Linked-Read Whole Genome Sequencing Solves a Double DMD Gene Rearrangement

 , , ,
, , ,  and
and 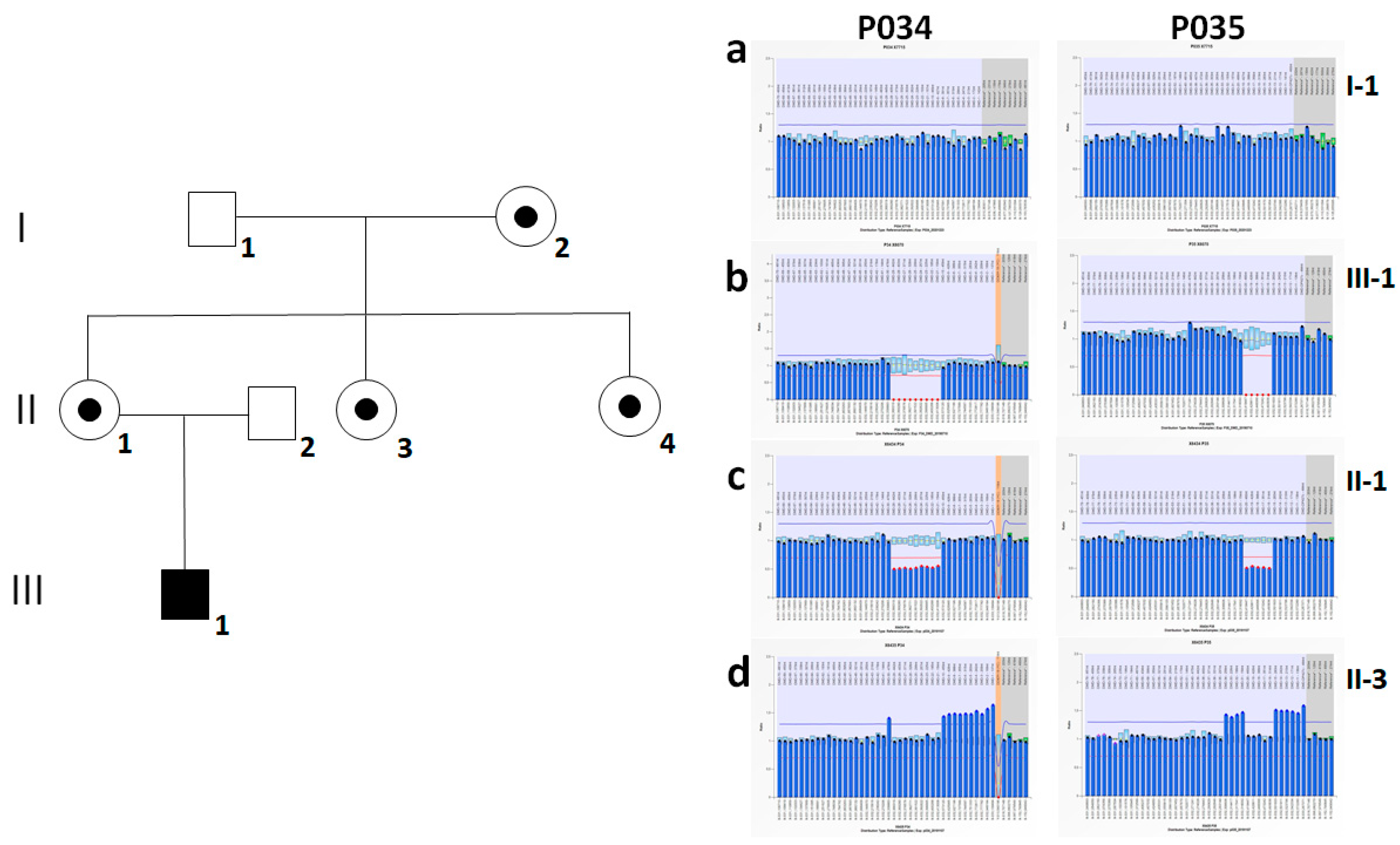{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Index Case
2.2. Sample Collection
2.3. Multiplex Ligation-Dependent Probe Amplification (MLPA)
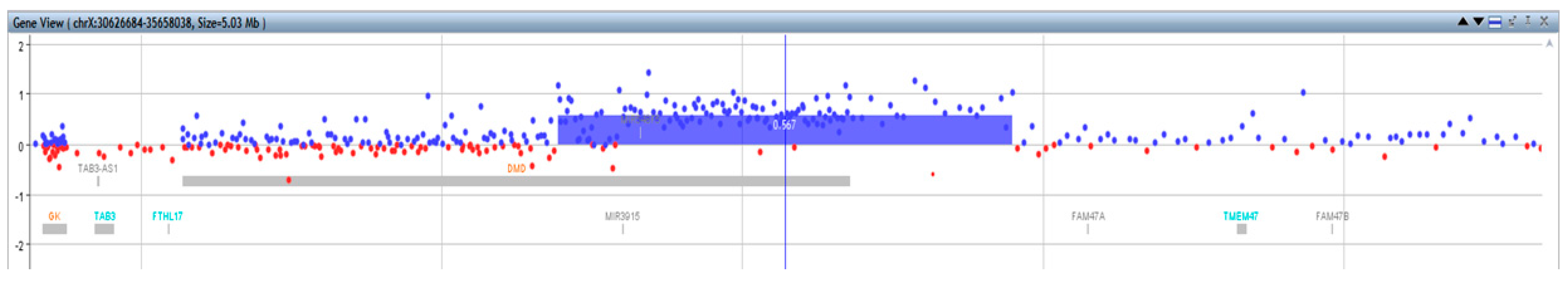
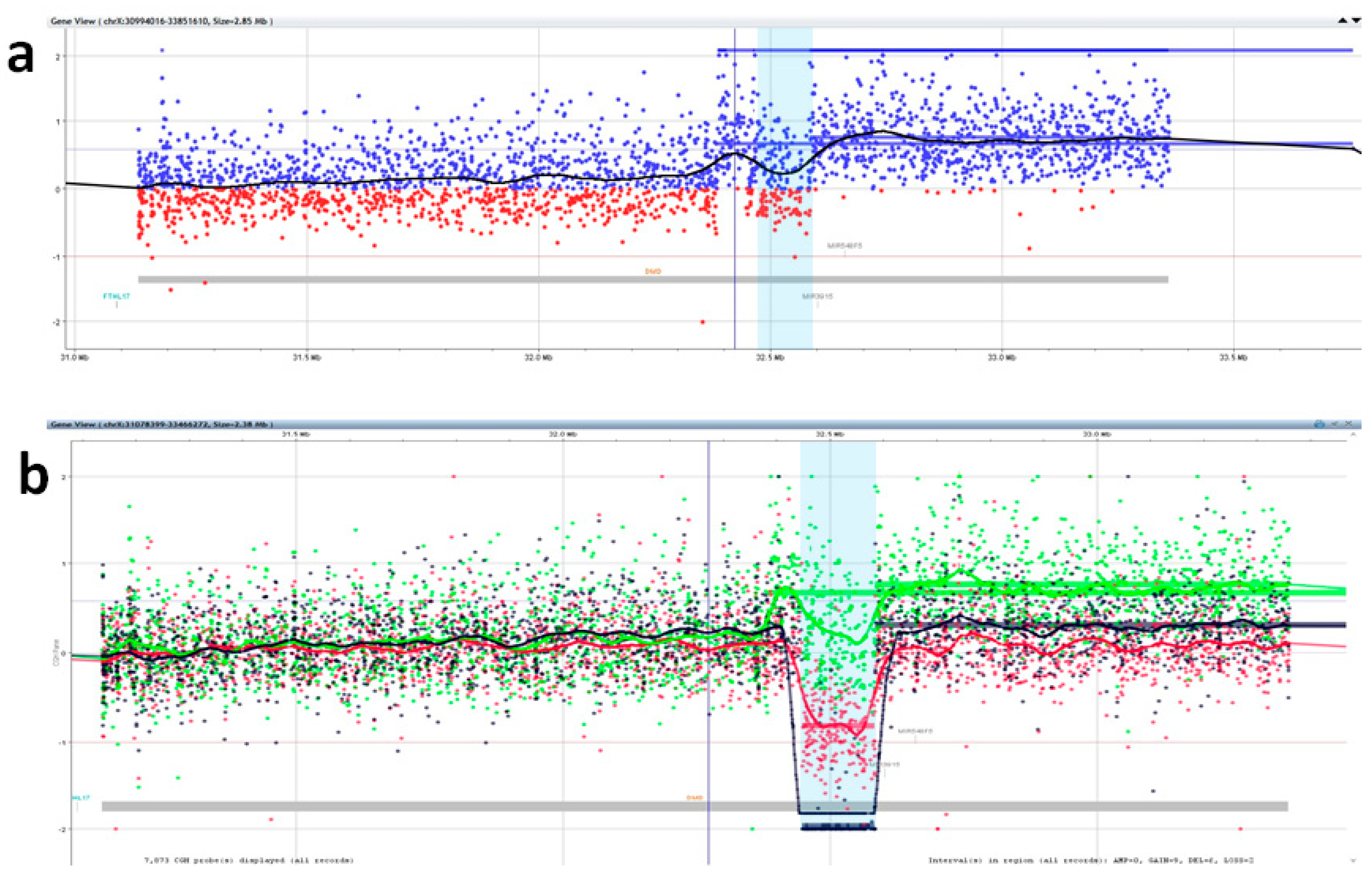
2.4. Array CGH and CGH+SNP
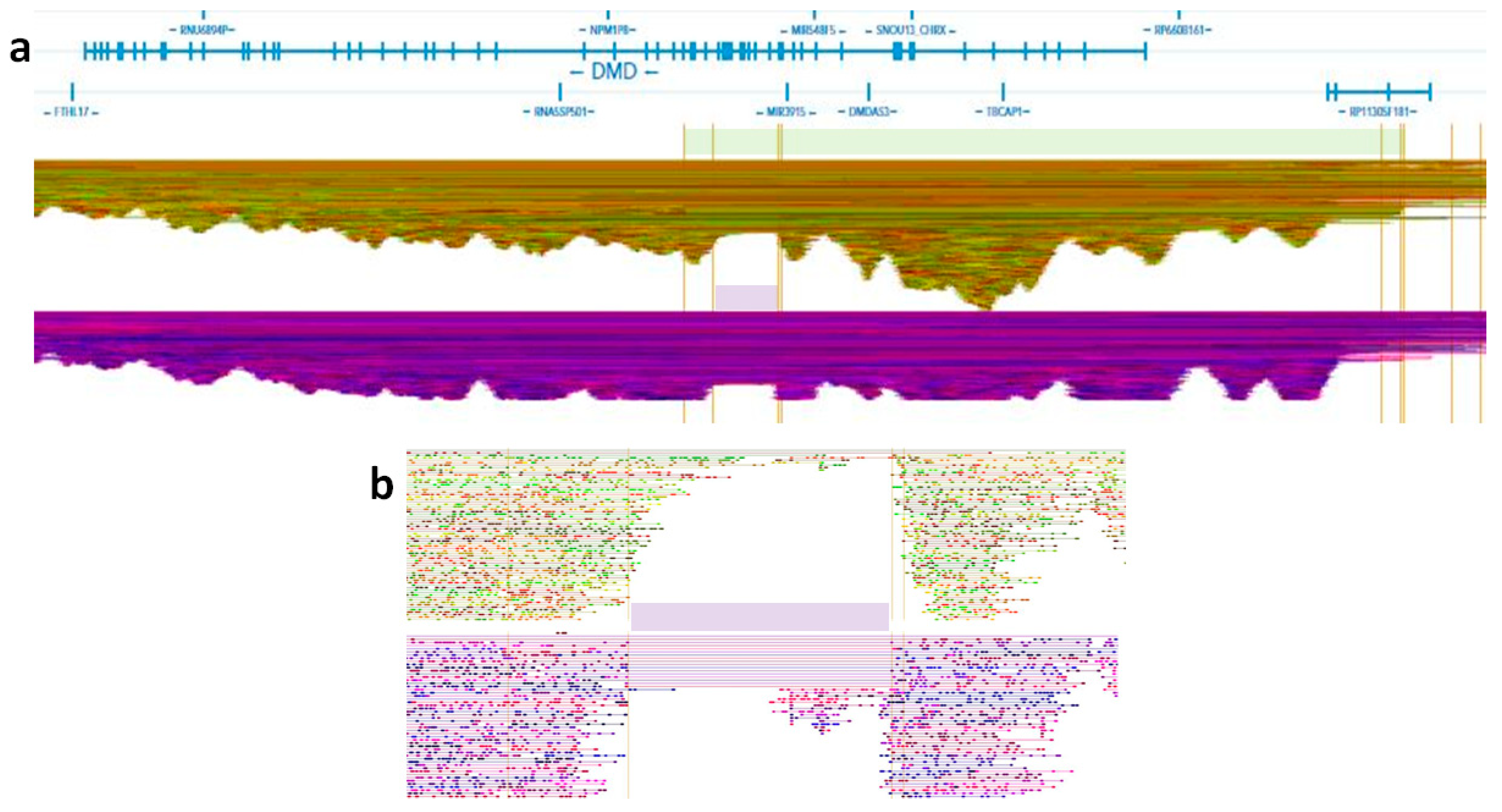
2.5. 10× Genomics Whole Genome Sequencing (10× WGS)
2.6. 1–10× Data Analysis and Visualization
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef]
- Torella, A.; Trimarco, A.; Blanco, F.D.V.; Cuomo, A.; Aurino, S.; Piluso, G.; Minetti, C.; Politano, L.; Nigro, V. One Hundred Twenty-One Dystrophin Point Mutations Detected from Stored DNA Samples by Combinatorial Denaturing High-Performance Liquid Chromatography. J. Mol. Diagn. 2010, 12, 65–73. [Google Scholar] [CrossRef]
- Torella, A.; Zanobio, M.; Zeuli, R.; Del Vecchio Blanco, F.; Savarese, M.; Giugliano, T.; Garofalo, A.; Piluso, G.; Politano, L.; Nigro, V. The position of nonsense mutations can predict the phenotype severity: A survey on the DMD gene. PLoS ONE 2020, 15, e0237803. [Google Scholar] [CrossRef]
- Baskin, B.; Stavropoulos, D.J.; Rebeiro, P.A.; Orr, J.; Li, M.; Steele, L.; Marshall, C.R.; Lemire, E.G.; Boycott, K.M.; Gibson, W.; et al. Complex genomic rearrangements in the dystrophin gene due to replication-based mechanisms. Mol. Genet. Genom. Med. 2014, 2, 539–547. [Google Scholar] [CrossRef] [Green Version]
- Zaum, A.-K.; Stüve, B.; Gehrig, A.; Kölbel, H.; Schara, U.; Kress, W.; Rost, S. Deep intronic variants introduce DMD pseudoexon in patient with muscular dystrophy. Neuromuscul. Disord. 2017, 27, 631–634. [Google Scholar] [CrossRef]
- Trimarco, A.; Torella, A.; Piluso, G.; Maria Ventriglia, V.; Politano, L.; Nigro, V. Log-PCR: A New Tool for Immediate and Cost-Effective Diagnosis of up to 85% of Dystrophin Gene Mutations. Clin. Chem. 2008, 54, 973–981. [Google Scholar] [CrossRef]
- Lalic, T.; Vossen, R.H.; Coffa, J.; Schouten, J.P.; Guc-Scekic, M.; Radivojevic, D.; Djurisic, M.; Breuning, M.H.; White, S.J.; den Dunnen, J.T. Deletion and duplication screening in the DMD gene using MLPA. Eur. J. Hum. Genet. 2005, 13, 1231–1234. [Google Scholar] [CrossRef]
- Falzarano, M.S.; Scotton, C.; Passarelli, C.; Ferlini, A. Duchenne Muscular Dystrophy: From Diagnosis to Therapy. Molecules 2015, 20, 18168–18184. [Google Scholar] [CrossRef] [Green Version]
- Nigro, V.; Piluso, G. Next generation sequencing (NGS) strategies for the genetic testing of myopathies. Acta Myol. 2012, 31, 196–200. [Google Scholar]
- Savarese, M.; Di Fruscio, G.; Torella, A.; Fiorillo, C.; Magri, F.; Fanin, M.; Ruggiero, L.; Ricci, G.; Astrea, G.; Passamano, L.; et al. The genetic basis of undiagnosed muscular dystrophies and myopathies: Results from 504 patients. Neurology 2016, 87, 71–76. [Google Scholar] [CrossRef] [Green Version]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Brumbaugh, D.; Case, L.E.; Clemens, P.R.; Hadjiyannakis, S.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018, 17, 251–267. [Google Scholar] [CrossRef] [Green Version]
- Lionel, A.C.; Costain, G.; Monfared, N.; Walker, S.; Reuter, M.S.; Hosseini, S.M.; Thiruvahindrapuram, B.; Merico, D.; Jobling, R.; Nalpathamkalam, T.; et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet. Med. 2018, 20, 435–443. [Google Scholar] [CrossRef] [Green Version]
- Marks, P.; Garcia, S.; Barrio, A.M.; Belhocine, K.; Bernate, J.; Bharadwaj, R.; Bjornson, K.; Catalanotti, C.; Delaney, J.; Fehr, A.; et al. Resolving the full spectrum of human genome variation using Linked-Reads. Genome Res. 2019, 29, 635–645. [Google Scholar] [CrossRef] [Green Version]
- Uguen, K.; Jubin, C.; Duffourd, Y.; Bardel, C.; Malan, V.; Dupont, J.-M.; El Khattabi, L.; Chatron, N.; Vitobello, A.; Rollat-Farnier, P.-A.; et al. Genome sequencing in cytogenetics: Comparison of short-read and linked-read approaches for germline structural variant detection and characterization. Mol. Genet. Genom. Med. 2020, 8, e1114. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Sun, C.; Zhang, S.; Liu, Y.; Yu, M.; Zheng, Y.; Meng, L.; Acharya, A.; Cornejo-Sanchez, D.M.; Wang, G.; et al. Long-read whole-genome sequencing for the genetic diagnosis of dystrophinopathies. Ann. Clin. Transl. Neurol. 2020, 7, 2041–2046. [Google Scholar] [CrossRef]
- Savarese, M.; Qureshi, T.; Torella, A.; Laine, P.; Giugliano, T.; Jonson, P.H.; Johari, M.; Paulin, L.; Piluso, G.; Auvinen, P.; et al. Identification and Characterization of Splicing Defects by Single-Molecule Real-Time Sequencing Technology (PacBio). J. Neuromuscul. Dis. 2020, 7, 477–481. [Google Scholar] [CrossRef]
- Merker, J.D.; Wenger, A.M.; Sneddon, T.; Grove, M.; Zappala, Z.; Fresard, L.; Waggott, D.; Utiramerur, S.; Hou, Y.; Smith, K.S.; et al. Long-read genome sequencing identifies causal structural variation in a Mendelian disease. Genet. Med. 2018, 20, 159–163. [Google Scholar] [CrossRef] [Green Version]
- Ott, A.; Schnable, J.C.; Yeh, C.T.; Wu, L.; Liu, C.; Hu, H.C.; Dalgard, C.L.; Sarkar, S.; Schnable, P.S. Linked read technology for assembling large complex and polyploid genomes. BMC Genom. 2018, 19, 651. [Google Scholar] [CrossRef]
- Giugliano, T.; Savarese, M.; Garofalo, A.; Picillo, E.; Fiorillo, C.; D’Amico, A.; Maggi, L.; Ruggiero, L.; Vercelli, L.; Magri, F.; et al. Copy Number Variants Account for a Tiny Fraction of Undiagnosed Myopathic Patients. Genes 2018, 9, 524. [Google Scholar] [CrossRef] [Green Version]
- Savarese, M.; Piluso, G.; Orteschi, D.; Di Fruscio, G.; Dionisi, M.; Blanco, F.D.V.; Torella, A.; Giugliano, T.; Iacomino, M.; Zollino, M.; et al. Enhancer Chip: Detecting Human Copy Number Variations in Regulatory Elements. PLoS ONE 2012, 7, e52264. [Google Scholar] [CrossRef] [Green Version]
- Elyanow, R.; Wu, H.T.; Raphael, B.J. Identifying structural variants using linked-read sequencing data. Bioinformatics 2018, 34, 353–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, V.; Halpern, A.L.; Axelrod, N.; Bafna, V. An MCMC algorithm for haplotype assembly from whole-genome sequence data. Genome Res. 2008, 18, 1336–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piluso, G.; Dionisi, M.; Del Vecchio Blanco, F.; Torella, A.; Aurino, S.; Savarese, M.; Giugliano, T.; Bertini, E.; Terracciano, A.; Vainzof, M.; et al. Motor Chip: A Comparative Genomic Hybridization Microarray for Copy-Number Mutations in 245 Neuromuscular Disorders. Clin. Chem. 2011, 57, 1584–1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mak, A.C.; Lai, Y.Y.; Lam, E.T.; Kwok, T.P.; Leung, A.K.; Poon, A.; Mostovoy, Y.; Hastie, A.R.; Stedman, W.; Anantharaman, T.; et al. Genome-Wide Structural Variation Detection by Genome Mapping on Nanochannel Arrays. Genetics 2016, 202, 351–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, C.M.; Lupski, J.R. Mechanisms underlying structural variant formation in genomic disorders. Nat. Rev. Genet. 2016, 17, 224–238. [Google Scholar] [CrossRef] [Green Version]
- Kumar, K.R.; Cowley, M.J.; Davis, R.L. Next-Generation Sequencing and Emerging Technologies. Semin. Thromb. Hemost. 2019, 45, 661–673. [Google Scholar] [CrossRef]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Giordano, F.; Ning, Z. Oxford Nanopore MinION Sequencing and Genome Assembly. Genom. Proteom. Bioinform. 2016, 14, 265–279. [Google Scholar] [CrossRef] [Green Version]
- Mitsuhashi, S.; Matsumoto, N. Long-read sequencing for rare human genetic diseases. J. Hum. Genet. 2020, 65, 11–19. [Google Scholar] [CrossRef]
- Fang, L.; Kao, C.; Gonzalez, M.V.; Mafra, F.A.; Da Silva, R.P.; Li, M.; Wenzel, S.-S.; Wimmer, K.; Hakonarson, H.; Wang, K. LinkedSV for detection of mosaic structural variants from linked-read exome and genome sequencing data. Nat. Commun. 2019, 10, 5585. [Google Scholar] [CrossRef] [Green Version]
- Greer, S.U.; Ji, H.P. Structural variant analysis for linked-read sequencing data with gemtools. Bioinformatics 2019, 35, 4397–4399. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, F.; Cau, M.; Ganau, A.; Congiu, R.; Arvedi, G.; Mateddu, A.; Marrosu, M.G.; Cianchetti, C.; Realdi, G.; Cao, A.; et al. Brief report: Deletion of the Dystrophin Muscle-Promoter Region Associated with X-Linked Dilated Cardiomyopathy. N. Engl. J. Med. 1993, 329, 921–925. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Onore, M.E.; Torella, A.; Musacchia, F.; D’Ambrosio, P.; Zanobio, M.; Del Vecchio Blanco, F.; Piluso, G.; Nigro, V. Linked-Read Whole Genome Sequencing Solves a Double DMD Gene Rearrangement. Genes 2021, 12, 133. https://doi.org/10.3390/genes12020133
Onore ME, Torella A, Musacchia F, D’Ambrosio P, Zanobio M, Del Vecchio Blanco F, Piluso G, Nigro V. Linked-Read Whole Genome Sequencing Solves a Double DMD Gene Rearrangement. Genes. 2021; 12(2):133. https://doi.org/10.3390/genes12020133
Chicago/Turabian StyleOnore, Maria Elena, Annalaura Torella, Francesco Musacchia, Paola D’Ambrosio, Mariateresa Zanobio, Francesca Del Vecchio Blanco, Giulio Piluso, and Vincenzo Nigro. 2021. "Linked-Read Whole Genome Sequencing Solves a Double DMD Gene Rearrangement" Genes 12, no. 2: 133. https://doi.org/10.3390/genes12020133