
Abstract
A new approach to reduce the chromium and aluminium concentrations in FeCrAl alloys without significantly impairing corrosion resistance is to alloy with 1–2 wt.% silicon. This paper investigates the “silicon effect” on oxidation by comparing the oxidation behavior and scale microstructure of two FeCrAl alloys, one alloyed with silicon and the other not, in dry and wet air at 600 °C and 800 °C. Both alloys formed thin protective oxide scales and the Cr-evaporation rates were small. In wet air at 800 °C the Si-alloyed FeCrAl formed an oxide scale containing mullite and tridymite together with α- and γ-alumina. It is suggested that the reported improvement of the corrosion resistance of Al- and Cr-lean FeCrAl’s by silicon alloying is caused by the appearance of Si-rich phases in the scale.
Similar content being viewed by others

Introduction
Ferritic Fe-base alloys containing chromium and aluminium, commonly referred to as FeCrAl alloys are frequently used as high temperature resistant materials. The ability of FeCrAl alloys to resist high temperature corrosion relies on the formation of a protective alumina scale. Alumina scales are often superior to chromia scales because they grow more slowly and form better barriers towards corrosive species in the environment [1]. In addition, the reactions of chromia with alkali to form alkali chromate and with O2 and H2O to form volatile chromium-oxy-hydroxide, are minimized. Alumina scale growth usually follows sub-parabolic kinetics [2].
Traditionally, FeCrAl alloys are used in the 900–1200 °C temperature range where protective α-alumina scales form [3]. However, recently there has been an increasing interest to use FeCrAl alloys in lower temperature applications such as biomass boilers [4], nuclear [5, 6] and SOFC balance-of-plant components [7], operating in the temperature range 500–800 °C. In this temperature range, FeCrAl alloys form oxide scales dominated by metastable forms of alumina which are less protective than α-alumina scales. However the “metastable” alumina scales are still superior to chromia scales. Using FeCrAl alloys at such relatively low temperatures gives rise to two major challenges: Firstly, the relatively slow diffusion of aluminium in the alloy and the corresponding slow supply of aluminium to the growing scale can hinder formation of a protective alumina scale at low temperature; secondly, the formation of significant amounts of brittle phases such as α′ and sigma phase has to be avoided in order to maintain ductility of the material [8, 9].
Normally, both the corrosion resistance and the ability to form an alumina scale are improved by increasing the concentration of aluminium and chromium. On the other hand, high levels of chromium and aluminium make the material more sensitive to embrittlement. Previous studies indicated that a composition around 10–12 wt.% chromium and 4–5 wt.% aluminium would be optimal for the studied temperature range, allowing the formation of an alumina scale without significantly impairing the ductility of the material by the formation of brittle phases [10]. However, it has been reported that, in the temperature range of interest, such Al- and Cr-lean alloys are unable to form a protective alumina scale in the presence of a combination of O2 and H2O [10, 11]. A recent study by Eklund et al. showed that alloying with 1–2 wt.% silicon improved the ability of Cr-lean FeCrAl alloys to form an alumina scale at 600 °C [11]. However, the reason for the improved oxidation resistance was not discovered and no enrichment of silicon was observed in the protective oxide scale.
Water vapour is known to play an important role in alumina scale growth. Thus, the oxidation rate of FeCrAl alloys has been reported to increase in the presence of water vapour. This effect has been attributed to the influence of water to slow down the transformation of metastable alumina into α-alumina [12, 13]. In a recent investigation at low oxygen activity, it was found that water is transported across the alumina scale, via the alumina grain boundaries [14].
When exposed to environments containing a combination of O2 and H2O, chromia-forming alloys suffer volatilization of chromium-oxy-hydroxide from the protective scale which can have major consequences for oxidation behaviour [15]. While the volatilization of chromium-oxy-hydroxide from FeCrAl alloys is expected to be much less than for chromia-forming alloys, some chromium-oxy-hydroxide volatilization is still expected, especially in the early, transient, oxidation stage. Accordingly, the rate of chromium-oxy-hydroxide formation from alumina-forming alloys in air with ≤ 12% H2O has been reported to be 1–3 orders of magnitudes lower than for chromia-formers in the 800–1000 °C range [16, 17]. In the current study, Cr-evaporation in air with 40% H2O at 600 °C and 800 °C is studied by a denuder technique [18].
In order to find the reasons behind the beneficial effect of silicon on the oxidation behaviour of Al- and Cr-lean FeCrAl’s in the presence of H2O and O2, the present study features exposures at higher temperature (800 °C) than the study by Eklund et al. [11]. The idea is to form a protective oxide scale which is thick enough to enable a detailed analysis of the oxidation microstructure and to allow using X-ray diffraction to identify crystalline phases in the scale. To enable comparison with protective alumina scales formed in the absence of silicon, the present study includes a “reference” alloy which is not added with silicon but which still forms a protective alumina scale in O2 + H2O environment because it features somewhat higher concentrations of aluminium and chromium.
The purpose of this study is to elucidate how silicon improves the ability to form a protective oxide scale by investigating oxidation behaviour in terms of scale morphology, chromium-oxy-hydroxide volatilization and oxidation rate.
Materials and Experimental
Materials
The chemical compositions of the alloys studied are shown in Table 1. The alloy designated Alkrothal 14 (A14) is slightly higher in chromium and aluminium while Alloy 197 (A197) has higher silicon content. Both alloys contain a small concentration of Reactive Elements (REs) which are present in the alloy as finely dispersed nanoparticles similar as in the alloy studied in [14]. The alloys were manufactured in full production batches by casting and hot rolling into 1 mm strip followed by annealing.
Sample coupons were cut to nominal dimension 12 × 12 × 1 mm. The coupons were prepared by grinding followed by polishing with 1 µm diamond paste on the faces while the edges were ground with 1000# mesh grit SiC paper in the final step. After polishing, the coupons were cleaned in an ultrasonic bath in two steps, first in acetone, then in ethanol.
Experimental Setup
The corrosion experiments were performed in horizontal tube furnaces equipped with 44 mm inner diameter fused silica reaction tubes. In the dry exposures, ambient air was dried and purified by passing the gas stream through potassium permanganate filters. The dew point of the gas was measured to be below −35 °C. In the wet exposures the purified air was bubbled through a heated water bath and then passed through a reflux condenser to achieve a dew point of 76.4 ± 0.3 °C, resulting in 40 vol.% of water in the gas. Dew point calibrations were performed prior to the exposures with a Michell Instruments Optidew Vision™ chilled mirror humidity sensor. A gas flow of 3,000 ± 5 ml/min (NTP) calibrated with a MesaLabs Defender™ 530 + flow meter was used, resulting in a gas flow velocity of 11.8 cm/s at 800 °C and 9.8 cm/s at 600 °C. Upstream of the samples, a flow restrictor made of porous ceramic monolith was placed to achieve a more homogenous gas flow and to increase the efficiency of the Cr-evaporation measurements.
The evaporation of chromium-oxy-hydroxide from the samples was measured using a method described in detail in [18], where the output gas from the reaction tube was passed through a fused silica denuder tube coated with Na2CO3 (s) which reacts with CrO2(OH)2(g) to form solid sodium chromate according to Reaction (1):
The denuder tube was then leached with water and the solution was analysed quantitively for chromate by photospectrometry using a Thermo Scientific Evolution™ 60S UV/vis spectrophotometer at a wavelength of 370 nm and using a Merck Certipur™ chromate solution as a standard.
After completing the exposures (20 h or 200 h), the sample mass change was recorded using a Sartorius™ 6-decimal balance.
Analysis Methods
Exposed samples were examined by means of Scanning Electron Microscopy equipped with an Energy Dispersive X-ray Spectrometer (SEM–EDX). Cross-sectional samples were prepared by Broad-Ion-Beam (BIB) milling to enable examination of an area which is not influenced by grinding and polishing. Top view analyses were performed using a FEI Quanta 200™ Field Emission Gun Scanning Electron Microscopy (FEG-SEM) while cross-sectional analyses were performed by a Zeiss Ultra 55™ FEG-SEM.
In-depth analyses of selected samples were performed using Scanning Transmission Electron Microscopy (STEM), where the area of interest was lift-out prepared using Focused-Ion-Beam (FIB) milling in a FEI Versa 3D™ workstation. Analysis was performed in an FEI Titan 80-300™ TEM.
The crystalline oxide phases formed were examined using Grazing Incidence X-ray Diffraction (GIXRD) using a Siemens D5000™ powder diffractometer equipped with a Göbel mirror. A Cu-Kα X-ray source was used, emitting X-rays with a wavelength of 1.5418 Å. The incidence angle was 0.2°.
Results
Exposures at 800 °C
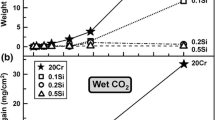
Figure 1 presents the net mass gains of the two alloys after 20 h and 200 h at 800 °C in dry air and in wet air, i.e. air with 40 vol% H2O. The figure shows that the oxidation kinetics are sub-parabolic. While all mass gains are small, A197 exhibits slightly larger mass gains than A14 after 200 h. Also, exposure in wet air resulted in slightly higher mass gains than dry air.

Net mass changes for A197 and A14 in dry and wet air after 20 h and 200 h exposures at 800 °C. The dashed line is a parabolic help line for comparison (y \(= \sqrt {4.5 \times 10^{ - 5} \times x} )\)
Plan view SEM-BSE images of the Si-alloyed and Cr- and Al-lean A197 after 20 h and 200 h exposure at 800 °C are presented in Fig. 2, showing that a smooth surface oxide (the “base oxide”) has formed in both wet and dry air. The bright spots seen at high magnification (insert) are rich in RE elements, corresponding to RE-particles in the alloy. After 200 h in wet air, the area in the immediate vicinity of the RE particles has become dark, corresponding to formation of a significantly thicker oxide layer in those areas. This is not the case in dry air. Another feature only observed after 200 h in wet air is a bright “halo” around the RE-particles. Figure 3 shows cross-sectional images of A197 after 20 h and 200 h in wet air, and after 200 h in dry air. Table 2 shows the measured oxide thicknesses and the corresponding values calculated from the mass gains. The fair agreement of calculated and measured scale thicknesses indicates that the cross sections are representative for the samples. The influence of chromia volatilization (see later in this paper) on calculated thickness was minimal and was not taken into account.

Top view SEM-BSE images of A197 samples exposed to a dry air 20 h, b dry air 200 h, c wet air 20 h and d wet air 200 h, at 800 °C

Cross-sectional SEM-SE images of A197 samples exposed to a wet air 20 h, b wet air 200 h, c dry air 200 h, at 800 °C. Dashed lines indicates the interface between the inner dense layer and the outer porous layer
The oxide scale is layered in all cases, consisting of a thin and dense inner layer and a thicker outer layer which is somewhat porous. Figure 4 shows cross section STEM-images and line scans across the oxide scale of A197 after 1 h, 20 h and 200 h in wet air. The line scans shows that the oxide scale is rich in both silicon and aluminium, silicon being predominant in the outer part while the scale interior is dominated by aluminium. By comparing the scales at the different exposure times it is seen that silicon is enriched in the outer part of the scale reaching > 60 at.% (cationic) after 20 h and > 90 at.% after 200 h. At the same time the aluminium concentration in the outer part is about 20–30 at.% after 20 h and < 10 at % after 200 h. The middle of the scale consist of a mixture of silicon and aluminium cations as gradients. The inner part of the scale consist mainly of aluminium cations with an aluminium concentration of 70–80 at.% after 20 h and > 90 at.% after 200 h. In the scale formed after 20 h a feature is observed at about 50 nm below the oxide surface where a sharp drop in silicon content and an increase in iron content is observed. The feature corresponds to a needle-shaped bright feature in the scale microstructure (indicated by an arrow). The chromium concentration in the oxide scale is quite low with a decreasing concentration with time. The chromium content observed is located close to the original metal/oxide interface.

STEM-images and line scans of A197 sample exposed to wet air 1, 20 and 200 h at 800 °C. The line scans show cation concentrations through the oxide indicating high amount of aluminium together with silicon
Figure 5 shows plan view SEM-images of the reference alloy, A14, after exposure at 800 °C. Compared to A197 the oxide is rougher, oxide ridges having formed at alloy grain boundaries, generating a dark contrast in the SEM-BSE images. Also, the oxide scale does not show the characteristic thickening at the RE-particles seen for A197. Cross-sectional images of A14, after 200 h in wet and dry air, are presented in Fig. 6. The image in Fig. 6b shows an oxide ridge at an alloy grain boundary, the scale thickness reaching about 530 nm. As for A197, the scale is layered, consisting of a dense inner part and a more porous outer part. In comparison to A197, the dense inner scale on A14 is thicker and the outer scale has more and larger pores.

Top view SEM-BSE images of A14 samples exposed to a dry air 20 h, b dry air 200 h, c wet air 20 h and d wet air 200 h, at 800 °C

Cross-sectional SEM-SE images of A14 samples exposed to a wet air 200 h, b dry air 200 h, at 800 °C, where the left side represents the general thickness and the right side represent oxide nodules/ridges. Dashed lines indicate the interface between the inner dense layer and the outer porous layer
The surprisingly large amounts of silicon detected in the scale formed on A197 in wet air by STEM/EDX (Fig. 4) prompted additional analyses by plan view SEM/EDX. In order to provide information on the elemental distribution as a function of distance from the scale surface, different acceleration voltages were used. The analysed areas were about 0.15 mm2. Figure 7 shows the spectra, including the aluminium Kα peak at 1.486 eV and silicon Kα peak at 1.739 eV, for three samples; A197 after 200 h in wet air, A197 after 200 h in dry air and for A14 after 200 h in wet air. Note that in the case of A197 exposed in wet air for 200 h, the silicon/aluminium peak ratio increases when the acceleration voltage is decreased. This in accordance with the STEM linescan results in Fig. 4, showing that silicon is indeed enriched in the oxide. In the two other cases no such increase in the silicon signal was observed, indicating that there was no significant enrichment of silicon in the scale in the other cases, i.e., for A197 in dry air or for A14 in wet air.

SEM-EDX examination from top view of a A197 exposed in wet air 200 h b A197 exposed in dry air 200 h c A14 exposed in wet air 200 h, at 800 °C. The two peaks show intensity for Al and Si normalized to Al for 3 different acceleration voltages; 5, 10 and 20 kV
X-ray diffractograms acquired from A197 and A14 after 200 h in dry and wet air are presented in Fig. 8. The diffractograms representing A14 in wet and dry air and A197 in dry air are similar, showing the presence of both α-alumina and γ-alumina. Diffraction peaks representing the substrate were also observed. In the case of A14 the γ-alumina peaks are slightly shifted compared to A197 in dry air, consistent with an increase of the lattice parameter a from 7.94 to 8.02 Å. The diffraction pattern generated by A197 in wet air is very different from the other three diffractograms, having multiple additional peaks, especially in the 2θ interval between 15° and 42°. Thus, in addition to α-alumina and γ-alumina, the diffraction pattern showed strong evidence for mullite (Al2(Al2+2xSi2−2x)O10−x) (PDF 04-016-1589). In addition, tridymite, a high temperature SiO2 polymorph (PDF 01-071-0261) was identified. A few of the peaks could not be attributed to a specific crystalline phase.

XRD-measurements from A14 and A197 coupons exposed in dry and wet air at 800 °C. Peaks are labeled as (M) mullite, (T) tridymite, (Al) metallic aluminium from mounting of the specimens, (S) substrate, (γ) γ-alumina, (α) α-alumina, (U) unidentified peak
Exposures at 600 °C
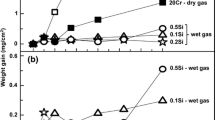
Figure 9 shows net mass changes for both alloys after exposures at 600 °C in dry and wet air. As expected, the mass changes are lower than at 800 °C (c.f. Figure 1). Both alloys exhibit strongly sub-parabolic mass gain kinetics, the mass gains in the wet air being slightly higher than in dry air.

Net mass changes measured for A197 and A14 in dry and wet air after 20 h and 200 h exposures at 600 °C. The dashed line is a parabolic help line for comparison (y \(= \sqrt {8 \times 10^{ - 7} \times x}\))
Plan view SEM-BSE images of A197 after exposure at 600 °C are presented in Fig. 10. The surface is covered by a smooth and uniform base oxide which includes frequent, relatively large, Fe-rich oxide nodules which appear in the vicinity of RE-particles. The Fe-rich oxide nodules formed in both dry and wet conditions but are slightly more frequent in wet air. The number density and size of the Fe-rich oxide nodules were similar after 20 h and 200 h of exposure, showing that they have formed during the first 20 h of exposure and that they do not grow with time. In contrast, a smooth and uniform oxide scale covered the A14 samples in all cases, no Fe-rich oxide nodules being observed, see SEM-BSE images in Fig. 11.

Top view SEM-BSE images of A197 samples exposed to a dry air 20 h, b dry air 200 h, c wet air 20 h and d wet air 200 h, at 600 °C

Top view SEM-BSE images of A14 samples exposed to a dry air 20 h, b dry air 200 h, c wet air 20 h and d wet air 200 h, at 600 °C
Figure 12 shows cross-sectional images of Alloy 197 after 200 h at 600 °C in wet air. The top image shows an area at an Fe-oxide nodule. The element mapping shows that the part of the oxide nodule which protrudes from the sample surface consists of relatively pure iron oxide. The lower part of the nodule forms an intrusion in the alloy substrate and is rich in the oxides of chromium and aluminium, in addition to iron oxide. RE-rich regions can be seen in the bottom of the intrusion. The bottom image shows the smooth and uniform base oxide, having a thickness of approximately 30 nm. Based on mass gain the calculated average oxide thickness is 80 nm for A197. The rather large discrepancy is attributed to the presence of the oxide nodules.

Cross-sectional image showing A197 after exposure in wet air for 200 h at 600 °C. Top image shows the area at a Fe-oxide nodule with element mapping. The lower image shows the base oxide
Chromium Evaporation
Figure 13 shows the rate of Cr-evaporation from the two alloys in wet air at 600 °C and 800 °C. The insert compares the rate of Cr-volatilization at 800 °C from the two alloys with the chromia-forming alloy 310S (25Cr20Ni), using identical exposure conditions. The figure shows that Cr-evaporation from the two alumina-forming alloys is 2–3 orders of magnitude lower than for 310S. A comparison of the two alumina-formers shows that Cr-evaporation from A197 is about \({\raise0.5ex\hbox{$\scriptstyle 1$} \kern-0.1em/\kern-0.15em \lower0.25ex\hbox{$\scriptstyle 3$}}\) of the value for A14. It should be noted that the rates of Cr-evaporation from A197 and A14 are close to the detection limit (about 0.002 µg/cm2h).

Cr2O3-evaporation rate in 200 h exposures in air/40 vol% H2O and a gas flow velocity of 11.8 cm/s at 800 °C and a gas flow velocity of 9.8 cm/s at 600 °C. The smaller diagram in top right corner shows Cr2O3-evaporation for the chromia-forming alloy 310S in same exposure conditions at 800 °C for comparison purpose
Discussion
The idea behind the present paper is to elucidate the beneficial effect of silicon on FeCrAl oxidation behaviour by characterizing the protective alumina scales formed by a Si-alloyed model alloy (designated A197) in humid air and in dry air and by comparing with the microstructure of oxidation of a similar FeCrAl (designated A14) which is not alloyed with silicon. A comparison of the two alloys revealed major differences in scale morphology which are discussed below.
As expected, all exposures resulted in quite small mass gains, both alloys forming protective oxide scales. Accordingly, the microstructural characterization reveals the formation of thin, continuous base oxides with good adherence to the substrate. However, the two alloys exhibit significant differences in oxidation behaviour. Thus, at 600 °C, A197 forms numerous iron oxide nodules during the early stages of oxidation which is not the case for the A14 material. It is suggested that the iron oxide nodules reflect a relatively slow transition from transient oxidation to protective oxidation which, in turn, is connected to the lower concentration of chromium and aluminium in A197.
While the protective oxide layers formed at 600 °C were too thin for detailed analysis, the base oxides formed at 800 °C were in the 200–400 nm range, allowing analysis of the oxide scales. All exposures at 800 °C resulted in similar base oxide morphologies, exhibiting a thin and dense inner layer and a thicker, somewhat porous outer layer (see Figs. 2, 3, 4 and 5). The oxide scale microstructure is similar to that reported in studies of the early oxidation of commercial FeCrAl alloys at 900 °C [13, 19, 20]. Those studies report that the inner dense oxide consists of inward-grown α-alumina while the outer layer is outward-grown and it was concluded that the interface between the inner and outer layers corresponds to the original alloy surface. This was supported by an Atom Probe Tomography study [20], showing the presence of Cr- and Fe-rich nanoparticles at the interface between the inner and outer layers in the oxide scale. The particles were concluded to be remnants from transient oxidation. In analogue, the chromium peak at the interface between the lower and upper oxide layers observed in the present study at 800 °C (see 20 h in Fig. 4), is interpreted to be a remnant of transient oxidation. Also, the outer and inner layers in the base oxide are considered to be outward-grown and inward-grown, respectively.
The greatest difference in the oxidation behaviour of the two alloys is seen in wet air at 800 °C. At this temperature, water vapour has very small effects on the oxidation behaviour of the reference alloy (A14). In contrast, the chemistry and phase composition of the oxide scale on A197 is fundamentally changed when water vapour is present. Thus, the oxide scale formed on A197 in wet air contained large amounts of silicon, while silicon was not detected in the scale formed in dry air (See SEM/EDX analysis in Fig. 7 and the line scan in Fig. 4). The silicon enrichment of the oxide scale on alloy A197 in the presence of water at 800 °C was confirmed by XRD analysis after 200 h which detected mullite (an aluminium silicate) and tridymite (a high temperature SiO2 polymorph) on A197, while no silicon-containing oxides were detected in the other cases (A14, dry and wet air 800 °C, A197 dry air 800 °C). To our knowledge, tridymite has not been reported as a corrosion product previously. Also, the temperature of 800 °C may seem low, considering that tridymite is commonly reported to be stable in the 870–1470 °C range. Yet, the reported preparation of tridymite by heating nanosilica at 800 °C in air with traces of NaCl [21] supports the present observation of tridymite. It may be noted that it is debated whether tridymite is a form of pure SiO2 or if it must contain small concentrations of e.g., alkali ions, hydroxonium ions or Al3+ [22, 23] All samples exposed at 800 °C showed evidence for α- and γ-alumina. Also, the lack of silicon in the oxide scale of A14 exposed to wet air is further evidence that the silicon in the scale of A197 originates from the alloy itself and not from external sources such as volatile species emanating from the silica reactor tube.
The phase composition of the Si-rich scale can be further elucidated by a comparison with the STEM line-scans in Fig. 4. Mullite is a solid solution with the formula Al2(Al2+2xSi2−2x)O10−x. According to Schneider et al. [24], its composition tends to fall in the range from x = 0.25 (3Al2O3.2SiO2) to x = 0.40 (2Al2O3.SiO2), corresponding to Al/Si ratios of 3 and 4, respectively. This corresponds to the Al/Si ratio about 100 nm beneath the surface of the scale after 20 h and 150 nm after 200 h, according to Fig. 4. Closer to the scale surface the silicon content is significantly higher, indicating the presence of SiO2 (tridymite). The aluminium-rich lower part of the scale is suggested to consist of a mixture of α-alumina, γ-alumina and mullite. Mullite is reported to dissolve up to 6 cationic % of Fe3+, substituting for Al3+ [24]. Accordingly, part of the iron detected in the Si-rich scale is suggested to be dissolved in mullite. However, to account for the high iron concentrations observed about 50 nm below the surface of the scale in Fig. 4 (20 h), it is necessary to presume the presence of an unidentified iron-rich mixed oxide. This phase probably corresponds to some of the unidentified peaks in the X-ray diffractogram.
Exposing chromia to humid air at high temperature results in chromia volatilization according to Reaction (2) [25]:
As expected, Cr-evaporation from the two alumina-forming alloys is only a small fraction of that of the chromia-forming alloy 310S, see Fig. 13. The chromium source is suggested to be the chromium enriched “band” in the oxide scale referred to above and the decreasing Cr-evaporation rate is attributed to the gradual depletion of chromium in the scale. An inspection of Fig. 13 shows that increasing the temperature from 600 to 800 °C resulted in a 3–5 time increase in Cr-evaporation from the reference alloy A14 in wet air (by logarithmic interpolation of the values measured at 800 °C). According to the thermodynamics of the volatilization reaction (Reaction 2) the rate of volatilization is expected to accelerate as temperature increases. Accordingly, Cr-evaporation from chromia-forming alloys is reported to increase by a factor of 2–3 per 100 °C in the temperature range studied [26, 27]. In contrast, the rate of Cr-evaporation from the Si-alloyed A197 was almost the same at 600 °C and 800 °C (by logarithmic interpolation of the measured values at 800 °C), suggesting that the transport of Cr3+ across the scale to the oxide/gas interface is impeded by the silicon-rich scale formed on A197 in humid air.
Silicon has been reported to improve the oxidation properties of high temperatures alloys [11, 28,29,30]. In the case of austenitic stainless steels (353MA™), exposure in dry and wet air at 700 °C and 900 °C was reported to result in formation of silica globules at the alloy/scale interface after 168 h [31]. In contrast, the present study reveals that silicon is accumulated in the outer part of the oxide scale. It is proposed that the beneficial effect of silicon alloying on the oxidation properties of Al- and Cr- lean FeCrAl’s in humid air 600 °C reported by [11] is caused by a Si-rich outer layer which is similar to that detected in the present study after exposure at 800 °C. It is suggested that the slower Cr-evaporation observed when a silicon-rich scale has formed may be a key to this effect. It has been suggested that the chromium-rich band in the lower part of the scale in FeCrAl’s (a result of transient oxidation) plays a role in the formation of a protective alumina scale [20]. This implies that the integrity of the alumina scale may be compromised by factors that deplete the oxide scale in chromia, e.g., chromia volatilization. Thus, it is hypothesized that the positive effect of silicon on FeCrAl oxidation reported by [11] is due to a slower transport of chromium ions across the Si-rich scale, from the Cr-enriched band towards the scale/gas interface.
Considering that large amounts of silicon are detected in the scale already after 20 h (see Fig. 4) it is suggested that the silicon enrichment observed in the scale in wet air is connected to the influence of water vapour on the early stages of oxidation. The observation of silicon, in the form of SiO2 and in mullite, in wet air and not under dry conditions implies that the transport of silicon across the scale involves water. Transport of silicon in the form of silicon monoxide is ruled out since SiO(g) formation would not depend on the availability of water. Because of the absence of large cracks and open porosity in the scale, transport by Si(OH)4(g) is also unlikely. This suggests that the transport of silicon occurs by a hydroxylated form of silica which is transported across the scale in the solid state, i.e. by grain boundary diffusion. This hypothesis is not supported by direct evidence and it is emphasized that more work is needed to understand both the mechanism of the “silicon effect” and its consequences for the ability of FeCrAl alloy’s to resist high temperature corrosion.
Summary
-
Exposure of a chromium-and aluminium-lean FeCrAl alloy containing 1.3 wt.% silicon to wet air at 800 °C resulted in the formation of a protective oxide scale which included the silicon-containing phases mullite and tridymite. Silicon was primarily found in the outer part of the scale. In addition, γ-Al2O3 and α-Al2O3 were also present in the scale.
-
Silicon did not enter into the protective oxide scale formed when the same alloy was exposed to dry air. In that case the scale consisted of a mixture of γ-Al2O3 and α-Al2O3 and closely resembled the protective alumina scales formed on a reference alloy which was not added with silicon and which featured higher levels of chromium and aluminium.
-
The previously reported effect of silicon alloying in improving the corrosion resistance of Cr-and Al- lean FeCrAl’s in wet air at medium temperature is attributed to the enrichment of silicon in the oxide scale.
-
Exposure of the two FeCrAl alloys in wet air at 600 °C and 800 °C caused volatilization of chromium-oxy-hydroxide, the rate of volatilization being 2–3 orders of magnitude lower than for an austenitic chromia-forming steel.
References
B. Jönsson and A. Westerlund, Oxidation Comparison of Alumina-Forming and Chromia-Forming Commercial Alloys at 1100 and 1200 A degrees C. Oxidation of Metals 88, (3–4), 2017 (315–326).
D. Naumenko, B. Gleeson, E. Wessel, L. Singheiser and W. J. Quadakkers, Correlation Between the Microstructure, Growth Mechanism, and Growth Kinetics of Alumina Scales on a FeCrAlY Alloy. Metallurgical and Materials Transactions a-Physical Metallurgy and Materials Science 38A, (12), 2007 (2974–2983).
S. E. Sadique, A. H. Mollah, M. S. Islam, M. M. Ali, et al., High-Temperature Oxidation Behavior of Iron–Chromium–Aluminum Alloys. Oxidation of Metals 54, (5), 2000 (385–400).
N. Israelsson, K. A. Unocic, K. Hellstrom, T. Jonsson, et al., A Microstructural and Kinetic Investigation of the KCl-Induced Corrosion of an FeCrAl Alloy at 600 degrees C. Oxidation of Metals 84, (1–2), 2015 (105–127).
J. Ejenstam, B. Jonsson and P. Szakalos, Optimizing the Oxidation Properties of FeCrAl Alloys at Low Temperatures. Oxidation of Metals 88, (3–4), 2017 (361–370).
S. Dryepondt, K. A. Unocic, D. T. Hoelzer, C. P. Massey and B. A. Pint, Development of Low-Cr ODS FeCrAl Alloys for Accident-Tolerant Fuel Cladding. Journal of Nuclear Materials 501, 2018 (59–71).
A. N. Aphale, B. X. Hu, M. Reisert, A. Pandey and P. Singh, Oxidation Behavior and Chromium Evaporation From Fe and Ni Base Alloys Under SOFC Systems Operation Conditions. Jom 71, (1), 2019 (116–123).
P. J. Grobner, 885 Degrees F (475 degrees C) embrittlement of ferritic stainless-steels. Metallurgical Transactions 4, (1), 1973 (251–260).
W. S. Spear and D. H. Polonis, Interstitial Precipitation in Fe-Cr-Al Alloys. Metallurgical and Materials Transactions a-Physical Metallurgy and Materials Science 25, (6), 1994 (1135–1146).
J. Lim, I. S. Hwang and J. H. Kim, Design of Alumina Forming FeCrAl Steels for Lead or Lead-Bismuth Cooled Fast Reactors. Journal of Nuclear Materials 441, (1–3), 2013 (650–660).
J. Eklund, B. Jonsson, A. Persdotter, J. Liske, et al., The Influence of Silicon on the Corrosion Properties of FeCrAl Model Alloys in Oxidizing Environments at 600 Degrees C. Corrosion Science 144, 2018 (266–276).
H. Götlind, F. Liu, J.-E. Svensson, M. Halvarsson and L.-G. Johansson, The Effect of Water Vapor on the Initial Stages of Oxidation of the FeCrAl Alloy Kanthal AF at 900 °C. Oxidation of Metals 67, (5), 2007 (251–266).
F. Liu, H. Josefsson, J. E. Svensson, L. G. Johansson and M. Halvarsson, TEM Investigation of the Oxide Scales Formed on a FeCrAlRE Alloy (Kanthal AF) at 900 Degrees C in Dry O-2 and O-2 with 40% H2O. Materials at High Temperatures 22, (3–4), 2005 (521–526).
N. Mortazavi, C. Geers, M. Esmaily, V. Babic, et al., Interplay of Water and Reactive Elements in Oxidation of Alumina-Forming Alloys. Nature Materials 17, (7), 2018 (610–617).
H. Asteman, J. E. Svensson, L. G. Johansson and M. Norell, Indication of Chromium Oxide Hydroxide Evaporation During Oxidation of 304L at 873 K in the Presence of 10% Water Vapor. Oxidation of Metals 52, (1), 1999 (95–111).
S. Bhowmick, G. Le, A. Verma, and P. Singh Assessment of chromium evaporation from chromia and alumina forming alloys, in Advances in Solid Oxide Fuel Cells Vii, eds. N. P. Bansal, et al. (2011). p. 115–124.
M. Stanislowski, E. Wessel, T. Markus, L. Singheiser and W. J. Quadakkers, Chromium Vaporization from Alumina-Forming and Aluminized Alloys. Solid State Ionics 179, (40), 2008 (2406–2415).
J. Froitzheim, H. Ravash, E. Larsson, L. G. Johansson and J. E. Svensson, Investigation of Chromium Volatilization from FeCr Interconnects by a Denuder Technique. Journal of The Electrochemical Society 157, (9), 2010 (B1295–B1300).
J. Engkvist, S. Canovic, F. Liu, H. Gotlind, et al., Oxidation of FeCrAl foils at 500–900 degrees C in dry O-2 and O-2 with 40% H2O. Materials at High Temperatures 26, (2), 2009 (199–210).
F. Liu, M. Halvarsson, K. Hellstrom, J. E. Svensson and L. G. Johansson, First Three-Dimensional Atomic Resolution Investigation of Thermally Grown Oxide on a FeCrAl Alloy. Oxidation of Metals 83, (5–6), 2015 (441–451).
S. H. Garofalini and A. D. Miller, Kinetics of tridymite formation. Journal of Crystal Growth 78, (1), 1986 (85–96).
S. J. Stevens, R. J. Hand and J. H. Sharp, Polymorphism of Silica. Journal of Materials Science 32, (11), 1997 (2929–2935).
D. K. Smith, Opal, Cristobalite, and Tridymite: Noncrystallinity Versus Crystallinity, Nomenclature of the Silica Minerals and Bibliography. Powder Diffraction 13, (1), 1998 (2–19).
H. Schneider, J. Schreuer and B. Hildmann, Structure and Properties of Mullite—A Review. Journal of the European Ceramic Society 28, (2), 2008 (329–344).
E. J. Opila, D. L. Myers, N. S. Jacobson, I. M. B. Nielsen, et al., Theoretical and Experimental Investigation of the Thermochemistry of CrO2(OH)2(g). The Journal of Physical Chemistry A 111, (10), 2007 (1971–1980).
H. Falk-Windisch, J. E. Svensson and J. Froitzheim, The Effect of Temperature on Chromium Vaporization and Oxide Scale Growth on Interconnect Steels for Solid Oxide Fuel Cells. Journal of Power Sources 287, 2015 (25–35).
T. Sand, C. Geers, Y. Cao, J. Svensson and L. Johansson, Effective Reduction of Chromium-oxy-hydroxide Evaporation from Ni-Base Alloy 690. Oxidation of Metals 92, 2019 (1–21).
S. W. Guan, J. Corkum, and W. Smeltzer, Oxidation behavior of Fe–Al–Si alloys at 1073-K and 1173-K. in, High Temperature Corrosion of Advanced Materials and Protective Coatings, eds. Y. Saito, B. Onay, and T. Maruyama (1992), p. 75–82.
G. C. Wood, J. A. Richardson, M. G. Hobby and J. Boustead, The Identification of Thin Healing Layers at the Base of Oxide Scales on FeCr Base Alloys. Corrosion Science 9, (9), 1969 (659–671).
F. H. Stott, G. J. Gabriel, F. I. Wei and G. C. Wood, The Development of Silicon-Containing Oxides During the Oxidation of Iron–Chromium-Base Alloys. Materials and Corrosion 38, (9), 1987 (521–531).
T. Jonsson, S. Canovic, F. Liu, H. Asteman, et al., Microstructural Investigation of the Effect of Water Vapour on the Oxidation of Alloy 353 MA in Oxygen at 700 and 900 Degrees C. Materials at High Temperatures 22, (3–4), 2005 (231–243).
Funding
Open Access funding provided by Chalmers University of Technology.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sand, T., Edgren, A., Geers, C. et al. Exploring the Effect of Silicon on the High Temperature Corrosion of Lean FeCrAl Alloys in Humid Air. Oxid Met 95, 221–238 (2021). https://doi.org/10.1007/s11085-020-10019-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11085-020-10019-2