
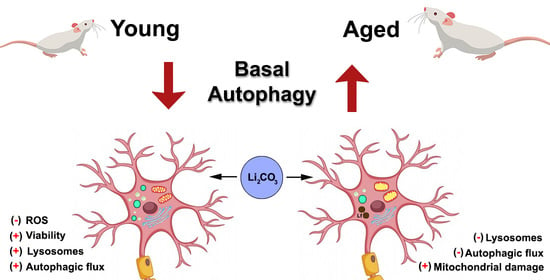
Lack of Autophagy Induction by Lithium Decreases Neuroprotective Effects in the Striatum of Aged Rats
 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Acute and in Loco Treatments in Striatal Slices
2.2.1. Brain Slices Preparation and Lithium Treatments
2.2.2. Western Blotting Analysis
2.2.3. Cell Viability and ROS Measurements
2.2.4. Oxygen Consumption Rate Measurement
2.3. Chronic and In Vivo Treatment and Ultrastructural Analysis
2.4. Statistical Analyses
3. Results
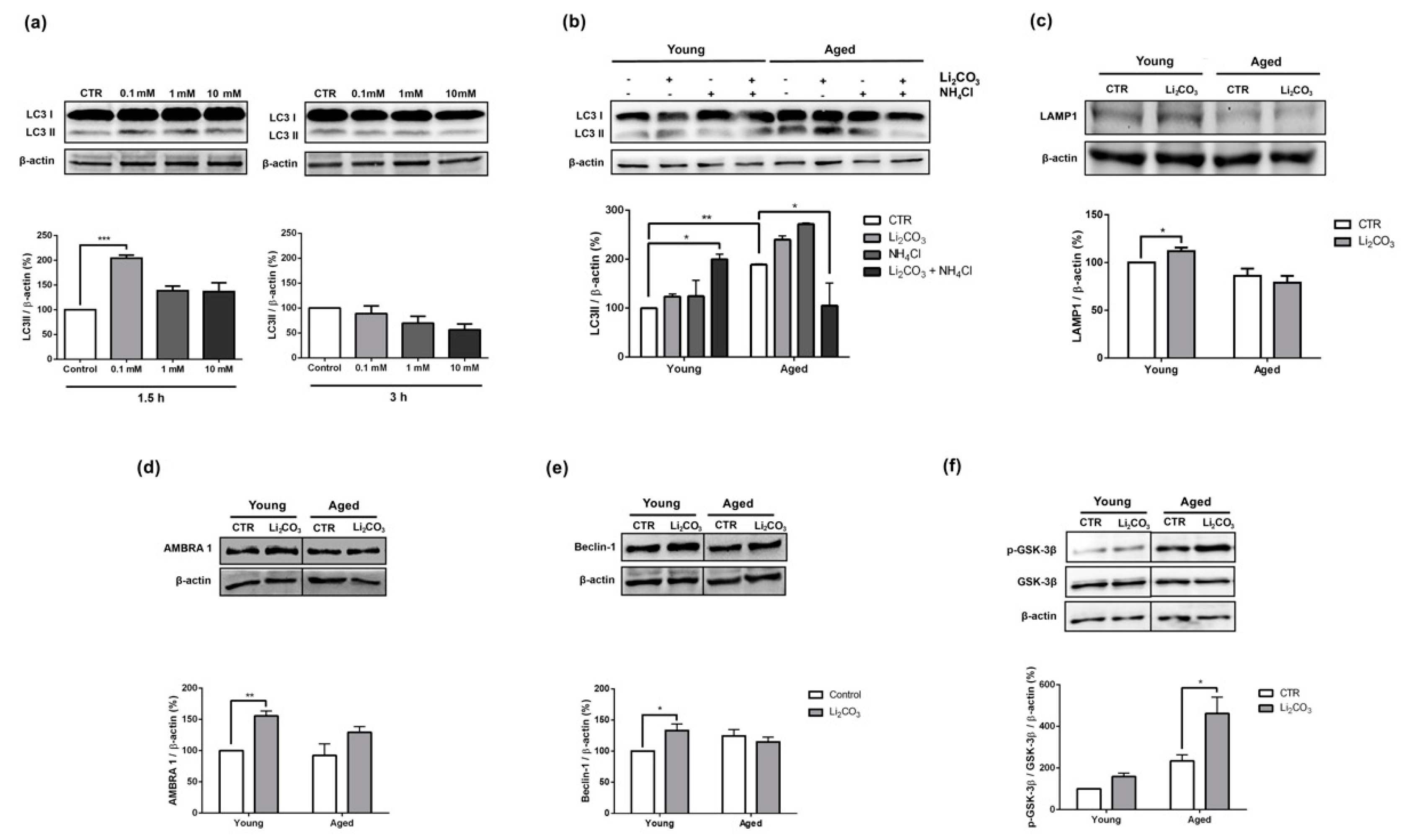
3.1. Acute Lithium Treatment Promotes Opposite Effects on Autophagy in Young and Aged Rat Striatum
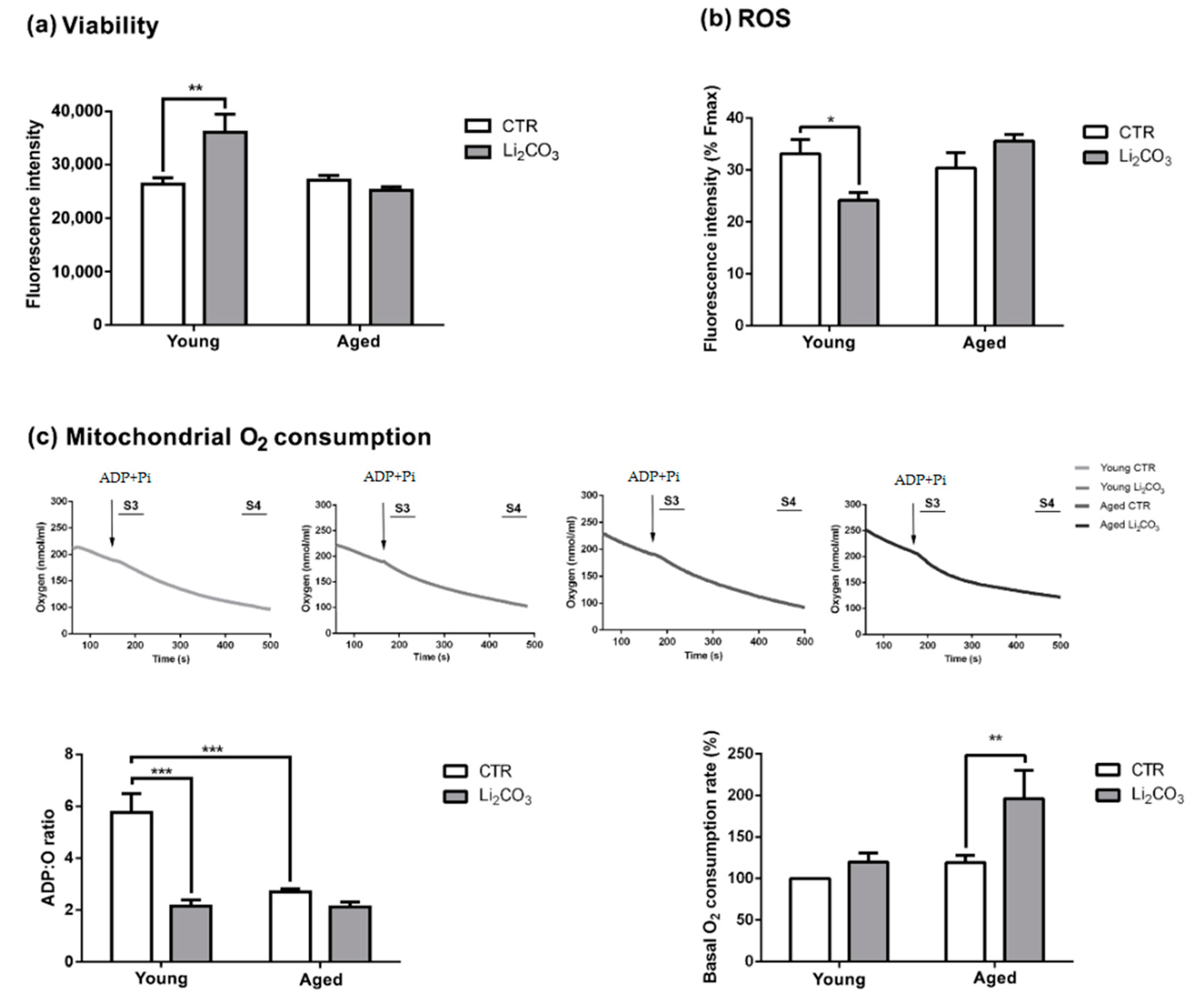
3.2. Acute Lithium Treatment Exerts an Age-Dependent Effect on Cell Viability and ROS Generation
3.3. Acute Lithium Treatment Increases O2 Consumption
3.4. Chronic Lithium Treatment Increases Organelle Damage in Aged Striatum
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goldberg, R.J. Tardive Dyskinesia in Elderly Patients. J. Am. Med. Dir. Assoc. 2003, 4, S32. [Google Scholar] [CrossRef]
- Morimoto, R.I.; Cuervo, A.M. Protein homeostasis and aging: Taking care of proteins from the cradle to the grave. J. Gerontol. A Biol. Sci. Med. Sci. 2009, 64, 167–170. [Google Scholar] [CrossRef] [Green Version]
- Ureshino, R.P.; Bertoncini, C.R.; Fernandes, M.J.S.; Abdalla, F.M.F.; Porto, C.S.; Hsu, Y.T.; Lopes, G.S.; Smaili, S.S. Alterations in calcium signaling and a decrease in Bcl-2 expression: Possible correlation with apoptosis in aged striatum. J. Neurosci. Res. 2010, 88, 438–447. [Google Scholar] [CrossRef]
- Ureshino, R.P.; Hsu, Y.T.; do Carmo, L.G.; Yokomizo, C.H.; Nantes, I.L.; Smaili, S.S. Inhibition of cytoplasmic p53 differentially modulates Ca2+ signaling and cellular viability in young and aged striata. Exp. Gerontol. 2014, 58, 120–127. [Google Scholar] [CrossRef]
- Terman, A. The effect of age on formation and elimination of autophagic vacuoles in mouse hepatocytes. Gerontology 1995, 41, 319–325. [Google Scholar] [CrossRef]
- Meléndez, A.; Tallóczy, Z.; Seaman, M.; Eskelinen, E.L.; Hall, D.H.; Levine, B. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 2003, 301, 1387–1391. [Google Scholar] [CrossRef] [Green Version]
- Vicencio, J.M.; Galluzzi, L.; Tajeddine, N.; Ortiz, C.; Criollo, A.; Tasdemir, E.; Morselli, E.; Ben Younes, A.; Maiuri, M.C.; Lavandero, S.; et al. Senescence, apoptosis or autophagy? When a damaged cell must decide its path—A mini-review. Gerontology 2008, 54, 92–99. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Mariño, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.P.; Liang, Z.Q.; Gu, Z.L.; Qin, Z.H. Molecular mechanism and regulation of autophagy. Acta Pharmacol. Sin. 2005, 26, 1421–1434. [Google Scholar] [CrossRef]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the Integrated Stress Response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [Green Version]
- Ohsumi, Y. Molecular dissection of autophagy: Two ubiquitin-like systems. Nat. Rev. Mol. Cell Biol. 2001, 2, 211–216. [Google Scholar] [CrossRef]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [Green Version]
- Kurz, T.; Terman, A.; Gustafsson, B.; Brunk, U.T. Lysosomes and oxidative stress in aging and apoptosis. Biochim. Biophys. Acta Gen. Subj. 2008, 1780, 1291–1303. [Google Scholar] [CrossRef]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.I.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef]
- Rubinsztein, D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006, 443, 780–786. [Google Scholar] [CrossRef]
- Williams, A.; Sarkar, S.; Cuddon, P.; Ttofi, E.K.; Saiki, S.; Siddiqi, F.H.; Jahreiss, L.; Fleming, A.; Pask, D.; Goldsmith, P.; et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat. Chem. Biol. 2008, 4, 295–305. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Davies, J.E.; Huang, Z.; Tunnacliffe, A.; Rubinsztein, D.C. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and α-synuclein. J. Biol. Chem. 2007, 282, 5641–5652. [Google Scholar] [CrossRef] [Green Version]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef] [Green Version]
- Shaldubina, A.; Agam, G.; Belmaker, R.H. The mechanism of lithium action: State of the art, ten years later. Prog. Neuropsychopharmacol. Biol. Psychiatry 2001, 25, 855–866. [Google Scholar] [CrossRef]
- Crespo-Biel, N.; Camins, A.; Pallàs, M.; Canudas, A.M. Evidence of calpain/cdk5 pathway inhibition by lithium in 3-nitropropionic acid toxicity in vivo and in vitro. Neuropharmacology 2009, 56, 422–428. [Google Scholar] [CrossRef]
- Yasuda, S.; Liang, M.H.; Marinova, Z.; Yahyavi, A.; Chuang, D.M. The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Mol. Psychiatry 2009, 14, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Sugawara, H.; Sakamoto, K.; Harada, T.; Ishigooka, J. Predictors of efficacy in lithium augmentation for treatment-resistant depression. J. Affect. Disord. 2010, 125, 165–168. [Google Scholar] [CrossRef]
- Riadh, N.; Allagui, M.S.; Bourogaa, E.; Vincent, C.; Croute, F.; Elfeki, A. Neuroprotective and neurotrophic effects of long term lithium treatment in mouse brain. BioMetals 2011, 24, 747–757. [Google Scholar] [CrossRef]
- Sarkar, S.; Floto, R.A.; Berger, Z.; Imarisio, S.; Cordenier, A.; Pasco, M.; Cook, L.J.; Rubinsztein, D.C. Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 2005, 170, 1101–1111. [Google Scholar] [CrossRef]
- Klein, P.S.; Melton, D.A. A molecular mechanism for the effect of lithium on development. Proc. Natl. Acad. Sci. USA 1996, 93, 8455–8459. [Google Scholar] [CrossRef] [Green Version]
- Ryves, W.J.; Harwood, A.J. Lithium inhibits glycogen synthase kinase-3 by competition for magnesium. Biochem. Biophys. Res. Commun. 2001, 280, 720–725. [Google Scholar] [CrossRef]
- Chalecka-Franaszek, E.; Chuang, D.M. Lithium activates the serine/threonine kinase Akt-1 and suppresses glutamate-induced inhibition of Akt-1 activity in neurons. Proc. Natl. Acad. Sci. USA 1999, 96, 8745–8750. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.Q.; Lewis, M.C.; Ketterman, J.K.; Clore, E.L.; Riley, M.; Richards, K.R.; Berry-Scott, E.; Liu, X.; Wagner, F.F.; Holson, E.B.; et al. AKT kinase activity is required for lithium to modulate mood-related behaviors in mice. Neuropsychopharmacology 2011, 36, 1397–1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, S.; Krishna, G.; Imarisio, S.; Saiki, S.; O’Kane, C.J.; Rubinsztein, D.C. A rational mechanism for combination treatment of Huntington’s disease using lithium and rapamycin. Hum. Mol. Genet. 2008, 17, 170–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Wang, Z.; Xia, Y.; Kuang, H.; Liu, S.; Li, L.; Tang, C.; Yin, D. The balance of apoptosis and autophagy via regulation of the AMPK signal pathway in aging rat striatum during regular aerobic exercise. Exp. Gerontol. 2019, 124, 110647. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Fu, R.; Wang, Z.; Liu, S.; Tang, C.; Li, L.; Yin, D. Regular Aerobic Exercise-Alleviated Dysregulation of CAMKIIα Carbonylation to Mitigate Parkinsonism via Homeostasis of Apoptosis With Autophagy. J. Neuropathol. Exp. Neurol. 2020, 79, 46–61. [Google Scholar] [CrossRef]
- Poulose, S.M.; Bielinski, D.F.; Shukitt-Hale, B. Walnut diet reduces accumulation of polyubiquitinated proteins and inflammation in the brain of aged rats. J. Nutr. Biochem. 2013, 24, 912–919. [Google Scholar] [CrossRef]
- Pereira, G.J.S.; Antonioli, M.; Hirata, H.; Ureshino, R.P.; Nascimento, A.R.; Bincoletto, C.; Vescovo, T.; Piacentini, M.; Fimia, G.M.; Smaili, S.S. Glutamate induces autophagy via the two-pore channels in neural cells. Oncotarget 2017, 8, 12730–12740. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Arozena, A.A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Fimia, G.M.; Stoykova, A.; Romagnoli, A.; Giunta, L.; Di Bartolomeo, S.; Nardacci, R.; Corazzari, M.; Fuoco, C.; Ucar, A.; Schwartz, P.; et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007, 447, 1121–1125. [Google Scholar] [CrossRef] [Green Version]
- Ouimet, C.C.; Greengard, P. Distribution of DARPP-32 in the basal ganglia: An electron microscopic study. J. Neurocytol. 1990, 19, 39–52. [Google Scholar] [CrossRef]
- Nonaka, S.; Chuang, D.M. Neuroprotective effects of chronic lithium on focal cerebral ischemia in rats. Neuroreport 1998, 9, 2081–2084. [Google Scholar] [CrossRef]
- Wei, H.; Qin, Z.H.; Senatorov, V.V.; Wei, W.; Wang, Y.; Qian, Y.; Chuang, D.M. Lithium suppresses excitotoxicity-induced striatal lesions in a rat model of Huntington’s disease. Neuroscience 2001, 106, 603–612. [Google Scholar] [CrossRef]
- Bianchi, P.; Ciani, E.; Contestabile, A.; Guidi, S.; Bartesaghi, R. Lithium restores neurogenesis in the subventricular zone of the ts65dn mouse, a model for down syndrome. Brain Pathol. 2010, 20, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Liu, H.; Cheng, J.; Zhu, Y.; Xiao, Q.; Bai, Y.; Tao, J. Neuroprotective effects of lithium on a chronic MPTP mouse model of Parkinson’s disease via regulation of α-synuclein methylation. Mol. Med. Rep. 2019, 19, 4989–4997. [Google Scholar] [CrossRef] [PubMed]
- Carlson, S.W.; Dixon, C.E. Lithium improves dopamine neurotransmission and increases dopaminergic protein abundance in the striatum after traumatic brain injury. J. Neurotrauma 2018, 35, 2827–2836. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Rane, A.; Lussier, S.; Andersen, J.K. Lithium protects against oxidative stress-mediated cell death in α-synuclein-overexpressing in vitro and in vivo models of Parkinson’s disease. J. Neurosci. Res. 2011, 89, 1666–1675. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.; Wang, Z.; Tchantchou, F.; Chiu, C.T.; Zhang, Y.; Chuang, D.M. Lithium ameliorates neurodegeneration, suppresses neuroinflammation, and improves behavioral performance in a mouse model of traumatic brain injury. J. Neurotrauma 2012, 29, 362–374. [Google Scholar] [CrossRef] [Green Version]
- Wix-Ramos, R.; Eblen-Zajjur, A. Time course of acute neuroprotective effects of lithium carbonate evaluated by brain impedanciometry in the global ischemia model. Can. J. Physiol. Pharmacol. 2011, 89, 753–758. [Google Scholar] [CrossRef]
- Chiu, C.T.; Liu, G.; Leeds, P.; Chuang, D.M. Combined treatment with the mood stabilizers lithium and valproate produces multiple beneficial effects in transgenic mouse models of huntington’s disease. Neuropsychopharmacology 2011, 36, 2406–2421. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M. Autophagy and aging: Keeping that old broom working. Trends Genet. 2008, 24, 604–612. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.; Takabatake, Y.; Kimura, T.; Takahashi, A.; Namba, T.; Matsuda, J.; Minami, S.; Kaimori, J.Y.; Matsui, I.; Kitamura, H.; et al. Time-dependent dysregulation of autophagy: Implications in aging and mitochondrial homeostasis in the kidney proximal tubule. Autophagy 2016, 12, 801–813. [Google Scholar] [CrossRef] [Green Version]
- Azoulay-Alfaguter, I.; Elya, R.; Avrahami, L.; Katz, A.; Eldar-Finkelman, H. Combined regulation of mTORC1 and lysosomal acidification by GSK-3 suppresses autophagy and contributes to cancer cell growth. Oncogene 2015, 34, 4613–4623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo-Quan, J.I.; Li, L.; Kinghorn, K.J.; Ivanov, D.K.; Tain, L.S.; Slack, C.; Kerr, F.; Nespital, T.; Thornton, J.; Hardy, J.; et al. Lithium Promotes Longevity through GSK3/NRF2-Dependent Hormesis. Cell Rep. 2016, 15, 638–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunk, U.T.; Terman, A. Lipofuscin: Mechanisms of age-related accumulation and influence on cell function. Free Radic. Biol. Med. 2002, 33, 611–619. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donovan, T.R.; Rajendran, S.; O’Reilly, S.; O’Sullivan, G.C.; McKenna, S.L. Lithium modulates autophagy in esophageal and colorectal cancer cells and enhances the efficacy of therapeutic agents in vitro and in vivo. PLoS ONE 2015, 10, e0134676. [Google Scholar] [CrossRef] [Green Version]
- Fabrizi, C.; De Vito, S.; Somma, F.; Pompili, E.; Catizone, A.; Leone, S.; Lenzi, P.; Fornai, F.; Fumagalli, L. Lithium improves survival of PC12 pheochromocytoma cells in high-density cultures and after exposure to toxic compounds. Int. J. Cell Biol. 2014. [Google Scholar] [CrossRef] [Green Version]
- Fiorentini, A.; Rosi, M.C.; Grossi, C.; Luccarini, I.; Casamenti, F. Lithium improves hippocampal neurogenesis, neuropathology and cognitive functions in APP mice. PLoS ONE 2010, 5, e14382. [Google Scholar] [CrossRef]
- Cui, J.; Shao, L.; Young, L.T.; Wang, J.F. Role of glutathione in neuroprotective effects of mood stabilizing drugs lithium and valproate. Neuroscience 2007, 144, 1447–1453. [Google Scholar] [CrossRef]
- Shao, L.; Cui, J.; Young, L.T.; Wang, J.F. The effect of mood stabilizer lithium on expression and activity of glutathione s-transferase isoenzymes. Neuroscience 2008, 151, 518–524. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Navarro, A.; Boveris, A. Brain mitochondrial dysfunction in aging, neurodegeneration, and Parkinson’s disease. Front. Aging Neurosci. 2010, 2, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.F.; Liu, H.; Ni, H.F.; Lv, L.L.; Zhang, M.H.; Zhang, A.H.; Tang, R.N.; Chen, P.S.; Liu, B.C. Improved mitochondrial function underlies the protective effect of pirfenidone against tubulointerstitial fibrosis in 5/6 nephrectomized rats. PLoS ONE 2013, 8, e83593. [Google Scholar] [CrossRef] [PubMed]
- Fornai, F.; Longone, P.; Cafaro, L.; Kastsiuchenka, O.; Ferrucci, M.; Manca, M.L.; Lazzeri, G.; Spalloni, A.; Bellio, N.; Lenzi, P.; et al. Lithium delays progression of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2008, 105, 2052–2057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, N.; Zielke, S.; Michaelis, J.B.; Linder, B.; Warnsmann, V.; Rakel, S.; Osiewacz, H.D.; Fulda, S.; Mittelbronn, M.; Münch, C.; et al. AT 101 induces early mitochondrial dysfunction and HMOX1 (heme oxygenase 1) to trigger mitophagic cell death in glioma cells. Autophagy 2018, 14, 1693–1709. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Shen, L.; Sun, L.; Zhang, X.; Cui, R.; Wang, L. Inhibition of PI3K/mTOR increased the sensitivity of hepatocellular carcinoma cells to cisplatin via interference with mitochondrial-lysosomal crosstalk. Cell Prolif. 2019, 52, e12609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thai, P.N.; Seidlmayer, L.K.; Miller, C.; Ferrero, M.; Dorn, G.W.; Schaefer, S.; Bers, D.M.; Dedkova, E.N. Mitochondrial quality control in aging and heart failure: Influence of ketone bodies and mitofusin-stabilizing peptides. Front. Physiol. 2019, 10, 382. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costa, A.J.; Erustes, A.G.; Sinigaglia, R.; Girardi, C.E.N.; Pereira, G.J.d.S.; Ureshino, R.P.; Smaili, S.S. Lack of Autophagy Induction by Lithium Decreases Neuroprotective Effects in the Striatum of Aged Rats. Pharmaceutics 2021, 13, 135. https://doi.org/10.3390/pharmaceutics13020135
Costa AJ, Erustes AG, Sinigaglia R, Girardi CEN, Pereira GJdS, Ureshino RP, Smaili SS. Lack of Autophagy Induction by Lithium Decreases Neuroprotective Effects in the Striatum of Aged Rats. Pharmaceutics. 2021; 13(2):135. https://doi.org/10.3390/pharmaceutics13020135
Chicago/Turabian StyleCosta, Angelica Jardim, Adolfo Garcia Erustes, Rita Sinigaglia, Carlos Eduardo Neves Girardi, Gustavo José da Silva Pereira, Rodrigo Portes Ureshino, and Soraya Soubhi Smaili. 2021. "Lack of Autophagy Induction by Lithium Decreases Neuroprotective Effects in the Striatum of Aged Rats" Pharmaceutics 13, no. 2: 135. https://doi.org/10.3390/pharmaceutics13020135