Calcium Signaling and Mitochondrial Function in Presenilin 2 Knock-Out Mice: Looking for Any Loss-of-Function Phenotype Related to Alzheimer’s Disease

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Primary Neuronal Cultures
- (1) The PS2–/– are homozygous deficient mice, where the PSEN2 gene was inactivated, replacing the mouse PS2 exon 5 with hygromycin cassette under the control of the PGK promoter [6];
- (2) The C57BL/6J WT mice share >90% genetic background of the other line.
- 3 × 105 cells/well, for colocalization experiments;
- 8 × 105 cells/well for FRET experiments;
- 3 × 105 cells/well for Seahorse experiments;
- 8 × 105 cells/well for tetramethyl rhodamine methyl ester (TMRM) experiments;
- 1.5 × 106 cells/well for Western Blot and Nonyl Acridine Orange (NAO) experiments.
2.3. Adeno-Associated Virus (AAV) Production
2.4. Ca2+ Imaging
- In Ca2+ experiments, upon metabotropic stimulation, neurons were perfused with: (1) mKRB, 60 s; (2) mKRB supplemented with EGTA (600 μM) for 20 s; (3) mKRB supplemented with CPA (20 μM), carbachol (500 μM); glutamate (100 μM) and ATP (100 μM).
- In Ca2+ experiments upon KCl stimulation, neurons were perfused with: (1) mKRB, 60 s; (2) mKRB supplemented with KCl (30 mM) for 170 s; (3) mKRB supplemented with EGTA (600 μM) for 200 s. KCl stimulation was performed in a modified mKRB to maintain the osmolarity. In particular, the KCl-based mKRB solution contains, in mM: 110 NaCl, 30 KCl, 2 MgCl2, 10 Hepes, 1 CaCl2, 5 glucose; pH 7.4 at 37 °C.
- In picrotoxin Ca2+ experiments, upon picrotoxin stimulation, neurons were continuously perfused with mKRB supplemented with picrotoxin (25 μM) for 100 s.
- In Ca2+ experiments, to evaluate mitochondrial resting Ca2+ level with 4mtGCaMP6f, 3–5 fields of view per coverslip were imaged for 1 min, keeping cells at 37 °C and in mKRB supplemented with 1 mM CaCl2.
2.5. Oxygen Consumption Rate (OCR) and Extracellular Acidification Rate (ECAR) Measurements
2.6. Preparation of Protein Extracts and Western Blot Analysis
2.7. TMRM Experiments
2.8. Immunofluorescence (IF) and Confocal Analysis
2.9. Nonyl Acridine Orange (NAO) Staining
2.10. Drugs
2.11. Antibodies
2.12. Statistical Analyses
3. Results
3.1. Ca2+ Handling in PS2–/– Neurons
3.1.1. Cytosolic Ca2+ Handling in PS2–/– Neurons
3.1.2. Neuronal Hyperexcitability in PS2–/– Neurons
3.1.3. Mitochondrial Ca2+ Handling in PS2–/– Neurons
3.1.4. ER–Mitochondria Apposition in PS2–/– Neurons
3.2. Mitochondrial Functionality in PS2–/– Neurons
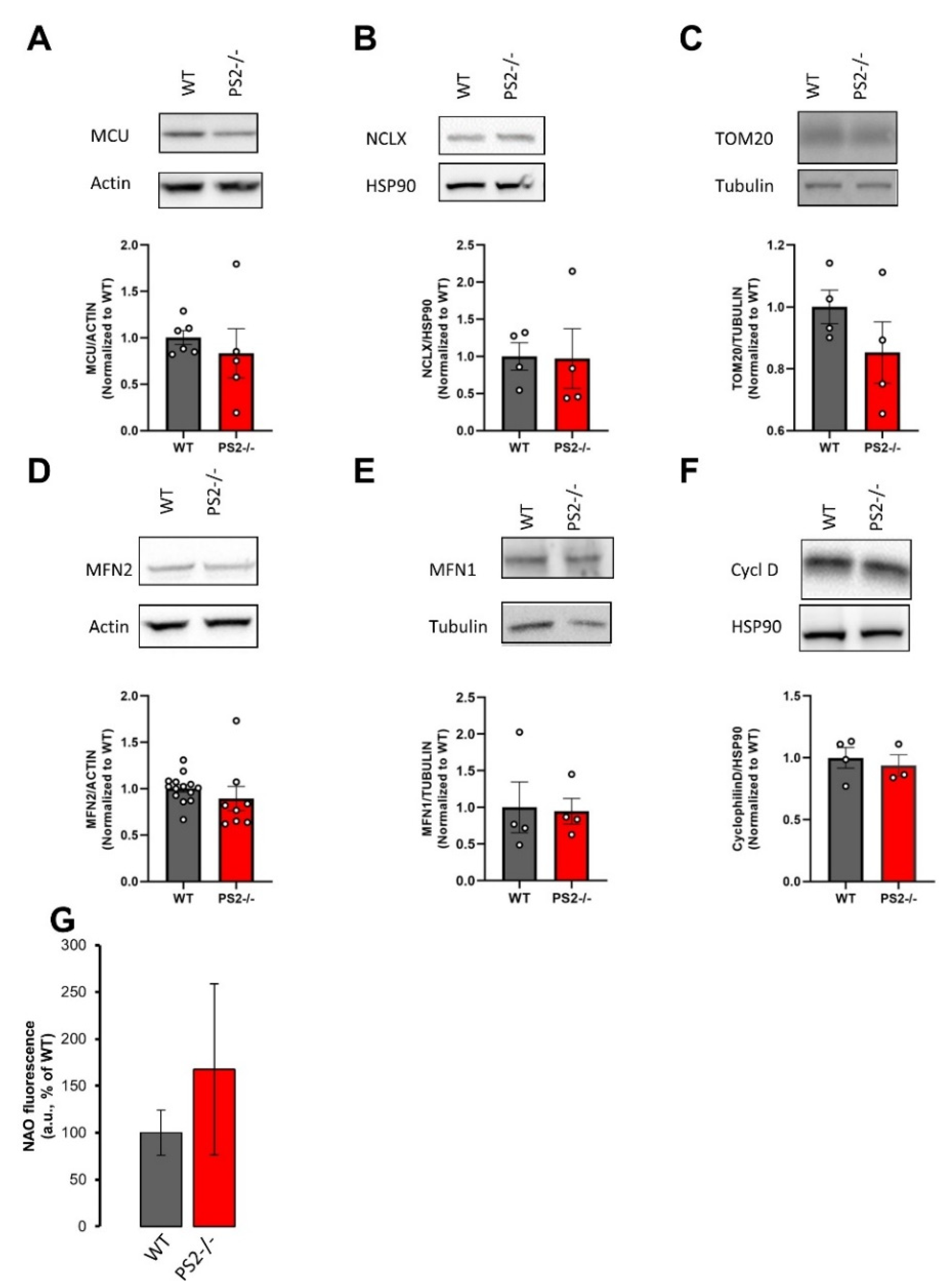
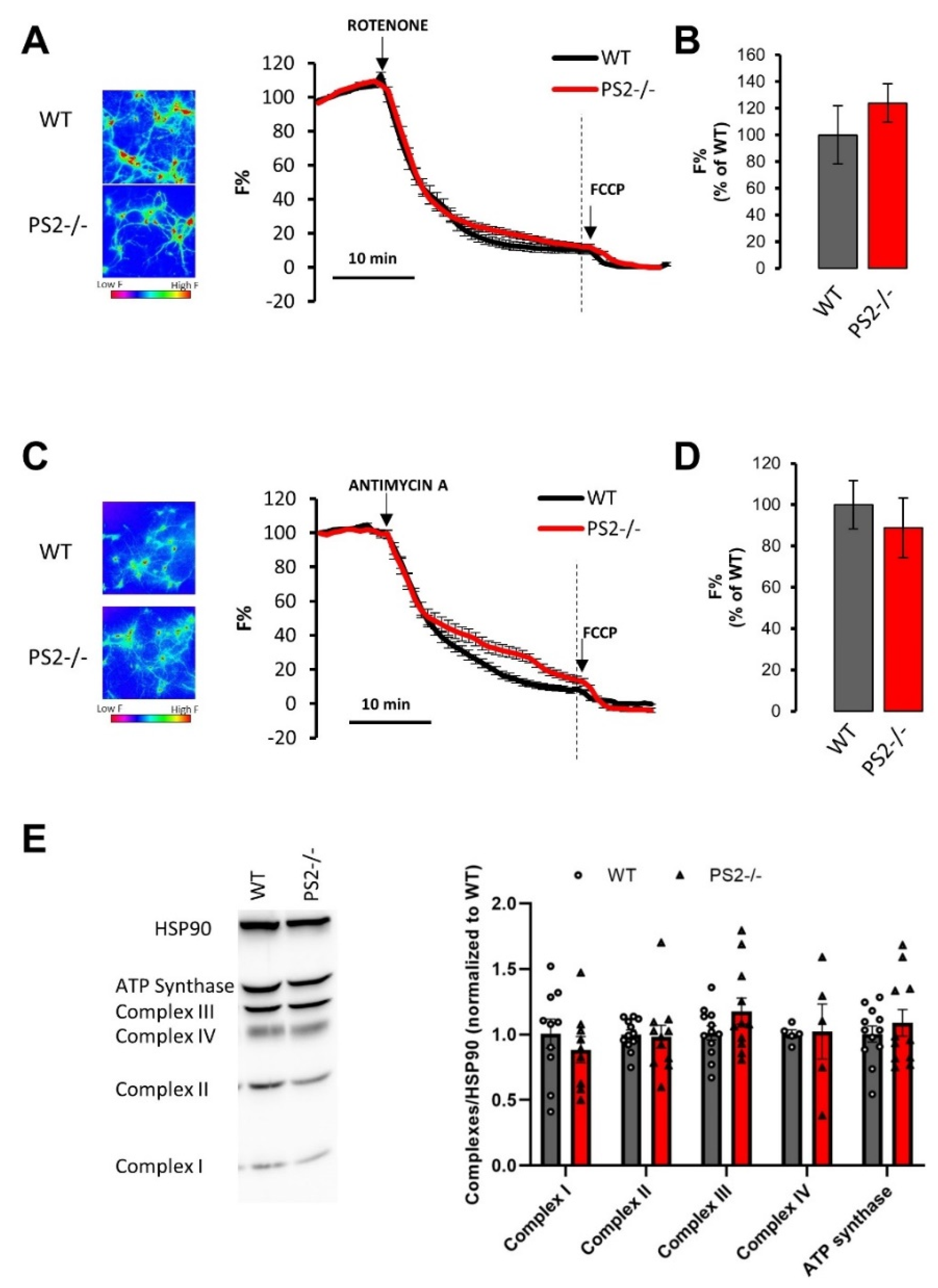
3.2.1. Mitochondrial Morphology and Mass in PS2–/– Neurons
3.2.2. Mitochondrial Membrane Potential
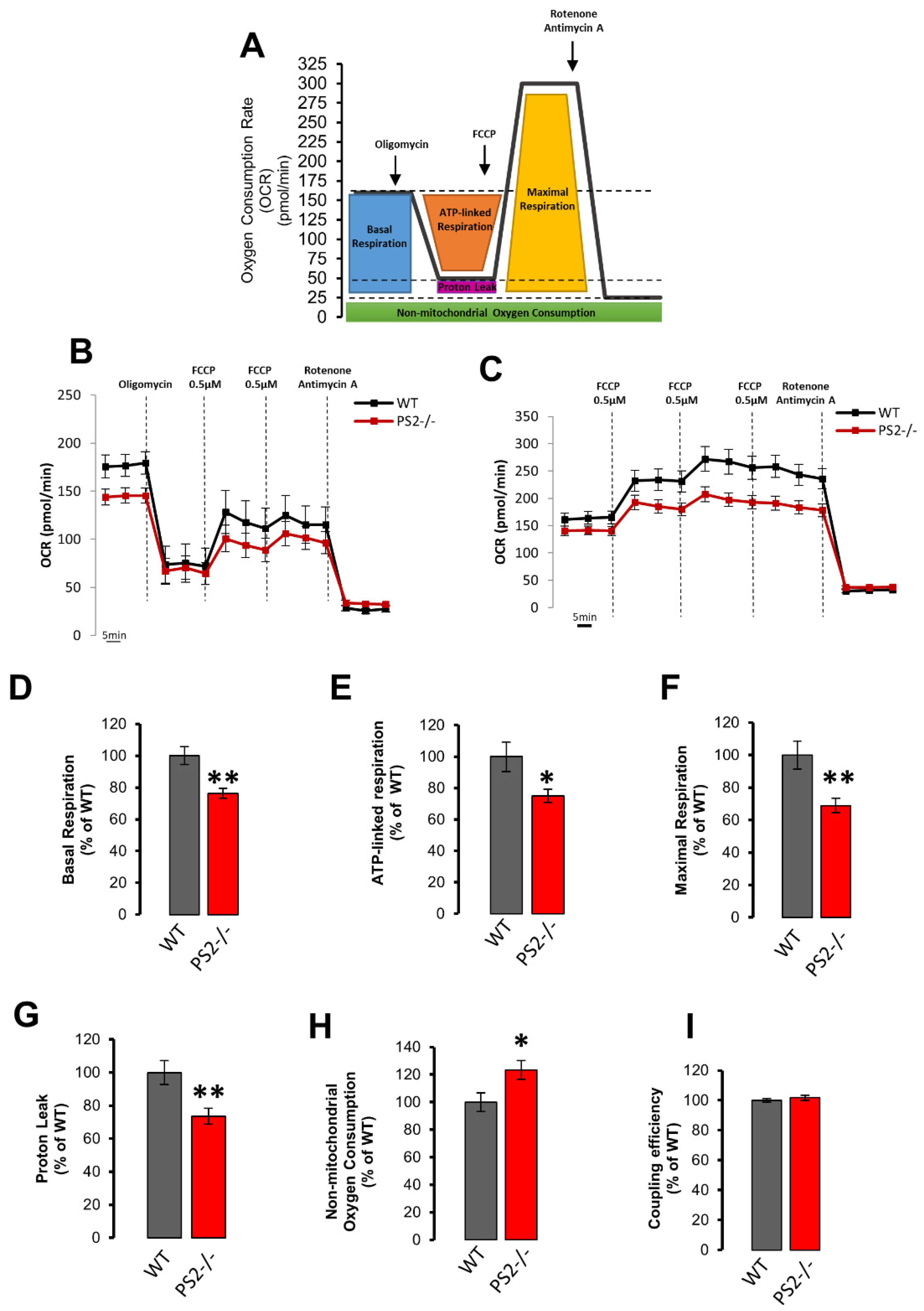
3.2.3. Mitochondrial Respiration, Glycolysis and ATP Production
3.2.4. Key Proteins in the Regulation of Glycolysis, Krebs Cycle and Oxidative Phosphorylation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bondi, M.W.; Edmonds, E.C.; Salmon, D.P. Alzheimer’s disease: Past, present, and future. J. Int. Neuropsychol. Soc. 2017, 23, 818–831. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Rosa-Neto, P.; Hsiung, G.Y.R.; Sadovnick, A.D.; Masellis, M.; Black, S.E.; Jia, J.; Gauthier, S. Early-onset familial alzheimer’s disease (EOFAD). Can. J. Neurol. Sci. 2012, 39, 436–445. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Ceyzériat, K.; Zilli, T.; Millet, P.; Frisoni, G.B.; Garibotto, V.; Tournier, B.B. Learning from the Past: A Review of Clinical Trials Targeting Amyloid, Tau and Neuroinflammation in Alzheimer’s Disease. Curr. Alzheimer Res. 2020, 17, 112–125. [Google Scholar] [CrossRef]
- Xia, D.; Watanabe, H.; Wu, B.; Lee, S.H.; Li, Y.; Tsvetkov, E.; Bolshakov, V.Y.; Shen, J.; Kelleher, R.J., 3rd. Presenilin-1 knockin mice reveal loss-of-function mechanism for familial Alzheimer’s disease. Neuron 2015, 85, 967–981. [Google Scholar] [CrossRef] [Green Version]
- Herreman, A.; Hartmann, D.; Annaert, W.; Saftig, P.; Craessaerts, K.; Serneels, L.; Umans, L.; Schrijvers, V.; Checler, F.; Vanderstichele, H.; et al. Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes in amyloid precursor protein processing but enhances the embryonic lethal phenotype of presenilin 1 deficiency. Proc. Natl. Acad. Sci. USA 1999. [Google Scholar] [CrossRef] [Green Version]
- Zatti, G.; Ghidoni, R.; Barbiero, L.; Binetti, G.; Pozzan, T.; Fasolato, C.; Pizzo, P. The presenilin 2 M239I mutation associated with familial Alzheimer’s disease reduces Ca2+ release from intracellular stores. Neurobiol. Dis. 2004, 15, 269–278. [Google Scholar] [CrossRef]
- Giacomello, M.; Barbiero, L.; Zatti, G.; Squitti, R.; Binetti, G.; Pozzan, T.; Fasolato, C.; Ghidoni, R.; Pizzo, P. Reduction of Ca2+ stores and capacitative Ca2+ entry is associated with the familial Alzheimer’s disease presenilin-2 T122R mutation and anticipates the onset of dementia. Neurobiol. Dis. 2005, 18, 638–648. [Google Scholar] [CrossRef]
- Zatti, G.; Burgo, A.; Giacomello, M.; Barbiero, L.; Ghidoni, R.; Sinigaglia, G.; Florean, C.; Bagnoli, S.; Binetti, G.; Sorbi, S.; et al. Presenilin mutations linked to familial Alzheimer’s disease reduce endoplasmic reticulum and Golgi apparatus calcium levels. Cell Calcium 2006, 39, 539–550. [Google Scholar] [CrossRef]
- Brunello, L.; Zampese, E.; Florean, C.; Pozzan, T.; Pizzo, P.; Fasolato, C. Presenilin-2 dampens intracellular Ca2+ stores by increasing Ca2+ leakage and reducing Ca2+ uptake. J. Cell. Mol. Med. 2009, 13, 3358–3369. [Google Scholar] [CrossRef] [Green Version]
- Zampese, E.; Fasolato, C.; Kipanyula, M.J.; Bortolozzi, M.; Pozzan, T.; Pizzo, P. Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc. Natl. Acad. Sci. USA 2011, 108, 2777–2782. [Google Scholar] [CrossRef] [Green Version]
- Kipanyula, M.J.; Contreras, L.; Zampese, E.; Lazzari, C.; Wong, A.K.C.; Pizzo, P.; Fasolato, C.; Pozzan, T. Ca2+ dysregulation in neurons from transgenic mice expressing mutant presenilin 2. Aging Cell 2012, 11, 885–893. [Google Scholar] [CrossRef] [Green Version]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Presenilin 2 Modulates Endoplasmic Reticulum-Mitochondria Coupling by Tuning the Antagonistic Effect of Mitofusin 2. Cell Rep. 2016, 15, 2226–2238. [Google Scholar] [CrossRef] [Green Version]
- Greotti, E.; Wong, A.; Pozzan, T.; Pendin, D.; Pizzo, P. Characterization of the ER-Targeted Low Affinity Ca(2+) Probe D4ER. Sensors 2016, 16, 1419. [Google Scholar] [CrossRef] [Green Version]
- Greotti, E.; Capitanio, P.; Wong, A.; Pozzan, T.; Pizzo, P.; Pendin, D. Familial Alzheimer’s disease-linked presenilin mutants and intracellular Ca2+ handling: A single-organelle, FRET-based analysis. Cell Calcium 2019, 79, 44–56. [Google Scholar] [CrossRef]
- Theurey, P.; Connolly, N.M.; Fortunati, I.; Basso, E.; Lauwen, S.; Ferrante, C.; Moreira Pinho, C.; Joselin, A.; Gioran, A.; Bano, D.; et al. Systems biology analysis identifies impairment of mitochondrial and glycolytic metabolism in a genetic model of Alzheimer’s disease. Aging Cell 2019, 18, e12924. [Google Scholar] [CrossRef] [Green Version]
- Rossi, A.; Rigotto, G.; Valente, G.; Giorgio, V.; Basso, E.; Filadi, R.; Pizzo, P. Defective Mitochondrial Pyruvate Flux Affects Cell Bioenergetics in Alzheimer’s Disease-Related Models. Cell Rep. 2020, 30, 2332–2348. [Google Scholar] [CrossRef] [Green Version]
- Arsic, N.; Zacchigna, S.; Zentilin, L.; Ramirez-Correa, G.; Pattarini, L.; Salvi, A.; Sinagra, G.; Giacca, M. Vascular endothelial growth factor stimulates skeletal muscle regeneration in Vivo. Mol. Ther. 2004, 10, 844–854. [Google Scholar] [CrossRef]
- Inagaki, K.; Fuess, S.; Storm, T.A.; Gibson, G.A.; Mctiernan, C.F.; Kay, M.A.; Nakai, H. Robust systemic transduction with AAV9 vectors in mice: Efficient global cardiac gene transfer superior to that of AAV8. Mol. Ther. 2006, 14, 45–53. [Google Scholar] [CrossRef]
- Ayuso, E.; Mingozzi, F.; Montane, J.; Leon, X.; Anguela, X.M.; Haurigot, V.; Edmonson, S.A.; Africa, L.; Zhou, S.; High, K.A.; et al. High AAV vector purity results in serotype- and tissue-independent enhancement of transduction efficiency. Gene Ther. 2010, 17, 503–510. [Google Scholar] [CrossRef] [Green Version]
- Zentilin, L.; Marcello, A.; Giacca, M. Involvement of Cellular Double-Stranded DNA Break Binding Proteins in Processing of the Recombinant Adeno-Associated Virus Genome. J. Virol. 2001, 75, 12279–12287. [Google Scholar] [CrossRef] [Green Version]
- Connolly, N.M.C.; Theurey, P.; Adam-Vizi, V.; Bazan, N.G.; Bernardi, P.; Bolanos, J.P.; Culmsee, C.; Dawson, V.L.; Deshmukh, M.; Duchen, M.R.; et al. Guidelines on experimental methods to assess mitochondrial dysfunction in cellular models of neurodegenerative diseases. Cell Death Differ. 2017. [Google Scholar] [CrossRef] [Green Version]
- Mookerjee, S.A.; Brand, M.D. Measurement and analysis of extracellular acid production to determine glycolytic rate. J. Vis. Exp. 2015, 2015. [Google Scholar] [CrossRef] [Green Version]
- Mookerjee, S.A.; Gerencser, A.A.; Nicholls, D.G.; Brand, M.D. Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J. Biol. Chem. 2017, 292, 7189–7207. [Google Scholar] [CrossRef] [Green Version]
- Filadi, R.; Leal, N.S.; Schreiner, B.; Rossi, A.; Dentoni, G.; Pinho, C.M.; Wiehager, B.; Cieri, D.; Cali, T.; Pizzo, P.; et al. TOM70 Sustains Cell Bioenergetics by Promoting IP3R3-Mediated ER to Mitochondria Ca(2+) Transfer. Curr. Biol. 2018, 28, 369–382.e6. [Google Scholar] [CrossRef] [Green Version]
- Ward, M.W.; Huber, H.J.; Weisová, P.; Düssmann, H.; Nicholls, D.G.; Prehn, J.H.M. Mitochondrial and plasma membrane potential of cultured cerebellar neurons during glutamate-induced necrosis, apoptosis, and tolerance. J. Neurosci. 2007, 27, 8238–8249. [Google Scholar] [CrossRef]
- Tottene, A.; Moretti, A.; Pietrobon, D. Functional diversity of P-type and R-type calcium channels in rat cerebellar neurons. J. Neurosci. 1996, 16, 6353–6363. [Google Scholar] [CrossRef] [Green Version]
- Koopman, W.J.H.; Verkaart, S.; Visch, H.J.; Van Der Westhuizen, F.H.; Murphy, M.P.; Van Den Heuvel, L.W.P.J.; Smeitink, J.A.M.; Willems, P.H.G.M. Inhibition of complex I of the electron transport chain causes O 2-.-mediated mitochondrial outgrowth. Am. J. Physiol. Cell Physiol. 2005, 288. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Pizzo, P.; Basso, E.; Filadi, R.; Greotti, E.; Leparulo, A.; Pendin, D.; Redolfi, N.; Rossini, M.; Vajente, N.; Pozzan, T.; et al. Presenilin-2 and Calcium Handling: Molecules, Organelles, Cells and Brain Networks. Cells 2020, 9, 2166. [Google Scholar] [CrossRef]
- Watanabe, H.; Iqbal, M.; Zheng, J.; Wines-Samuelson, M.; Shen, J. Partial loss of presenilin impairs age-dependent neuronal survival in the cerebral cortex. J. Neurosci. 2014, 34, 15912–15922. [Google Scholar] [CrossRef] [Green Version]
- Greotti, E.; Fortunati, I.; Pendin, D.; Ferrante, C.; Galla, L.; Zentilin, L.; Giacca, M.; Kaludercic, N.; Di Sante, M.; Mariotti, L.; et al. mCerulean3-Based Cameleon Sensor to Explore Mitochondrial Ca2+ Dynamics In Vivo. iScience 2019, 16, 340–355. [Google Scholar] [CrossRef] [Green Version]
- Kügler, S.; Meyn, L.; Holzmüller, H.; Gerhardt, E.; Isenmann, S.; Schulz, J.B.; Bähr, M. Neuron-specific expression of therapeutic proteins: Evaluation of different cellular promoters in recombinant adenoviral vectors. Mol. Cell. Neurosci. 2001, 17, 78–96. [Google Scholar] [CrossRef]
- Kipanyula, M.J. Ca2+ Homeostasis in Familial Alzheimer’s Disease: A View from Intracellular Ca2+ Stores. Ph.D. Thesis, University of Padua, Padova, Italy, 2011. [Google Scholar]
- Zott, B.; Busche, M.A.; Sperling, R.A.; Konnerth, A. What happens with the circuit in Alzheimer’s disease in mice and humans? Annu. Rev. Neurosci. 2018, 41, 277–297. [Google Scholar] [CrossRef]
- Palop, J.J.; Mucke, L. Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2016, 17, 777–792. [Google Scholar] [CrossRef]
- Busche, M.A.; Konnerth, A. Neuronal hyperactivity—A key defect in Alzheimer’s disease? BioEssays 2015, 37, 624–632. [Google Scholar] [CrossRef]
- Ozmen, L.; Albientz, A.; Czech, C.; Jacobsen, H. Expression of transgenic APP mRNA is the key determinant for beta-amyloid deposition in PS2APP transgenic mice. Neurodegener. Dis. 2009, 6, 29–36. [Google Scholar] [CrossRef]
- Richards, J.G.; Higgins, G.A.; Ouagazzal, A.M.; Ozmen, L.; Kew, J.N.; Bohrmann, B.; Malherbe, P.; Brockhaus, M.; Loetscher, H.; Czech, C.; et al. PS2APP transgenic mice, coexpressing hPS2mut and hAPPswe, show age-related cognitive deficits associated with discrete brain amyloid deposition and inflammation. J. Neurosci. 2003, 23, 8989–9003. [Google Scholar] [CrossRef] [Green Version]
- Cieri, D.; Vicario, M.; Giacomello, M.; Vallese, F.; Filadi, R.; Wagner, T.; Pozzan, T.; Pizzo, P.; Scorrano, L.; Brini, M.; et al. SPLICS: A split green fluorescent protein-based contact site sensor for narrow and wide heterotypic organelle juxtaposition. Cell Death Differ. 2018, 25, 1131–1145. [Google Scholar] [CrossRef] [Green Version]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc. Natl. Acad. Sci. USA 2015, 112, E2174–E2181. [Google Scholar] [CrossRef] [Green Version]
- Mammucari, C.; Gherardi, G.; Zamparo, I.; Raffaello, A.; Boncompagni, S.; Chemello, F.; Cagnin, S.; Braga, A.; Zanin, S.; Pallafacchina, G.; et al. The mitochondrial calcium uniporter controls skeletal muscle trophism in vivo. Cell Rep. 2015, 10, 1269–1279. [Google Scholar] [CrossRef]
- Tosatto, A.; Sommaggio, R.; Kummerow, C.; Bentham, R.B.; Blacker, T.S.; Berecz, T.; Duchen, M.R.; Rosato, A.; Bogeski, I.; Szabadkai, G.; et al. The mitochondrial calcium uniporter regulates breast cancer progression via HIF-1alpha. EMBO Mol. Med. 2016, 8, 569–585. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pozzan, T. Microdomains of intracellular Ca2+: Molecular determinants and functional consequences. Physiol. Rev. 2006, 86, 369–408. [Google Scholar] [CrossRef]
- Giacomello, M.; Drago, I.; Bortolozzi, M.; Scorzeto, M.; Gianelle, A.; Pizzo, P.; Pozzan, T. Ca2+ hot spots on the mitochondrial surface are generated by Ca2+ mobilization from stores, but not by activation of store-operated Ca2+ channels. Mol. Cell 2010, 38, 280–290. [Google Scholar] [CrossRef]
- Cardenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgo, J.; Muller, M.; Vais, H.; Cheung, K.H.; Yang, J.; Parker, I.; et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef] [Green Version]
- Ruas, J.S.; Siqueira-Santos, E.S.; Amigo, I.; Rodrigues-Silva, E.; Kowaltowski, A.J.; Castilho, R.F. Underestimation of the maximal capacity of the mitochondrial electron transport system in oligomycin-treated cells. PLoS ONE 2016, 11, e0150967. [Google Scholar] [CrossRef] [Green Version]
- Rossi, A.; Pizzo, P.; Filadi, R. Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1068–1078. [Google Scholar] [CrossRef]
- Pendin, D.; Fasolato, C.; Basso, E.; Filadi, R.; Greotti, E.; Galla, L.; Gomiero, C.; Leparulo, A.; Redolfi, N.; Scremin, E.; et al. Familial Alzheimer’s disease presenilin-2 mutants affect Ca2+ homeostasis and brain network excitability. Aging Clin. Exp. Res. 2019, 1–4. [Google Scholar] [CrossRef]
- Galla, L.; Redolfi, N.; Pozzan, T.; Pizzo, P.; Greotti, E. Intracellular Calcium Dysregulation by the Alzheimer’s Disease-Linked Protein Presenilin 2. Int. J. Mol. Sci. 2020, 21, 770. [Google Scholar] [CrossRef] [Green Version]
- Behbahani, H.; Shabalina, I.G.; Wiehager, B.; Concha, H.; Hultenby, K.; Petrovic, N.; Nedergaard, J.; Winblad, B.; Cowburn, R.F.; Ankarcrona, M. Differential role of Presenilin-1 and -2 on mitochondrial membrane potential and oxygen consumption in mouse embryonic fibroblasts. J. Neurosci. Res. 2006, 84, 891–902. [Google Scholar] [CrossRef]
- Contino, S.; Porporato, P.E.; Bird, M.; Marinangeli, C.; Opsomer, R.; Sonveaux, P.; Bontemps, F.; Dewachter, I.; Octave, J.N.; Bertrand, L.; et al. Presenilin 2-dependent maintenance of mitochondrial oxidative capacity and morphology. Front. Physiol. 2017, 8, 796. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [Green Version]
- Lanzillotta, C.; Di Domenico, F.; Perluigi, M.; Butterfield, D.A. Targeting Mitochondria in Alzheimer Disease: Rationale and Perspectives. CNS Drugs 2019, 33, 957–969. [Google Scholar] [CrossRef]
- Albensi, B.C. Dysfunction of mitochondria: Implications for Alzheimer’s disease. In International Review of Neurobiology; Academic Press Inc.: Cambridge, MA, USA, 2019; Volume 145, pp. 13–27. ISBN 9780128172247. [Google Scholar]
- Rossi, A.; Pizzo, P. Mitochondrial bioenergetics and neurodegeneration: A paso doble. Neural Regen. Res. 2021, 16, 686–687. [Google Scholar] [CrossRef]
- Baldelli, P.; Meldolesi, J. The Transcription Repressor REST in Adult Neurons: Physiology, Pathology, and Diseases. eNeuro 2015, 2. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Aron, L.; Zullo, J.; Pan, Y.; Kim, H.; Chen, Y.; Yang, T.H.; Kim, H.M.; Drake, D.; Liu, X.S.; et al. REST and stress resistance in ageing and Alzheimer’s disease. Nature 2014, 507, 448–454. [Google Scholar] [CrossRef] [Green Version]
- Keller, J.N.; Guo, Q.; Holtsberg, F.W.; Bruce-Keller, A.J.; Mattson, M.P. Increased sensitivity to mitochondrial toxin-induced apoptosis in neural cells expressing mutant presenilin-1 is linked to perturbed calcium homeostasis and enhanced oxyradical production. J. Neurosci. 1998, 18, 4439–4450. [Google Scholar] [CrossRef] [Green Version]
- Johnston, J.M.; Burnett, P.; Thomas, A.P.; Tezapsidis, N. Calcium oscillations in type-1 astrocytes, the effect of a presenilin 1 (PS1) mutation. Neurosci. Lett. 2006, 395, 159–164. [Google Scholar] [CrossRef]
- Kilb, W.; Hartmann, D.; Saftig, P.; Luhmann, H.J. Altered morphological and electrophysiological properties of Cajal-Retzius cells in cerebral cortex of embryonic Presenilin-1 knockout mice. Eur. J. Neurosci. 2004, 20, 2749–2756. [Google Scholar] [CrossRef]
- Huang, H.M.; Fowler, C.; Xu, H.; Zhang, H.; Gibson, G.E. Mitochondrial function in fibroblasts with aging in culture and/or Alzheimer’s disease. Neurobiol. Aging 2005, 26, 839–848. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim. Biophys. Acta 2014, 1842, 1219–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontana, R.; Agostini, M.; Murana, E.; Mahmud, M.; Scremin, E.; Rubega, M.; Sparacino, G.; Vassanelli, S.; Fasolato, C. Early hippocampal hyperexcitability in PS2APP mice: Role of mutant PS2 and APP. Neurobiol. Aging 2017, 50, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Leparulo, A.; Mahmud, M.; Scremin, E.; Pozzan, T.; Vassanelli, S.; Fasolato, C. Dampened Slow Oscillation Connectivity Anticipates Amyloid Deposition in the PS2APP Mouse Model of Alzheimer’s Disease. Cells 2019, 9, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Bronson, R.T.; Chen, D.F.; Xia, W.; Selkoe, D.J.; Tonegawa, S. Skeletal and CNS defects in Presenilin-1-deficient mice. Cell 1997, 89, 629–639. [Google Scholar] [CrossRef] [Green Version]
- Wong, P.C.; Zheng, H.; Chen, H.; Becher, M.W.; Sirinathsinghji, D.J.S.; Trumbauer, M.E.; Chen, H.Y.; Price, D.L.; Van Der Ploeg, L.H.T.; Sisodia, S.S. Presenilin 1 is required for Notch 1 and Dll1 expression in the paraxial mesoderm. Nature 1997, 387, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, D.; De Strooper, B.; Saftig, P. Presenilin-1 deficiency leads to loss of Cajal-Retzius neurons and cortical dysplasia similar to human type 2 lissencephaly. Curr. Biol. 1999, 9, 719–727. [Google Scholar] [CrossRef] [Green Version]
- De Strooper, B.; Saftig, P.; Craessaerts, K.; Vanderstichele, H.; Guhde, G.; Annaert, W.; Von Figura, K.; Van Leuven, F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 1998, 391, 387–390. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PS2N141I | PS2N141I APPswe | MEF PS2−/− | PS2−/−(This Paper) | ||
|---|---|---|---|---|---|
| Cytosolic Ca2+ handling | Basal levels | unchanged [32] | unchanged [32] | ND | unchanged |
| Store Ca2+ content | reduced [12] | reduced [12] | ND | unchanged | |
| KCl response | unchanged [12] | unchanged [12] | ND | unchanged | |
| Neuronal hyper-excitability | increased [12] | increased [12] | ND | increased | |
| MitochondrialCa2+handling | Basal levels | ND | ND | ND | unchanged |
| ER–mitochondria Ca2+ transfer | reduced [12] | reduced [12] | ND | reduced | |
| KCl response | ND | ND | ND | reduced | |
| ER–mitochondria contacts | increased [12] | increased[12] | ND | reduced | |
| Mitochondrial morphology | unchanged | unchanged [16] | defective cristae [53] | unchanged | |
| Mitochondrial mass | unchanged [17] | unchanged [16] | ND | unchanged | |
| Mitochondrial membrane potential | Basal | unchanged [12] | unchanged [12, 16] | unchanged [52,53] | ND |
| Complex I inhibition | faster decay [17] | unchanged [16] | ND | unchanged | |
| Complex III inhibition | ND | unchanged [16] | ND | unchanged | |
| OxygenConsumptionRate (OCR) | Basal | reduced [17] | unchanged [16] | reduced [52,53] | reduced |
| Maximal | reduced [17] | reduced [16] | reduced [52,53] | reduced | |
| ATP-linked respiration | reduced [17] | unchanged [16] | reduced [52,53] | reduced | |
| Proton leak | unchanged [17] | unchanged [16] | ND | reduced | |
| Non-mitochondrial OCR | unchanged [17] | unchanged [16] | ND | increased | |
| Glycolysis(ECAR) | Basal | unchanged [17] | reduced [16] | increased [52,53] | increased |
| Maximal Glycolysis | unchanged [17] | reduced [16] | ND | increased | |
| Respiratory chain complexes (levels of expression) | unchanged [17] | unchanged [16] | reduced CI, CII, CIV [53] | unchanged | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossi, A.; Galla, L.; Gomiero, C.; Zentilin, L.; Giacca, M.; Giorgio, V.; Calì, T.; Pozzan, T.; Greotti, E.; Pizzo, P. Calcium Signaling and Mitochondrial Function in Presenilin 2 Knock-Out Mice: Looking for Any Loss-of-Function Phenotype Related to Alzheimer’s Disease. Cells 2021, 10, 204. https://doi.org/10.3390/cells10020204
Rossi A, Galla L, Gomiero C, Zentilin L, Giacca M, Giorgio V, Calì T, Pozzan T, Greotti E, Pizzo P. Calcium Signaling and Mitochondrial Function in Presenilin 2 Knock-Out Mice: Looking for Any Loss-of-Function Phenotype Related to Alzheimer’s Disease. Cells. 2021; 10(2):204. https://doi.org/10.3390/cells10020204
Chicago/Turabian StyleRossi, Alice, Luisa Galla, Chiara Gomiero, Lorena Zentilin, Mauro Giacca, Valentina Giorgio, Tito Calì, Tullio Pozzan, Elisa Greotti, and Paola Pizzo. 2021. "Calcium Signaling and Mitochondrial Function in Presenilin 2 Knock-Out Mice: Looking for Any Loss-of-Function Phenotype Related to Alzheimer’s Disease" Cells 10, no. 2: 204. https://doi.org/10.3390/cells10020204