W.; Zea, S.; Silva, M.; Briceño, I.; Gómez, A.; Rodríguez, J.V. Diversidad del ADN mitocondrial en restos óseos prehispánicos asociados al Templo del Sol en los Andes orientales colombianos. Biomédica 2017, 37, 548–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xavier, C.; Eduardoff, M.; Strobl, C.; Parson, W. SD quants—Sensitive detection tetraplex-system for nuclear and mitochondrial DNA quantification and degradation inference. Forensic Sci. Int. Genet. 2019, 42, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.M.; Kubacka, I.; Chinnery, P.F.; Lightowlers, R.N.; Turnbull, D.M.; Howell, N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 1999, 23, 147. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2012, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Bandelt, H.J.; Parson, W. Consistent treatment of length variants in the human mtDNA control region: A reappraisal. Int. J. Leg. Med. 2008, 122, 11–21. [Google Scholar] [CrossRef]
- Parson, W.; Bandelt, H.J. Extended guidelines for mtDNA typing of population data in forensic science. Forensic Sci. Int. Genet. 2007, 1, 13–19. [Google Scholar] [CrossRef]
- Parson, W.; Gusmão, L.; Hares, D.R.; Irwin, J.A.; Mayr, W.R.; Morling, N.; Pokorak, E.; Prinz, M.; Salas, A.; Schneider, P.M.; et al. DNA Commission of the International Society for Forensic Genetics: Revised and extended guidelines for mitochondrial DNA typing. Forensic Sci. Int. Genet. 2014, 13, 134–142. [Google Scholar] [CrossRef]
- Parson, W.; Dür, A. EMPOP—A forensic mtDNA database. Forensic Sci. Int. Genet. 2007, 1, 88–92. [Google Scholar] [CrossRef]
- Huber, N.; Parson, W.; Dür, A. Next generation database search algorithm for forensic mitogenome analyses. Forensic Sci. Int. Genet. 2018, 37, 204–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jónsson, H.; Ginolhac, A.; Schubert, M.; Johnson, P.L.F.; Orlando, L. mapDamage2.0: Fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 2013, 29, 1682–1684. [Google Scholar] [CrossRef] [PubMed]
- Xavier, C.; de la Puente, M.; Mosquera-Miguel, A.; Freire-Aradas, A.; Kalamara, V.; Vidaki, A.; Gross, T.; Revoir, A.; Pośpiech, E.; Kartasińska, E.; et al. Development and validation of the VISAGE AmpliSeq basic tool to predict appearance and ancestry from DNA. Forensic Sci. Int. Genet. 2020, 48, 102336. [Google Scholar] [CrossRef] [PubMed]
- Chaitanya, L.; Breslin, K.; Zuniga, S.; Wirken, L.; Pospiech, E.; Kukla-Bartoszek, M.; Sijen, T.; Knijff, P.; Liu, F.; Branicki, W.; et al. The HIrisPlex-S system for eye, hair and skin colour prediction from DNA: Introduction and forensic developmental validation. Forensic Sci. Int. Genet. 2018, 35, 123–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, S.; Liu, F.; Ballantyne, K.N.; van Oven, M.; Lao, O.; Kayser, M. IrisPlex: A sensitive DNA tool for accurate prediction of blue and brown eye colour in the absence of ancestry information. Forensic Sci. Int. Genet. 2011, 5, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.; Liu, F.; Wollstein, A.; Kovatsi, L.; Ralf, A.; Kosiniak-Kamysz, A.; Branicki, W.; Kayser, M. The HIrisPlex system for simultaneous prediction of hair and eye colour from DNA. Forensic Sci. Int. Genet. 2013, 7, 98–115. [Google Scholar] [CrossRef] [Green Version]
- Rohland, N.; Siedel, H.; Hofreiter, M. A rapid column-based ancient DNA extraction method for increased sample throughput. Mol. Ecol. Resour. 2010, 10, 677–683. [Google Scholar] [CrossRef]
- Briggs, A.W.; Stenzel, U.; Johnson, P.L.; Green, R.E.; Kelso, J.; Prüfer, K.; Meyer, M.; Krause, J.; Ronan, M.T.; Lachmann, M.; et al. Patterns of damage in genomic DNA sequences from a Neandertal. Proc. Natl. Acad. Sci. USA 2007, 104, 14616–14621. [Google Scholar] [CrossRef] [Green Version]
- Orlando, L.; Ginolhac, A.; Raghavan, M.; Vilstrup, J.; Rasmussen, M.; Magnussen, K.; Steinmann, K.E.; Kapranov, P.; Thompson, J.F.; Zazula, G.; et al. True single-molecule DNA sequencing of a pleistocene horse bone. Genome Res. 2011, 21, 1705–1719. [Google Scholar] [CrossRef] [Green Version]
- Dabney, J.; Meyer, M.; Pääbo, S. Ancient DNA damage. Cold Spring Harb. Perspect. Biol. 2013, 5, a012567. [Google Scholar] [CrossRef]
- Bennett, E.A.; Massilani, D.; Lizzo, G.; Daligault, J.; Geigl, E.M.; Grange, T. Library construction for ancient genomics: Single strand or double strand? Biotechniques 2014, 56, 289–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wales, N.; Carøe, C.; Sandoval-Velasco, M.; Gamba, C.; Barnett, R.; Samaniego, J.A.; Madrigal, J.R.; Orlando, L.; Gilbert, M.T. New insights on single-stranded versus double-stranded DNA library preparation for ancient DNA. Biotechniques 2015, 59, 368–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gansauge, M.T.; Meyer, M. A Method for Single-Stranded Ancient DNA Library Preparation. Methods Mol. Biol. Clifton N.J. 2019, 1963, 75–83. [Google Scholar] [CrossRef]
- Sidstedt, M.; Steffen, C.R.; Kiesler, K.M.; Vallone, P.M.; Rådström, P.; Hedman, J. The impact of common PCR inhibitors on forensic MPS analysis. Forensic Sci. Int. Genet. 2019, 40, 182–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xavier, C.; Eduardoff, M.; Bertoglio, B.; Amory, C.; Berger, C.; Casas-Vargas, A.; Pallua, J.; Parson, W. Evaluation of DNA Extraction Methods Developed for Forensic and Ancient DNA Applications Using Bone Samples of Different Age. Genes 2021, 12, 146. https://doi.org/10.3390/genes12020146
Xavier C, Eduardoff M, Bertoglio B, Amory C, Berger C, Casas-Vargas A, Pallua J, Parson W. Evaluation of DNA Extraction Methods Developed for Forensic and Ancient DNA Applications Using Bone Samples of Different Age. Genes. 2021; 12(2):146. https://doi.org/10.3390/genes12020146
Chicago/Turabian StyleXavier, Catarina, Mayra Eduardoff, Barbara Bertoglio, Christina Amory, Cordula Berger, Andrea Casas-Vargas, Johannes Pallua, and Walther Parson. 2021. "Evaluation of DNA Extraction Methods Developed for Forensic and Ancient DNA Applications Using Bone Samples of Different Age" Genes 12, no. 2: 146. https://doi.org/10.3390/genes12020146