Regulation of COX Assembly and Function by Twin CX9C Proteins—Implications for Human Disease



1
Center for Molecular Medicine and Genetics, Wayne State University School of Medicine, Detroit, MI 48201, USA
2
Perinatology Research Branch, Division of Obstetrics and Maternal-Fetal Medicine, Division of Intramural Research, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, U.S. Department of Health and Human Services, Bethesda, Maryland and Detroit, MI 48201, USA
*
Author to whom correspondence should be addressed.
Cells 2021, 10(2), 197; https://doi.org/10.3390/cells10020197
Submission received: 21 December 2020
/
Revised: 11 January 2021
/
Accepted: 12 January 2021
/
Published: 20 January 2021
(This article belongs to the Collection Regulation of Eukaryotic Cytochrome c Oxidase)
Abstract
:Oxidative phosphorylation is a tightly regulated process in mammals that takes place in and across the inner mitochondrial membrane and consists of the electron transport chain and ATP synthase. Complex IV, or cytochrome c oxidase (COX), is the terminal enzyme of the electron transport chain, responsible for accepting electrons from cytochrome c, pumping protons to contribute to the gradient utilized by ATP synthase to produce ATP, and reducing oxygen to water. As such, COX is tightly regulated through numerous mechanisms including protein–protein interactions. The twin CX9C family of proteins has recently been shown to be involved in COX regulation by assisting with complex assembly, biogenesis, and activity. The twin CX9C motif allows for the import of these proteins into the intermembrane space of the mitochondria using the redox import machinery of Mia40/CHCHD4. Studies have shown that knockdown of the proteins discussed in this review results in decreased or completely deficient aerobic respiration in experimental models ranging from yeast to human cells, as the proteins are conserved across species. This article highlights and discusses the importance of COX regulation by twin CX9C proteins in the mitochondria via COX assembly and control of its activity through protein–protein interactions, which is further modulated by cell signaling pathways. Interestingly, select members of the CX9C protein family, including MNRR1 and CHCHD10, show a novel feature in that they not only localize to the mitochondria but also to the nucleus, where they mediate oxygen- and stress-induced transcriptional regulation, opening a new view of mitochondrial-nuclear crosstalk and its involvement in human disease.
1. Introduction
Mitochondria are the major source of cellular energy that is required to sustain life. They are double-membrane organelles in which the process of cellular respiration and ATP production takes place. This process, oxidative phosphorylation, occurs at the electron transport chain (ETC), a series of four protein complexes embedded in the inner mitochondrial membrane (IM). The complexes create a proton gradient by pumping protons from the matrix to the intermembrane space (IMS), which is coupled with electron transfer down the chain. The electrochemical proton gradient thereby produced is used by ATP synthase (complex V) to generate ATP from ADP and phosphate.
Complex IV, or cytochrome c oxidase (COX), is the terminal enzyme of the ETC and is responsible for reducing oxygen to water. Physiologically, the mammalian complex is a dimer, with each monomer composed of 13 tightly bound subunits embedded in the IM, an assembly supported by several crystal structures resolved from COX in bovine heart [1,2]. However, more recently, monomeric crystal structures of COX were also published [3,4] and monomeric COX was also reported in a supercomplex [5]. It is therefore possible that an equilibrium exists between dimeric and monomeric COX, which could be subject to regulation. In addition, a 14th subunit has been proposed—NDUFA4—which was originally believed to be a subunit of complex I [6,7]. A structural study showed that NDUFA4 appears to be a subunit in the COX monomer, likely adding to the stability of the complex [7]. NDUFA4 as part of the COX monomer is located at the interface of the dimeric complex, where it would prevent or interfere with dimer formation and which could be a reason that the protein was never detected in the dimeric crystal structure. The validity of NDUFA4′s role as a true subunit has been questioned and it was argued that, because NDUFA4 may bind to both complexes I and IV and is not consistently found in COX preparations, it may function as an assembly factor for the respirasome [8].
The three largest subunits are encoded by the mitochondrial genome whereas the other subunits are encoded by the nuclear genome. Among the mitochondrial-encoded subunits, subunits I and II contain the catalytic centers. The latter consist of metal centers that are involved in the electron acceptance from complex III via cytochrome c and the pathway of the electron through the complex itself: electrons received from cytochrome c first reach the CuA center in subunit II, are then transferred to heme a in subunit I, and finally reach the heme a3-CuB site of subunit I, where oxygen is reduced to water.
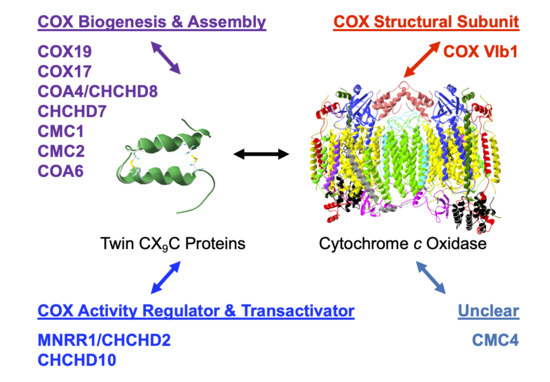
There are various modes of regulation of COX activity [1], summarized in Table 1. The purpose of this review is to explore the regulation of COX through the interaction with proteins of the twin CX9C family. Members of this protein family have been shown to be important in COX complex assembly and function, as well as for direct regulation of the oxidase [9] (Table 2). Note that the 13 tightly bound COX subunits are traditionally distinguished by Roman numerals introduced by the Kadenbach lab, whereas auxiliary proteins are designated with Arabic numerals (yeast nomenclature can be found in Table 2).
The twin CX9C family of proteins is characterized by its unique motif of two cysteines separated by usually nine amino acid residues. This motif is found in the coiled-coil-helix-coiled-coil-helix (CHCH) domain, where pairs of cysteines form a helix turn helix fold by forming disulfide bonds with one another [10,11,12]. Another family of proteins, called the “small Tim” proteins, contains a similar but shorter twin CX3C motif and plays chaperone roles in the TIM22 pathway for insertion of proteins into the IMS-facing side of the inner membrane (IM) [13]. The CHCH domain is important for the import of the proteins into the intermembrane space (IMS) of the mitochondria. IMS import is facilitated through the Mia40/CHCHD4 redox mechanism [14,15]. The first studies of this family of proteins took place in Saccharomyces cerevisiae, where a detailed study found that 13 of the 14 yeast family members were conserved across species [16]. A follow-up study contained a genome-wide analysis to determine family member functions, with six of the CX9C proteins determined to be involved in COX assembly [9]. Recently, more information has become available through further research into the function of twin CX9C family members.
2. COX Regulation through Assembly
The biogenesis and maturation of COX is critical for its proper function. There are multiple steps in this tightly regulated process: the insertion of metal groups in COX I and COX II, the import and folding of nuclear encoded subunits, and the proper assembly of the subunits into the complex. Over 30 auxiliary proteins are involved in the biogenesis of the core enzyme composed of COX I, COX II, and COX III [17]. The hypothesized assembly pathway favors a modular–linear assembly, where subunits are first assembled into module intermediates and then these modules are assembled into the COX monomer (Figure 1) [18,19]. The first step of monomer assembly is the synthesis of mitochondrially encoded COX I, including the insertion of heme a, which is followed by its association with the COX IV and COX Va module [18,20]. The COX II module, which requires the insertion of the CuA center into the subunit before assembly can continue [21,22,23,24], forms a complex intermediate with COX VIc, COX VIIb, COX VIIc, and COX VIIIa. The COX III module consists of COX III, COX VIa, COX VIb, and COX VIIa. These modules are then assembled in a linear fashion, upon which NDUFA4 interacts to assist in the stabilization of the COX monomer [18]. Detailed COX biogenesis and assembly has been reviewed elsewhere [19]; however, summaries of assembly will be provided where necessary.
It is important to note that copper metabolism and homeostasis in the mitochondria are important in aerobic respiration of the cell (for detailed reviews, see [25,26]). All of the assembly proteins discussed in this section have been shown to be involved or associated with copper transport and moiety insertion into appropriate subunits during COX assembly, which is an absolute necessity for the complex to function. Chaperones are very important in this process because of the redox sensitivity of the transition metal that, if left unchecked, can become a source of ROS (for review, see [27]).
Most of the twin CX9C proteins involved in COX assembly were first identified in yeast in a screen to identify proteins that affect OXPHOS; the proteins that were shown to affect OXPHOS were COX17, COX23, COX19, CMC1, and CMC2 [16]. COA4 and COA6 were identified in separate studies [28,29].
2.1. COX Subunit I Module Assembly
COX I is simultaneously synthesized from mtDNA and inserted into the mitochondrial inner membrane. Early membrane insertion is important as this subunit is highly hydrophobic, with 12 transmembrane helices, and the process is likely guided by chaperones COX14 and COX25/COA3 [30,31,32,33]. CMC1 has also been shown to stabilize this sub-assembly [34]. Before COX I can mature, three moieties must be inserted into the subunit, two hemes (heme a and heme a3) and one copper center (CuB). COX11, a copper chaperone required for COX subunit I maturation [35], is a membrane anchored protein with an exposed copper binding site facing the IMS, allowing it to receive copper from COX17, a twin CX9C protein [10].
2.1.1. COX17
COX17 and COX19 are involved in copper transport to the copper insertion chaperones during COX I assembly and are well studied in both yeast and mammals. COX17 has been extensively studied both functionally and structurally. It was first studied in S. cerevisiae, where it was identified in a screen for proteins that affect OXPHOS and was later shown to be a metallochaperone that localized to the cytosol and IMS [10,16,36]. However, a subsequent study showed that cycling from the cytosol to the IMS was unnecessary for COX17 function [37]. This was done by creating a SCO2/COX17 fusion protein such that the domain of SCO2 that interacts with copper was replaced by COX17 [37]. SCO2 is an inner membrane-bound protein facing the IMS that is involved in COX I assembly. IM tethered COX17 protein was found to still be able to bind copper and participate in COX biogenesis, suggesting that its copper transport function is solely within the mitochondria. COX17 functions as a copper ion transporter in the IMS, carrying copper ions to SCO1/SCO2 and COX11, which are critical assembly factors with the role of delivering copper to the redox-active metal center CuA that contains two copper ions and the single copper CuB site of the COX catalytic centers [10,38,39,40]. In fact, respiratory-deficient yeast containing a COX17 mutant are unable to grow on non-fermentable substrates unless supplemented with copper salts [10].
COX17 function is conserved across species; however, the protein sequence identity between yeast and human is only 34%. Despite this low conservation, all of the cysteines are conserved, illustrating their importance for the function of the protein (Figure 2). The cysteines of COX17 are not only important for its import, stability, and localization to the IMS, but also for its metallochaperone function. Initial functional studies in yeast determined that three cysteine residues in the CHCH domain of the protein are critical for its Cu1+ binding function (Figure 2) [41]. The residues are in a unique CCXC motif and, in in vitro experiments, the wildtype protein was able to bind three Cu1+ ions. However, when any of the cysteine residues was mutated to a serine, the result was a non-functioning COX complex, despite retaining the ability to bind copper and localize to the IMS [41]. The ability of COX17 to bind copper ions was only lost when cysteine residues 23 and 24 were mutated to serine. Evidence supporting the exact function of C26 is lacking although it is hypothesized, given its close proximity to the copper binding cysteines, to be involved in ligand transfer [41].
The evidence that COX17 could still maintain the ability to bind copper if any one of the cysteines was mutated but would still result in respiratory-deficient yeast was an interesting finding. The data suggested that more than just the CCXC motif was necessary for COX17-mediated copper transport that would result in proper functional maturation of COX. Further studies determined that COX17 exists in a dimer/tetramer equilibrium. COX17 proteins with any one of the critical CCXC motif cysteines mutated failed to form the higher molecular weight tetramer, suggesting that the ability for COX17 to form oligomers is functionally important in addition to binding copper [42]. The concentration of COX17 within the mitochondria exceeds 20 µM, suggesting that the protein exists primarily as a tetramer due to concentration dependence [42].
The COX17 function of copper transport for use in the copper centers of COX is conserved in mammals. Attempts to knock out COX17 in mice resulted in embryonic lethality [43]. As in yeast, the knockdown of COX17 results in defective COX activity [44] as well as decreased levels of mtDNA encoded COX subunits I and II [44,45]. In cell culture, COX17 retains its dual localization to the cytosol and the mitochondria, with protein levels enriched in the mitochondria [44]. It was originally thought that the protein is able to shuttle copper from the cytosol to the mitochondria [36]; however, functional studies suggest that the protein operates only in the mitochondria in mammals [46,47] due to the redox state of the protein as the cysteines are involved in metal binding, IMS import, and proper protein folding. In vitro, fully reduced COX17 is able to bind four copper ions, whereas partially oxidized COX17 with two disulfide bridges binds one metal ion with low specificity, and the fully oxidized form with three disulfide bonds is unable to interact with metals [46]. A dynamic NMR structural study further clarified that the functional form of COX17 resides in the IMS with the cysteines within the twin CX9C motif oxidized to retain its structure [47]. This oxidation state leaves the metal binding cysteines (residues 22 and 23 of the CCXC motif; Figure 2, arrows) reduced and able to bind one copper ion for the delivery to COX during biogenesis. When the protein is fully oxidized, the third disulfide bond forms between cysteines 22 and 23, effectively blocking the metal binding site [47].
COX17 may also play a role in the formation of supercomplexes [44,45]. The association with COX17 knockdown and COX depletion in supercomplexes likely hinges upon the oxidase not having the copper sites assembled in the holoenzyme, leading to degradation of the complex. HEK 293 cells with depleted levels of COX17 also display swollen mitochondria with disordered cristae [45]. Isolated mitochondria show roughly a 20% reduction in copper levels [45], which is different from yeast, despite having a conserved matrix copper pool from lower to higher eukaryotes [48,49]. However, given the swollen mitochondria, it may be that the reduced level in isolated mitochondria results from the isolation procedure.
2.1.2. COX19
Another well-studied copper chaperone in the twin CX9C family is COX19. COX19 was first identified in yeast, where knockout or mutation of the protein resulted in COX deficiency and an inability to grow on medium containing non-fermentable carbon sources [52,53,54]. COX19 retains one of the cysteines (C30) involved in the copper transport function of COX17 (C26) and localizes to the IMS and cytosol (Figure 3) [52]. Recombinant COX19 protein expressed in bacteria with copper salt supplementation purified as a monomer with copper bound [53]. In vitro experiments also showed that Cu1+ is bound to COX19 with a 1:1 stoichiometry and that excess amounts of Cu1+ resulted in a reduced spectral signal [53]. The authors suggested that, in the presence of excess Cu1+, there is a structural change in COX19 resulting in defective copper binding. In the same study, it was determined that both apoCOX19 and CuCOX19 exist as a dimer and that, when excess apoCOX19 is added, higher molecular weight oligomers can form. However, unlike COX17 deficiency, the addition of copper salts to growth medium does not rescue COX function and the ability for growth on non-fermentable carbon sources.
Recombinant COX19 purified directly from yeast showed that copper was bound; however, the amount bound varied across samples, which is likely due to the cysteine residues in the protein being oxidized to varying extents in vivo, although follow-up studies to confirm this hypothesis have not yet been performed [53,55]. Cysteine to alanine substitution in COX19 showed that a mutant carrying the single C30A substitution was compromised for non-fermentable carbon growth, and that the C30A/C63A double mutant was even more so. These mutants were also deficient in copper binding [53]. Although these studies show the potential for cysteine residues of COX19 to bind metal ions in vitro, it is important to perform studies regarding the redox status and potential metal binding of the protein in vivo. Until more evidence is available, it seems much more likely that the cysteine residues play a key role in the structure of the protein rather than metal binding.
In 2015, a stable isotope labeling with amino acids in cell culture (SILAC)—mass spectrometry approach identified COX11 as a COX19 interaction partner and the apparent transient interaction was confirmed by further co-purification assays [54]. COX11 is a COX assembly factor that is important for copper insertion into COX subunit I [39,56,57,58]. When COX11 or COX19 was deleted in yeast, there was deficient COX activity and assembly [54]. Furthermore, when COX11 was knocked down, COX19 levels were depleted, suggesting that COX11 is required for COX19 accumulation or stability in the IMS. In rescue experiments, the overexpression of the proteins in opposite mutants, i.e., COX19 mutant overexpressing COX11, did not rescue the growth defect seen on non-fermentable media. The interaction of the two proteins was also shown to be dependent on the redox state of COX11, where the oxidized form of the protein has a stronger affinity for the interaction with COX19 [54]. Additionally, the same study showed that a function of COX19 is to maintain the cysteine residues at positions 208 and 210 of COX11 in an oxidized functional state through its interaction with the cysteine residue in position 111.
In addition to the cysteine residues, yeast COX19 has a hydrophobic YL motif within each alpha helix, contained within the CX9C motif (CX6YLXC) (Figure 3, arrows) that is critical for the import of the protein into mitochondria [54]. However, only the second YL motif is conserved in humans. A COX19 mutant containing glutamates at the YL positions (COX19EE) was unable to localize to the IMS; even when the mutant was forced into the mitochondria via an added IMS targeting signal, it was unable to rescue COX function, remained in a reduced state, and did not interact with COX11 [54]. The defective interaction between COX19EE and COX11 suggests that this hydrophobic region is important for the ability of COX19 to interact with its binding partner.
The COX19 protein sequence is evolutionarily conserved between yeast and humans, with 35% identity. The redox-sensitive cysteines are conserved as well as one of the two YL motifs within the CX6YLXC motif. The difference in the YL residues between yeast and humans is potentially interesting (Figure 3, arrows). In the human protein, the YL motif is evolutionarily conserved only in the second CX9C motif, suggesting that the residues in the second alpha helix play a critical role in COX19 function. It would be interesting to explore whether the YL position in the human protein gives the same results when mutated to glutamates as in the above study in yeast. Despite these interesting observations, a mutational screen of 53 patients with COX deficiency in 2004 did not reveal any mutations in the COX19 gene [59].
As in yeast, human COX19 localizes to the mitochondria [60,61]. However, a study by Leary and others in 2012 [61] suggested an additional non-COX assembly factor role for the protein. They showed that steady-state levels of COX19 were decreased in patient fibroblasts harboring a SCO1 or SCO2 mutation, with the decrease more pronounced in the former. The cellular copper levels did not affect COX19 protein levels but did affect the localization of the protein, with the bulk of COX19 localized to the cytosol if cytosolic copper was high [61]. These findings suggest that COX19 plays a role in redox-sensitive signaling during disrupted copper homeostasis. The knockdown of COX19 in control cells resulted in approximately 40% depletion of total copper levels, and completely knocking down the protein in SCO2 mutant cells resulted in a roughly 60% increase in copper levels [61]. A surprising finding of this study was that the knockdown of COX19 in control cells did not cause a significant change in COX activity, suggesting that the perturbation of the protein does not directly affect COX. It is possible that in humans, COX19 plays a role in SCO1-mediated signal transduction that regulates copper flux and homeostasis, rather than copper transport, which is its suggested role in yeast [61].
2.1.3. CHCHD7/Cox23p
CHCHD7/Cox23p is a less studied protein of the family. Cox23p deletion in yeast resulted in OXPHOS deficiency with decreased levels of mitochondrial encoded subunits [62]. As with COX19, Cox23p retained one of the three cysteines found in COX17 (C26). The sequence homology of the proteins suggests that its role may be similar to that of COX17: copper transport to chaperones for maturation of COX. However, as with COX19, more protein studies are needed to support this hypothesis. OXPHOS was rescued in Cox23p-deficient yeast cells when COX17 was overexpressed in the presence of copper salts [62]. The same study showed that the proteins have similar localization to the cytosol and the IMS and that they do not interact with one another. In a screen to identify COX23 respiratory deficiency suppressors, a COX subunit I gain of function mutation was found to rescue respiration in COX23 mutants [63]. The authors suggested that Cox23p may be involved in the biogenesis of COX I, although the exact function of the protein remains unknown.
Cox23p is the yeast homolog of CHCHD7 and is also homologous to COX17. However, the protein identity is only 27%, with the CHCH domain conserved (Figure 4) [9,16,62]. In yeast, the CHCH domain is located at the C-terminus of the 151-residue protein, whereas in humans this domain is located at the N-terminus of the 85-residue sequence [64]. Additionally, the putative N-terminal mitochondrial targeting sequence, which is used to direct to the mitochondrial matrix, is retained in yeast but not in humans, although both proteins have been shown to localize to the IMS. Whether the targeting sequence of the yeast protein is actually needed for targeting to mitochondria remains to be tested.
Human CHCHD7 has five isoforms, some of which retain the CHCH domain [9]. Lack of the CHCH domain suggests that isoforms 3, 4, and 5 do not localize to the IMS for function in the mitochondria. Translocations and splicing events in CHCHD7 transcripts are associated with some cancers in humans (Table 3) [65,66]. An NMR structural study showed that CHCHD7 is similar in structure to that of COX17, which contains a CHCH domain that has low hydrophobicity [64]. The study also found CHCHD7 to have high hydrophobicity in a unique, extended second alpha-helix and third alpha-helix, which is not structurally similar to COX17. However, more functional studies are required to identify the role of CHCHD7 in COX assembly.
2.1.4. CMC1
CMC1 was identified in a screen seeking novel candidate proteins involved in copper metalation of COX and SOD1, as well as in copper trafficking in the mitochondria [67]. The study further revealed that CMC1 in yeast is capable of binding Cu1+ in a similar manner to COX17 and COX19 in vitro and that perturbation of the gene results in decreased respiration, defective COX assembly, and inability to grow on non-fermentable substrates. However, the defect could be rescued by supplementation with copper. CMC1 is localized to the mitochondrial inner membrane, facing the IMS [67]. The yeast protein has an unpaired cysteine in its C-terminal region (Figure 5). However, mutation of this cysteine does not result in disrupted mitochondrial import or a growth defect on non-fermentable substrates as compared to mutations of the CX9C cysteines; thus, the functional ramifications of the unpaired residue are not at present clear in yeast [68].
CMC1 is conserved, with a 22% sequence identity between humans and yeast (Figure 5). Human CMC1 has two unpaired cysteines: one is located directly next to the first CX9C motif, near the middle of the protein sequence, and the other at the N-terminal end. Human CMC1 localizes to the mitochondria and has been shown to interact directly or indirectly with COX via co-IP experiments [34,67,68,69]. CMC1 knockout in HEK 293T cells reduced the stability of COX subunit I, decreased cellular respiration, COX activity, and level of supercomplexes containing COX [34]. All of these data suggest that CMC1 plays a role in COX assembly, stability, or maturation. In the same study, COX I was shown to form an early COX assembly intermediate with CMC1, COA3, and COX14, steps that are upstream of the incorporation of subunits IV and Va and are important in the post-translational stabilization of the subunit [34]. In yeast, the expression of COX I is tightly regulated though complex IV assembly; however, in humans, this does not seem to be the case. When COX biogenesis was disrupted in various backgrounds (COX I, COX II, COX III cybrids, COX Va, and COX IV silenced, and knockout of COX20, a COX II chaperone) the CMC1-COX I-COA3-COX14 complex still formed, resulting in stabilized COX I [34]. Taken together, human CMC1 appears to be a non-essential COX I chaperone that assists in stabilizing the newly synthesized subunit in an intermediate complex with COA3 and COX14 prior to downstream COX assembly events. This suggests that mutations causing functional disruptions in CMC1 could lead to destabilization of COX I and downstream complex IV assembly. Additional studies need to be performed in order to understand the association between CMC1 mutations and human disease.
2.1.5. CMC2
CMC2 shares 50% sequence identity with CMC1, is evolutionarily conserved, and has a similar localization pattern to the inner mitochondrial membrane [70]. A CMC2-null yeast strain cannot grow on non-fermentable carbon sources and cannot be rescued by overexpression of CMC1; it has a very low level of cellular respiration, depleted COX activity, and lower levels of complex subunits, with COX subunits I and II being undetectable [70]. The study also showed that production of COX I was severely diminished in CMC2-null yeast cells, suggesting that the perturbation of the protein slows down mitochondrial translation of the core subunits due to deficient COX assembly, as has generally been reported for mutations in COX assembly proteins [30]. A portion of CMC2 was shown to co-sediment and co-IP with CMC1, and additionally, CMC1 levels were increased in the CMC2 KO strain [70]. It is thus clear that the two proteins interact in some capacity and that, in the absence of CMC2, CMC1 may be upregulated or stabilized as a compensatory mechanism. However, unlike CMC1 mutant strains, when CMC2-null yeast cells are supplemented with copper salts, they are still unable to grow on non-fermentable carbon sources, even in the presence of overexpressed CMC1, COX11, or SCO1 [70]. These data suggest that CMC1 and CMC2 perform non-overlapping but cooperative functions in the trafficking of copper for use in COX biogenesis in yeast.
As noted above, CMC2 is evolutionarily conserved from yeast to humans, with 32% sequence identity (Figure 6), although the human form of the protein is smaller than the yeast homolog. As in yeast, human CMC2 shares considerable sequence overlap with human CMC1, with cysteines in the corresponding positions and similar localization to the mitochondria in a cell culture system [70]. Although one could suggest that the human protein performs a similar function as its yeast homolog, the example of COX19 serves as a reminder that this may not necessarily be true until proven experimentally. Because such data are lacking in humans with regard to CMC2 and its function in COX assembly, a number of questions remain unanswered.
2.1.6. COA5/Pet191p
Pet191p was first studied in yeast [71,72], and later an ortholog was identified in humans (C2orf64; COA5). The sequences between species have 32% sequence identity (Figure 7). Instead of having a true twin CX9C motif, the protein has a CX10C-CX9C motif. Pet191p was shown to localize to the mitochondrial IMS side of the IM in yeast [73]. In PET191 mutant yeast, COX activity was completely diminished, as was the ability of the mutant strain to grow on non-fermentable media [72,73]. Cysteine to alanine mutational studies indicate that most of the cysteines are of some importance to facilitate growth on non-fermentable media, including C5 and C56, where no growth was seen, and C15, C32, and C46, where growth was partially inhibited [73]. As with all twin CX9C family members, this suggests that the cysteines are important for the formation of disulfide bonds (Figure 7). Additionally, the dual expression of HA- and Myc-tagged Pet191p with pulldown from mitochondrial lysates showed that the protein interacts with itself since both tags were present in the bound fraction [73].
In humans, COA5 has been implicated in early COX assembly, where 2D-blue native gel analysis shows the protein in an intermediate step containing COX I, but not the other core subunits or subunits Vb or IV [74]. In the same study, a rare mutation in COA5 was shown to be associated with hypertrophic cardiomyopathy, where both patients (siblings) died shortly after birth. Both patients were homozygous for the A53P mutation discovered in the protein (Figure 7, arrow). Cultured fibroblasts from each patient showed COX deficiency both in activity and levels of both mitochondrial and nuclear encoded subunits [74]. When the patient fibroblasts harboring the A53P mutation were infected with a retrovirus expressing WT COA5, COX subunit levels were restored. Although the exact mechanism of COA5 in COX I assembly has yet to be shown, this study does suggest that mutations within the CX9C motif can interfere with protein function, likely through misfolding, as postulated by the authors.
2.2. COX Subunit II Module Assembly
COX II assembly is another important module in COX complex assembly (Figure 1). Copper insertion into the CuA site, which is required for the maturation of COX II, has been shown to take place in the mitochondrial IMS [19,75]. COX17 has also been shown to deliver copper to the binuclear CuA site, which is facilitated by the transfer of the ion to SCO2 via SCO1 [24,76].
COA6
COA6 is not a classic twin CX9C family member as it contains a single CX9C motif and a CX10C motif (Figure 8). It was first described in yeast as a novel Mia40 substrate and COX assembly factor [29]. When COA6 was deleted, yeast colonies were unable to grow under aerobic conditions, mitochondrial encoded COX subunit levels were decreased compared to WT, and overall complex IV levels were reduced in supercomplexes [29,77]. Copper supplementation [77] or treatment with elesclomal (an anticancer drug that can mimic a copper metallochaperone) [78] were able to rescue COX assembly in COA6 knockout yeast, suggesting that the protein is involved in the assembly of the copper sites either directly or indirectly.
Numerous studies have shown that COA6 and newly synthesized COX II interact with one another [79,80]. Pathogenic mutations of COA6 resulted in abrogation of this interaction [79,81], possibly due to the mislocalization of the protein within the mitochondria [79] or misfolding of the protein [80]. COA6 was also shown to interact with the copper insertion machinery for COX II, specifically the SCO1 [80] and SCO2 [79] proteins. A later study provided insight into the interaction with the SCO proteins, showing that COA6 preferentially interacts with SCO1 in the presence of both SCO proteins [82]. Additionally, the levels of mitochondrial COA6 are dependent on copper availability in yeast [81], further supporting its role in the copper chaperone machinery. In addition to interacting with SCO2, COA6 was also shown to interact with COX12/COX VIb and all three of the proteins had overlapping but non-redundant roles in COX II maturation [81]. Interestingly, purified recombinant COA6 is able to bind copper [79,80] despite its lack of the CXCC motif found in other copper binding twin CX9C proteins. Structural studies have shown that recombinant WT COA6 can bind Cu1+ at a 1:1 stoichiometry and that the binding interface is dependent upon cysteines C58 and C90 [83]. This is likely because COA6 was shown to exist within the mitochondria in a partially oxidized state [80]. However, later studies determined that the binding of copper by COA6 may not be physiological [82], instead showing that COA6 acts as a thiol-disulfide reductase when interacting with SCO1, SCO2, and COX II [82,84]. The thiol-disulfide reductase activity of the protein would allow the cysteines of SCO1, SCO2, and COX II to be modified as the copper is passed along from chaperone to chaperone until finally reaching its site in COX subunit II.
In humans, missense mutations in COA6 were identified in two unrelated patients who died from neonatal hypertrophic cardiomyopathy with reduced COX activity in the heart [80,85,86]. One patient had a compound heterozygous mutation, resulting in amino acid changes that reside in the CX9C-CX10C domain of the protein: W59C and E87X (truncation) [85]. The missense mutation at position 59 creates an extra cysteine within the CX9C motif and the nonsense mutation in the CX10C motif results in truncation and loss of a critical domain for mitochondrial import. The second patient, who also displayed muscle hypotonia and lactic acidosis, harbored a homozygous missense mutation resulting in a W66R change shown to cause decreased COX activity in fibroblasts [86]. The human point mutations were unable to fully rescue COX deficiency in COA6 knockout yeast strains, suggesting that the mutations are pathogenic, perhaps by disrupting COA6 import into the IMS [77] or protein–protein interactions necessary for its function [82]. Additionally, the crystal structure of W59C displayed differences compared to WT, including oligomerization of the mutant and differences in charge distribution, which may account for the changes seen in protein function [83].
In order to understand the importance of COA6 in heart development, a study in zebrafish embryos looked for cardiac defects in a COA6 depleted background [77]. Indeed, pericardiac edema and reduced heart rate were observed in COA6 knockdown embryos, as were reduced COX II levels. In mammalian cell culture, the knockdown of COA6 resulted in reduced oxygen consumption and COX activity, supporting its role in COX biogenesis in mammals [79,80,84]. Interestingly, exogenous copper supplementation in mammalian cells does not rescue COA6 deficiency [86] as it does in yeast, suggesting a more complex mechanism in the mammalian system.
2.3. Undetermined
CHCHD8/COA4
COA4 is conserved from yeast to humans (Figure 9). It is an inner membrane-associated protein facing the IMS that was shown to affect COX assembly in yeast but the overexpression of cytochrome c rescued COX activity defects of COA4 mutant cells [28]. The findings of the study did not reveal a direct interaction of COA4 and COX but instead suggested that COA4 helps to stabilize the membrane association of cytochrome c in these cells. This agrees with other studies showing that cytochrome c is implicated in the assembly and stability of COX [87,88]. A later study in yeast confirmed that knockout of COA4 results in a reduced oxygen consumption rate, reduced COX activity, reduced levels of the COX core subunits, and increased hydrogen peroxide production [89]. More studies are required, however, including in the mammalian system, to better understand the role of COA4 in COX assembly.
3. COX Structural Subunits
COX VIb1/Cox12p
Although not a true twin CX9C protein, this protein is often considered an “unofficial” family member in the context of COX due to structural similarity and its role in complex IV function. The nuclear encoded Cox12p/COX VIb1 has one CX9C motif and one CX10C motif. Cox12p in yeast was first shown to be a novel subunit that is required for proper COX activity but not complex assembly [90]. Cox12p protein is associated specifically with COX II biogenesis, where interaction studies show its physical interaction with COX II, COA6, and SCO1/2 [81]. The COX6B gene was mapped in human heart and three pseudogenes were identified and subsequently characterized [91,92]. COX6B1 is ubiquitously expressed across tissues whereas its isoform, COX6B2, is testes-specific [93]. COX VIb1 shares 45% sequence identity with Cox12p (Figure 10). In 1997, the dimeric complex of COX was crystallized from bovine heart and its structure determination confirmed that COX VIb1 is part of the COX complex, sitting on top of the dimer and seemingly forming a bridge on the IMS side of COX that allows the COX monomers to interact [2]. The crystal structure shows that COX VIb1 interacts with core subunits II and III [2] and, based on computer modeling [94], is part of the cytochrome c binding site of COX.
Some mutations in COX6B1 have been associated with COX deficiency disease. Two individuals with early-onset leukodystrophic encephalopathy, myopathy, and growth retardation carried a homozygous mutation in COX6B1 resulting in an amino acid residue replacement of arginine to histidine at the N-terminus of the protein [95]. Another missense mutation, resulting in an arginine to cysteine change at the same position, was mapped in an individual with encephalomyopathy, hydrocephaly, and cardiomyopathy [96].
4. COX Regulation through Direct Interaction
4.1. MNRR1 (CHCHD2)/Mix17p
MNRR1 and its isoform CHCHD10 (see next section) have a common ancestor in yeast called Mix17p (formerly Mic17p), which was initially shown to localize to the nucleus [97]. However, as interest in IMS protein import increased, it was determined that Mix17p also localized to the IMS via the Mia40 pathway [98]. In a screen looking to determine which CX9C proteins affect OXPHOS in yeast, the knockdown of Mix17p resulted in the reduction of oxygen consumption to approximately 50% of WT [16]. The protein was later shown to be stress-sensitive using agents that induce DNA replication distress, a stimulus that also led to the characterization of changes in protein localization [99].
Mix17p and MNRR1 have 36% sequence identity, with a centrally located hydrophobic domain, which is largely conserved (Figure 11). This overlap suggests that the hydrophobic central domain plays an important role in the function of the protein. Additionally, the cysteines in the CHCH domain are conserved in a twin CX9C motif. As in yeast, mammalian MNRR1 was determined to affect OXPHOS in a computational screen coupled with functional assays in human cell lines [100]. MNRR1 is also a stress-sensitive protein and displays dual localization to the mitochondria and the nucleus [101,102]. During 20% oxygen tension growth in cell culture, most cellular MNRR1 is in the mitochondria; however, at more physiological 4% oxygen tension, MNRR1 levels rapidly turn over in the mitochondria and increase in the nucleus [102].
In the mitochondria, MNRR1 has been shown to interact with COX and regulate its activity [102,103]. MNRR1 knockdown in cells affected multiple mitochondrial processes including a ~50% reduction of oxygen consumption rate, an increase in reactive oxygen species (ROS), a reduction in mitochondrial membrane potential, slower growth [102], and fragmented mitochondria, which increasingly form during oxidative stress [104,105,106]. A large-scale protein–protein interaction study showed that MNRR1 interacts with two COX subunits on the IMS-facing side of the IM (COX VIc and COX VIa1), as well as cytochrome c, further suggesting that MNRR1 is a regulator of COX activity [107]. However, further work will be needed as the interaction study, which was carried out with total cell lysate, also identified an interaction with wholly matrix-localized COX subunits yet MNRR1 has not been detected in the matrix [108].
The interaction between MNRR1 and COX is promoted when MNRR1 is phosphorylated at tyrosine residue 99 (Figure 11, arrow) by Abl2/Arg kinase [103]. When Y99 is mutated to glutamate to mimic phosphorylation, an increase in oxygen consumption is detected compared to WT. MNRR1 has been shown to have another phosphorylation site, in the retained (non-cleaved due to IMS localization) putative mitochondrial targeting sequence of the protein.
Serine residue 41 has been shown to be phosphorylated in breast cancer and levels of MNRR1 are also increased with tumor grade, seemingly aiding the aggressiveness of breast cancer cells [109,110]. Studies have shown that oxygen consumption increases with rising levels of MNRR1, as does tumor grade [109]. MNRR1 is also associated with lung and liver cancer [111,112], and the neurodegenerative disorders Parkinson’s disease (PD) [113], Huntington’s disease [114], and lissencephaly (a neuronal migration disorder) [115]. MNRR1 has been shown to regulate cell migration [116], suggesting that it could be important for metastasis in cancer and affecting migration in lissencephaly. However, in most cases, the mechanistic involvement of MNRR1 in the disease is unknown. Of the disease-associated mutations of MNRR1, T61I (PD) is perhaps the best studied. In HEK 293 cells, overexpression of the T61I mutation results in a greater binding affinity for endogenous CHCHD10 and endogenous MNRR1 interaction [117]. However, it is important to note that the presence of increased CHCHD10 in the pulldown performed could be due to the protein binding to the endogenous WT MNRR1 in the pulldown, and not necessarily due to interaction with the overexpressed mutant. This would suggest an alteration in MNRR1 function in the mitochondria, including COX regulation.
In the nucleus, MNRR1 acts as a transcriptional activator by binding to RBPJκ at the highly conserved oxygen responsive element (ORE) in the COX4I2 promoter, stimulating the expression of the COX IV-2 subunit isoform, which is highly expressed in the lung, trachea, and carotid body [101,102,118,119,120]. Expression of COX IV-2 is maximal at 4% oxygen tension [101,102,118,119]. When COX IV-2 is present in the complex isolated from lung, COX activity was twofold higher than the activity of COX isolated from liver with only COX IV-1 present [121]. Other COX subunits and chaperones contain the ORE within their gene promoters, including COX VIIa2 and COA4, as well as other proteins discussed in this review (Table 4). Based on this information, MNRR1 regulates COX activity with a bi-organellar approach: both through physical interaction with the complex in the mitochondria and through stress-induced transcription in the nucleus.
MNRR1 and CHCHD10 are the only members of the twin CX9C family of proteins shown to dually localize to the mitochondria and the nucleus. Interestingly, CMC1 and CMC2 also have putative nuclear localization signals on the C-terminal end of the proteins; however, these putative signals have yet to be tested by mutations and, for CMC1 and CMC2, it has yet to be shown that they localize to the nucleus. Additionally, the structures of CMC1 and CMC2 are unknown, which makes it difficult to determine if the putative NLS would be structurally accessible for nuclear localization.
4.2. CHCHD10/Mix17p
Mix17p and CHCHD10 have a 33% sequence identity (Figure 11) and, like MNRR1, the central hydrophobic domain and CHCH domain are largely conserved between the two proteins. Due to the sequence homology between MNRR1 and CHCHD10 (56%) and recent common ancestor divergence (Figure 12), it was suggested that the two proteins perform similar roles within the cell [9]. Further studies provided evidence that this hypothesis was true in the mitochondria but not in the nucleus.
CHCHD10 has been studied in the mitochondria and the nucleus [122,123,124]. Like MNRR1, CHCHD10 also localizes to the nucleus; however, at least for the COX4I2 promoter, the protein acts as a transcriptional repressor at the ORE by binding to transcription factor CXXC5 at 8% oxygen tension in cell culture [124]. It is possible that the accumulation of MNRR1 in the nucleus at 4% oxygen displaces CXXC5 and CHCHD10 to activate ORE-mediated transcription; however, additional work is needed to characterize the mechanism in more detail. In the mitochondria, CHCHD10 localizes to the IMS and its knockdown results in decreased COX activity and ATP levels [122,125]. Like MNRR1, CHCHD10 co-purifies with COX at sub-stochiometric levels, suggesting an interaction with the complex. Experimental evidence supports an indirect interaction through MNRR1 [124]. MNRR1 knockout results in defective CHCHD10-COX binding as well as decreased COX activity [124]. In the same study, knockdown of CHCHD10 resulted in less phosphorylation of MNRR1, suggesting that CHCHD10 regulates COX through the recruitment of Arg/Abl2 kinase for MNRR1 phosphorylation. CHCHD10 has also been shown to interact with COX subunits (COX Va, COX VIc, COX VIa1, COX VIIa2) and COX assembly factor COA3 [107].
Much like MNRR1, CHCHD10 is associated with various neurodegenerative diseases: spinal muscular atrophy (Jokela type) (SMAJ) [126], amyotrophic lateral sclerosis (ALS) and/or frontotemporal dementia (FTD) [125,127], lower motor neuron syndrome [128], and autosomal dominant mitochondrial myopathy [123]. In all these cases, specific mutations in CHCHD10 are associated with each disease. Missense mutations S59L, P34S, and P80L are found in patients with FTD and/or ALS. Double missense mutation R15S and G58R is associated with autosomal dominant mitochondrial myopathy. Missense mutations R15L and G66V are associated with lower motor neuron syndrome and G66V is also associated with SMAJ and Charcot-Marie-Tooth disease type 2 [129]. A recent paper showed that both G66V and P80L were functionally defective in both the nucleus and the mitochondria, thereby characterizing the pathogenicity of the two mutations [124].
5. CX9C Proteins of Unknown Function
CMC4
CMC4 is a CX9C family member that localizes to the mitochondrial IMS [16,98]. In yeast, CMC4 has eight cysteine residues, four of which comprise the twin CX9C motif. Seven of the eight cysteines are conserved in the human homolog (Figure 13). The overall sequence identity is 30%, with the yeast protein being slightly larger than the human. The limited studies in yeast show that CMC4 knockdown produces no defect in respiration [16]. However, an affinity purification–mass spectrometry proximity labeling study determined that CMC4 interacts with SCO1 in human cell cultures [130]. This suggests that CMC4 plays a non-essential role in COX II sub-assembly. One hypothesis is that CMC4 has a redundant function with another SCO1 interacting protein and somehow compensates when the other assembly factor is dysfunctional. CMC4 is associated with two diseases: T cell leukemia with translocation (X:14), where overexpression of the protein is observed [131,132,133,134,135], and frataxin mRNA deficiency [136].
6. Concluding Remarks
The evolutionary conservation from yeast to human of the twin CX9C proteins indicates that the proteins play important and conserved roles (Figure 12). This family of proteins has recently piqued the interest of investigators in the field of mitochondrial biology because (a) many family members were until recently still classified as “orphan” proteins of unknown function, and (b) the limited functional studies showed that they play important roles in the regulation of oxidative phosphorylation, seemingly through their activities relating to COX regulation. Although some of the family members are better studied than others, much remains unknown about the functional aspects of the proteins, including their underlying roles in disease.
Over the years, many diseases have been studied that are associated with defective COX biogenesis, which often involve brain, heart, and skeletal muscle (extensively reviewed in [137,138,139]). Most of these diseases involve assembly factors, chaperones, and other regulatory proteins because COX biogenesis and activity are tightly regulated processes [140]. Mutations in SCO1/SCO2 [22,141,142,143,144], COX10 [145,146], COX15 [147], and SURF1 [21,143,148,149] have all been associated with COX dysfunction. Studies have also shown that mutations in members of the twin CX9C family can affect COX activity and are associated with various diseases including neurological disorders, cancers, and cardiomyopathy (Table 3). However, in most cases, the exact molecular pathogenic mechanism causing COX deficiency is unknown. Understanding the function of these proteins and the mutations that result in functional defects is critical to understanding disease etiology, which remains unknown in most cases.
Although the proteins in this family display diverse functions, one commonality is their import into the IMS. The proteins are able to localize to the IMS through the Mia40/CHCHD4-Erv1/ALR import pathway, which relies on redox sensitivity [14,15,150,151]. An interesting consideration is that IM-bound Mia40/CHCHD4, which is also a family member and an oxidoreductase, may act as an indirect regulator of COX activity through the active or inhibited import of twin CX9C proteins. The IMS import machinery works through a disulfide relay system, where unfolded (reduced) twin CX9C proteins are brought into the IMS via the translocase of the outer membrane (TOM). The oxidized cysteine residues of Mia40/CHCHD4 then capture the reduced incoming proteins by forming disulfide bridges. Once the disulfide bonds are modified, Mia40/CHCHD4 releases the properly folded (oxidized) substrate protein into the IMS and is re-oxidized by Erv1/ALR [151]. Additionally, Erv1 has been shown to transfer electrons to Cytc [152], suggesting that OXPHOS is regulated at multiple levels by the IMS import machinery.
One understudied area is that of post-translational modifications (PTMs) of the twin CX9C proteins. The addition or removal of PTMs affects protein activity, lifetime, and interactions. Better understanding of PTMs as regulators of COX would provide a layer of new knowledge with regard to regulation and dysregulation. A prime example is that of MNRR1, which has been shown to be phosphorylated at residue Y99 through the interaction with CHCHD10 and Abl2 kinase [103]. Phosphorylation at this site increases the activity of COX compared to WT MNRR1. However, in a family with Charcot-Marie-Tooth disease type 1A, a Q112H mutation resulted in decreased COX activity, no interaction with COX, and decreased phosphorylation of MNRR1 [103]. Another example is that of phosphorylation at serine 41, which has been associated with breast cancer in two high-throughput studies [110]. Other PTMs have also been identified for twin CX9C proteins although most of them derive from high-throughput studies and require functional follow-up [153].
In addition to enzymatic PTM additions to proteins, non-enzymatic PTMs can occur due to oxidative stress. For example, cysteine residues can be modified by reactive oxygen species. This is particularly true in the mitochondria, which are a central hub for redox signaling (see [154] for comprehensive review on cysteine-mediated redox signaling in the mitochondria). Alterations of the cysteine residues of twin CX9C proteins due to oxidative stress could affect IMS import, misfolding, and thereby stability and/or protein–protein interactions, and copper interaction for copper binding protein family members. In fact, tissue-specific cysteine modifications have recently been mapped in young and old mice, laying a foundational landscape upon which to study cysteine oxidation [155]. The members of the twin CX9C family that were annotated in this study as having cysteine oxidation are MNRR1, CHCHD7, CMC1, COA6, and COX VIb1. This is yet another area ready for investigation to further build upon the mechanistic activities of twin CX9C proteins during aging, stress, and disease.
The twin CX9C proteins are a unique family of proteins that are emerging in the field of mitochondrial biology as important regulators of COX through assembly, structure, and direct interaction. Some of the proteins have expanded roles in mitochondrial function, such as MNRR1 and CHCHD10. These two proteins, in addition to directly regulating COX activity, also localize in the nucleus, where they function as transcription factors, modulating mitochondrial–nuclear crosstalk. In fact, many genes involved in COX biogenesis have the minimal ORE (to which MNRR1 and CHCHD10 bind) within 500 bp upstream of the transcriptional start site (Table 4). COX dysfunction is associated with numerous diseases. The “why” of COX dysfunction translating to disease is easy enough to understand: COX is the terminal enzyme of the ETC. If the complex is not working properly, the result is disrupted mitochondrial homeostasis. It is the “how” that has an important role to play in treating disease, as the assembly and regulation of the complex is finely tuned. Of course, this is a simplified way of thinking about a complex problem. Further understanding this protein family’s involvement in maintaining mitochondrial homeostasis during both normal, stress, and disease conditions will greatly add to the understanding of the regulation of COX activity and the family’s direct involvement in disease pathophysiology.
Author Contributions
S.G., writing original draft; S.G, S.A., M.H., L.I.G., revising and editing of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by the Office of the Assistant Secretary of Defense for Health Affairs through the Peer-Reviewed Medical Research Program under Awards W81XWH-16–1-0516 (to L.I.G. and M.H.), U.S. National Institutes of Health grant R01 GM116807 (to M.H.), the Perinatology Research Branch, Division of Obstetrics and Maternal-Fetal Medicine, Division of Intramural Research, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, U.S. Department of Health and Human Services (NICHD/NIH/DHHS); and, in part, with Federal funds from NICHD/NIH/DHHS under Contract No. HHSN275201300006C (to S.A. and L.I.G.), and the Henry L. Brasza endowment at Wayne State University (to L.I.G.). The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the Department of Defense or the US Government.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the collection, analyses or interpretation of data; in the writing of the manuscript, or in the decision to publish the review.
References
- Kadenbach, B.; Hüttemann, M. The subunit composition and function of mammalian cytochrome c oxidase. Mitochondrion 2015, 24, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Tsukihara, T.; Aoyama, H.; Yamashita, E.; Tomizaki, T.; Yamaguchi, H.; Shinzawa-Itoh, K.; Nakashima, R.; Yaono, R.; Yoshikawa, S. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 Å. Science 1996, 272, 1136. [Google Scholar] [CrossRef] [PubMed]
- Osuda, Y.; Shinzawa-Itoh, K.; Tani, K.; Maeda, S.; Yoshikawa, S.; Tsukihara, T.; Gerle, C. Two-dimensional crystallization of monomeric bovine cytochrome c oxidase with bound cytochrome c in reconstituted lipid membranes. J. Electron Microsc. 2016, 65, 263–267. [Google Scholar] [CrossRef] [Green Version]
- Shinzawa-Itoh, K.; Sugimura, T.; Misaki, T.; Tadehara, Y.; Yamamoto, S.; Hanada, M.; Yano, N.; Nakagawa, T.; Uene, S.; Yamada, T.; et al. Monomeric structure of an active form of bovine cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 2019, 116, 19945–19951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Gu, J.; Guo, R.; Huang, Y.; Yang, M. Structure of mammalian respiratory supercomplex I2III2IV2. Cell 2016, 167, 1598–1609.e10. [Google Scholar] [CrossRef] [Green Version]
- Balsa, E.; Marco, R.; Perales-Clemente, E.; Szklarczyk, R.; Calvo, E.; Landazuri, M.O.; Enriquez, J.A. NDUFA4 is a subunit of complex IV of the mammalian electron transport chain. Cell Metab. 2012, 16, 378–386. [Google Scholar] [CrossRef] [Green Version]
- Zong, S.; Wu, M.; Gu, J.; Liu, T.; Guo, R.; Yang, M. Structure of the intact 14-subunit human cytochrome c oxidase. Cell Res. 2018, 28, 1026–1034. [Google Scholar] [CrossRef] [Green Version]
- Kadenbach, B. Regulation of mammalian 13-subunit cytochrome c oxidase and binding of other proteins: Role of NDUFA4. Trends Endocrinol. Metab. 2017, 28, 761–770. [Google Scholar] [CrossRef]
- Cavallaro, G. Genome-wide analysis of eukaryotic twin CX9C proteins. Mol. Biosyst. 2010, 6, 2459–2470. [Google Scholar] [CrossRef]
- Glerum, D.M.; Shtanko, A.; Tzagoloff, A. Characterization of COX17, a yeast gene involved in copper metabolism and assembly of cytochrome oxidase. J. Biol. Chem. 1996, 271, 14504–14509. [Google Scholar] [CrossRef] [Green Version]
- Abajian, C.; Yatsunyk La Fau-Ramirez, B.E.; Ramirez Be Fau-Rosenzweig, A.C.; Rosenzweig, A.C. Yeast Cox17 solution structure and Copper(I) binding. J. Biol. Chem. 2004, 279, 53584–53592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnesano, F.; Balatri, E.; Banci, L.; Bertini, I.; Winge, D.R. Folding studies of Cox17 reveal an important interplay of cysteine oxidation and copper binding. Structure 2005, 13, 713–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koehler, C.M. The small Tim proteins and the twin Cx3C motif. Trends Biochem. Sci. 2004, 29, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Terziyska, N.; Lutz, T.; Kozany, C.; Mokranjac, D.; Mesecke, N.; Neupert, W.; Herrmann, J.M.; Hell, K. Mia40, a novel factor for protein import into the intermembrane space of mitochondria is able to bind metal ions. FEBS Lett. 2005, 579, 179–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chacinska, A.; Pfannschmidt, S.; Wiedemann, N.; Kozjak, V.; Sanjuan Szklarz, L.K.; Schulze-Specking, A.; Truscott, K.N.; Guiard, B.; Meisinger, C.; Pfanner, N. Essential role of Mia40 in import and assembly of mitochondrial intermembrane space proteins. EMBO J. 2004, 23, 3735–3746. [Google Scholar] [CrossRef] [Green Version]
- Longen, S.; Bien, M.; Bihlmaier, K.; Kloeppel, C.; Kauff, F.; Hammermeister, M.; Westermann, B.; Herrmann, J.M.; Riemer, J. Systematic analysis of the twin CX9C protein family. J. Mol. Biol. 2009, 393, 356–368. [Google Scholar] [CrossRef] [PubMed]
- Barrientos, A.; Barros, M.H.; Valnot, I.; Rotig, A.; Rustin, P.; Tzagoloff, A. Cytochrome oxidase in health and disease. Gene 2002, 286, 53–63. [Google Scholar] [CrossRef]
- Vidoni, S.; Harbour, M.E.; Guerrero-Castillo, S.; Signes, A.; Ding, S.; Fearnley, I.M.; Taylor, R.W.; Tiranti, V.; Arnold, S.; Fernandez-Vizarra, E.; et al. MR-1S interacts with PET100 and PET117 in module-based assembly of human cytochrome c oxidase. Cell Rep. 2017, 18, 1727–1738. [Google Scholar] [CrossRef] [Green Version]
- Timon-Gomez, A.; Nyvltova, E.; Abriata, L.A.; Vila, A.J.; Hosler, J.; Barrientos, A. Mitochondrial cytochrome c oxidase biogenesis: Recent developments. Semin. Cell Dev. Biol. 2018, 76, 163–178. [Google Scholar] [CrossRef]
- Stiburek, L.; Vesela, K.; Hansikova, H.; Pecina, P.; Tesarova, M.; Cerna, L.; Houstek, J.; Zeman, J. Tissue-specific cytochrome c oxidase assembly defects due to mutations in SCO2 and SURF1. Biochem. J. 2005, 392, 625–632. [Google Scholar] [CrossRef] [Green Version]
- Williams, S.L.; Valnot, I.; Rustin, P.; Taanman, J.W. Cytochrome c oxidase subassemblies in fibroblast cultures from patients carrying mutations in COX10, SCO1, or SURF1. J. Biol. Chem. 2004, 279, 7462–7469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaksch, M.; Ogilvie, I.; Yao, J.; Kortenhaus, G.; Bresser, H.G.; Gerbitz, K.D.; Shoubridge, E.A. Mutations in SCO2 are associated with a distinct form of hypertrophic cardiomyopathy and cytochrome c oxidase deficiency. Hum. Mol. Genet. 2000, 9, 795–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaksch, M.; Paret, C.; Stucka, R.; Horn, N.; Muller-Hocker, J.; Horvath, R.; Trepesch, N.; Stecker, G.; Freisinger, P.; Thirion, C.; et al. Cytochrome c oxidase deficiency due to mutations in SCO2, encoding a mitochondrial copper-binding protein, is rescued by copper in human myoblasts. Hum. Mol. Genet. 2001, 10, 3025–3035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leary, S.C.; Kaufman, B.A.; Pellecchia, G.; Guercin, G.H.; Mattman, A.; Jaksch, M.; Shoubridge, E.A. Human SCO1 and SCO2 have independent, cooperative functions in copper delivery to cytochrome c oxidase. Hum. Mol. Genet. 2004, 13, 1839–1848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horn, D.; Barrientos, A. Mitochondrial copper metabolism and delivery to cytochrome c oxidase. Iubmb. Life 2008, 60, 421–429. [Google Scholar] [CrossRef] [Green Version]
- Leary, S.C.; Winge, D.R.; Cobine, P.A. Pulling the plug on cellular copper: The role of mitochondria in copper export. Biochim. Biophys Acta 2009, 1793, 146–153. [Google Scholar] [CrossRef] [Green Version]
- Valentine, J.S.; Wertz, D.L.; Lyons, T.J.; Liou, L.L.; Goto, J.J.; Gralla, E.B. The dark side of dioxygen biochemistry. Curr. Opin. Chem. Biol. 1998, 2, 253–262. [Google Scholar] [CrossRef]
- Bestwick, M.; Jeong, M.Y.; Khalimonchuk, O.; Kim, H.; Winge, D.R. Analysis of Leigh syndrome mutations in the yeast SURF1 homolog reveals a new member of the cytochrome oxidase assembly factor family. Mol. Cell. Biol. 2010, 30, 4480–4491. [Google Scholar] [CrossRef] [Green Version]
- Vogtle, F.N.; Burkhart, J.M.; Rao, S.; Gerbeth, C.; Hinrichs, J.; Martinou, J.C.; Chacinska, A.; Sickmann, A.; Zahedi, R.P.; Meisinger, C. Intermembrane space proteome of yeast mitochondria. Mol. Cell. Proteom. 2012, 11, 1840–1852. [Google Scholar] [CrossRef] [Green Version]
- Barrientos, A.; Zambrano, A.; Tzagoloff, A. Mss51p and Cox14p jointly regulate mitochondrial Cox1p expression in Saccharomyces cerevisiae. EMBO J. 2004, 23, 3472–3482. [Google Scholar] [CrossRef] [Green Version]
- Fontanesi, F.; Clemente, P.; Barrientos, A. Cox25 teams up with Mss51, Ssc1, and Cox14 to regulate mitochondrial cytochrome c oxidase subunit 1 expression and assembly in Saccharomyces cerevisiae. J. Biol. Chem. 2011, 286, 555–566. [Google Scholar] [CrossRef] [Green Version]
- Mick, D.U.; Vukotic, M.; Piechura, H.; Meyer, H.E.; Warscheid, B.; Deckers, M.; Rehling, P. Coa3 and Cox14 are essential for negative feedback regulation of COX1 translation in mitochondria. J. Cell. Biol. 2010, 191, 141–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clemente, P.; Peralta, S.; Cruz-Bermudez, A.; Echevarria, L.; Fontanesi, F.; Barrientos, A.; Fernandez-Moreno, M.A.; Garesse, R. hCOA3 stabilizes cytochrome c oxidase 1 (COX1) and promotes cytochrome c oxidase assembly in human mitochondria. J. Biol. Chem. 2013, 288, 8321–8331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourens, M.; Barrientos, A. A CMC1-knockout reveals translation-independent control of human mitochondrial complex IV biogenesis. Embo. Rep. 2017, 18, 477–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, A.K.; Smith, D.; Gray, J.; Carr, H.S.; Liu, A.; Winge, D.R.; Hosler, J.P. Mutagenic analysis of Cox11 of Rhodobacter sphaeroides: Insights into the assembly of Cu(B) of cytochrome c oxidase. Biochemistry 2010, 49, 5651–5661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beers, J.; Glerum, D.M.; Tzagoloff, A. Purification, characterization, and localization of yeast Cox17p, a mitochondrial copper shuttle. J. Biol. Chem. 1997, 272, 33191–33196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maxfield, A.B.; Heaton, D.N.; Winge, D.R. Cox17 is functional when tethered to the mitochondrial inner membrane. J. Biol. Chem. 2004, 279, 5072–5080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glerum, D.M.; Shtanko, A.; Tzagoloff, A. SCO1 and SCO2 act as high copy suppressors of a mitochondrial copper recruitment defect in Saccharomyces cerevisiae. J. Biol. Chem. 1996, 271, 20531–20535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horng, Y.C.; Cobine, P.A.; Maxfield, A.B.; Carr, H.S.; Winge, D.R. Specific copper transfer from the Cox17 metallochaperone to both Sco1 and Cox11 in the assembly of yeast cytochrome c oxidase. J. Biol. Chem. 2004, 279, 35334–35340. [Google Scholar] [CrossRef] [Green Version]
- Hiser, L.; Di Valentin, M.; Hamer, A.G.; Hosler, J.P. Cox11p is required for stable formation of the Cu(B) and magnesium centers of cytochrome c oxidase. J. Biol. Chem. 2000, 275, 619–623. [Google Scholar] [CrossRef] [Green Version]
- Heaton, D.; Nittis, T.; Srinivasan, C.; Winge, D.R. Mutational analysis of the mitochondrial copper metallochaperone Cox17. J. Biol. Chem. 2000, 275, 37582–37587. [Google Scholar] [CrossRef] [Green Version]
- Heaton, D.N.; George, G.N.; Garrison, G.; Winge, D.R. The mitochondrial copper metallochaperone Cox17 exists as an oligomeric, polycopper complex. Biochemistry 2001, 40, 743–751. [Google Scholar] [CrossRef]
- Takahashi, Y.; Kako, K.; Kashiwabara, S.; Takehara, A.; Inada, Y.; Arai, H.; Nakada, K.; Kodama, H.; Hayashi, J.; Baba, T.; et al. Mammalian copper chaperone Cox17p has an essential role in activation of cytochrome c oxidase and embryonic development. Mol. Cell. Biol. 2002, 22, 7614–7621. [Google Scholar] [CrossRef] [Green Version]
- Oswald, C.; Krause-Buchholz, U.; Rödel, G. Knockdown of Human COX17 Affects assembly and supramolecular organization of cytochrome c oxidase. J. Mol. Biol. 2009, 389, 470–479. [Google Scholar] [CrossRef]
- Vanisova, M.; Burska, D.; Krizova, J.; Danhelovska, T.; Dosoudilova, Z.; Zeman, J.; Stiburek, L.; Hansikova, H. Stable COX17 downregulation leads to alterations in mitochondrial ultrastructure, decreased copper content and impaired cytochrome c oxidase biogenesis in HEK293 cells. Folia Biol. 2019, 65, 181–187. [Google Scholar]
- Palumaa, P.; Kangur, L.; Voronova, A.; Sillard, R. Metal-binding mechanism of Cox17, a copper chaperone for cytochrome c oxidase. Biochem. J. 2004, 382, 307–314. [Google Scholar] [CrossRef] [Green Version]
- Banci, L.; Bertini, I.; Ciofi-Baffoni, S.; Janicka, A.; Martinelli, M.; Kozlowski, H.; Palumaa, P. A structural-dynamical characterization of human Cox17. J. Biol. Chem. 2008, 283, 7912–7920. [Google Scholar] [CrossRef] [Green Version]
- Cobine, P.A.; Ojeda, L.D.; Rigby, K.M.; Winge, D.R. Yeast contain a non-proteinaceous pool of copper in the mitochondrial matrix. J. Biol. Chem. 2004, 279, 14447–14455. [Google Scholar] [CrossRef] [Green Version]
- Cobine, P.A.; Pierrel, F.; Bestwick, M.L.; Winge, D.R. Mitochondrial matrix copper complex used in metallation of cytochrome oxidase and superoxide dismutase. J. Biol. Chem. 2006, 281, 36552–36559. [Google Scholar] [CrossRef] [Green Version]
- Horvath, R.; Lochmuller, H.; Stucka, R.; Yao, J.; Shoubridge, E.A.; Kim, S.H.; Gerbitz, K.D.; Jaksch, M. Characterization of human SCO1 and COX17 genes in mitochondrial cytochrome c oxidase deficiency. Biochem. Biophys. Res. Commun. 2000, 276, 530–533. [Google Scholar] [CrossRef]
- Sacconi, S.; Salviati, L.; Sue, C.M.; Shanske, S.; Davidson, M.M.; Bonilla, E.; Naini, A.B.; De Vivo, D.C.; DiMauro, S. Mutation screening in patients with isolated cytochrome c oxidase deficiency. Pediatr. Res. 2003, 53, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Nobrega, M.P.; Bandeira, S.C.; Beers, J.; Tzagoloff, A. Characterization of COX19, a widely distributed gene required for expression of mitochondrial cytochrome oxidase. J. Biol. Chem. 2002, 277, 40206–40211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigby, K.; Zhang, L.; Cobine, P.A.; George, G.N.; Winge, D.R. characterization of the cytochrome c oxidase assembly factor Cox19 of Saccharomyces cerevisiae. J. Biol. Chem. 2007, 282, 10233–10242. [Google Scholar] [CrossRef] [Green Version]
- Bode, M.; Woellhaf, M.W.; Bohnert, M.; van der Laan, M.; Sommer, F.; Jung, M.; Zimmermann, R.; Schroda, M.; Herrmann, J.M. Redox-regulated dynamic interplay between Cox19 and the copper-binding protein Cox11 in the intermembrane space of mitochondria facilitates biogenesis of cytochrome c oxidase. Mol. Biol. Cell. 2015, 26, 2385–2401. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Horn, S.; Belkacemi, A.; Kojer, K.; Petrungaro, C.; Habich, M.; Ali, M.; Kuttner, V.; Bien, M.; Kauff, F.; et al. Protein import and oxidative folding in the mitochondrial intermembrane space of intact mammalian cells. Mol. Biol. Cell. 2013, 24, 2160–2170. [Google Scholar] [CrossRef] [PubMed]
- Tzagoloff, A.; Capitanio, N.; Nobrega, M.P.; Gatti, D. Cytochrome oxidase assembly in yeast requires the product of COX11, a homolog of the P. denitrificans protein encoded by ORF3. EMBO J. 1990, 9, 2759–2764. [Google Scholar] [CrossRef]
- Carr, H.S.; George, G.N.; Winge, D.R. Yeast Cox11, a protein essential for cytochrome c oxidase assembly, is a Cu(I)-binding protein. J. Biol. Chem. 2002, 277, 31237–31242. [Google Scholar] [CrossRef] [Green Version]
- Banci, L.; Bertini, I.; Cantini, F.; Ciofi-Baffoni, S.; Gonnelli, L.; Mangani, S. Solution structure of Cox11, a novel type of beta-immunoglobulin-like fold involved in CuB site formation of cytochrome c oxidase. J. Biol. Chem. 2004, 279, 34833–34839. [Google Scholar] [CrossRef] [Green Version]
- Tay, S.K.; Nesti, C.; Mancuso, M.; Schon, E.A.; Shanske, S.; Bonilla, E.; Davidson, M.M.; Dimauro, S. Studies of COX16, COX19, and PET191 in human cytochrome c oxidase deficiency. Arch. Neurol. 2004, 61, 1935–1937. [Google Scholar] [CrossRef] [Green Version]
- Sacconi, S.; Trevisson, E.; Pistollato, F.; Baldoin, M.C.; Rezzonico, R.; Bourget, I.; Desnuelle, C.; Tenconi, R.; Basso, G.; DiMauro, S.; et al. hCOX18 and hCOX19: Two human genes involved in cytochrome c oxidase assembly. Biochem. Biophys. Res. Commun. 2005, 337, 832–839. [Google Scholar] [CrossRef]
- Leary, S.C.; Cobine, P.A.; Nishimura, T.; Verdijk, R.M.; de Krijger, R.; de Coo, R.; Tarnopolsky, M.A.; Winge, D.R.; Shoubridge, E.A. COX19 mediates the transduction of a mitochondrial redox signal from SCO1 that regulates ATP7A-mediated cellular copper efflux. Mol. Biol. Cell. 2013, 24, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Barros, M.H.; Johnson, A.; Tzagoloff, A. COX23, a homologue of COX17, is required for cytochrome oxidase assembly. J. Biol. Chem. 2004, 279, 31943–31947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dela Cruz, R.; Jeong, M.Y.; Winge, D.R. Cox1 mutation abrogates need for Cox23 in cytochrome c oxidase biogenesis. Microb. Cell. 2016, 3, 275–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banci, L.; Bertini, I.; Ciofi-Baffoni, S.; Jaiswal, D.; Neri, S.; Peruzzini, R.; Winkelmann, J. Structural characterization of CHCHD5 and CHCHD7: Two atypical human twin CX9C proteins. J. Struct. Biol. 2012, 180, 190–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asp, J.; Persson, F.; Kost-Alimova, M.; Stenman, G. CHCHD7-PLAG1 and TCEA1-PLAG1 gene fusions resulting from cryptic, intrachromosomal 8q rearrangements in pleomorphic salivary gland adenomas. Genes Chromosom. Cancer 2006, 45, 820–828. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.N.; Choi, P.S.; de Waal, L.; Sharifnia, T.; Imielinski, M.; Saksena, G.; Pedamallu, C.S.; Sivachenko, A.; Rosenberg, M.; Chmielecki, J.; et al. A pan-cancer analysis of transcriptome changes associated with somatic mutations in U2AF1 reveals commonly altered splicing events. PLoS ONE 2014, 9, e87361. [Google Scholar] [CrossRef] [Green Version]
- Horn, D.; Al-Ali, H.; Barrientos, A. Cmc1p is a conserved mitochondrial twin CX9C protein involved in cytochrome c oxidase biogenesis. Mol. Cell. Biol. 2008, 28, 4354–4364. [Google Scholar] [CrossRef] [Green Version]
- Bourens, M.; Dabir, D.V.; Tienson, H.L.; Sorokina, I.; Koehler, C.M.; Barrientos, A. Role of twin Cys-Xaa9-Cys motif cysteines in mitochondrial import of the cytochrome c oxidase biogenesis factor Cmc1. J. Biol. Chem. 2012, 287, 31258–31269. [Google Scholar] [CrossRef] [Green Version]
- Mick, D.U.; Dennerlein, S.; Wiese, H.; Reinhold, R.; Pacheu-Grau, D.; Lorenzi, I.; Sasarman, F.; Weraarpachai, W.; Shoubridge, E.A.; Warscheid, B.; et al. MITRAC links mitochondrial protein translocation to respiratory-chain assembly and translational regulation. Cell 2012, 151, 1528–1541. [Google Scholar] [CrossRef] [Green Version]
- Horn, D.; Zhou, W.; Trevisson, E.; Al-Ali, H.; Harris, T.K.; Salviati, L.; Barrientos, A. The conserved mitochondrial twin CX9C protein Cmc2 is a Cmc1 homologue essential for cytochrome c oxidase biogenesis. J. Biol. Chem. 2010, 285, 15088–15099. [Google Scholar] [CrossRef] [Green Version]
- McEwen, J.E.; Hong, K.H.; Park, S.; Preciado, G.T. Sequence and chromosomal localization of two PET genes required for cytochrome c oxidase assembly in Saccharomyces cerevisiae. Curr. Genet. 1993, 23, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Baganz, F.; Hayes, A.; Farquhar, R.; Butler, P.R.; Gardner, D.C.; Oliver, S.G. Quantitative analysis of yeast gene function using competition experiments in continuous culture. Yeast 1998, 14, 1417–1427. [Google Scholar] [CrossRef]
- Khalimonchuk, O.; Rigby, K.; Bestwick, M.; Pierrel, F.; Cobine, P.A.; Winge, D.R. Pet191 is a cytochrome c oxidase assembly factor in Saccharomyces cerevisiae. Eukaryot Cell. 2008, 7, 1427–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huigsloot, M.; Nijtmans, L.G.; Szklarczyk, R.; Baars, M.J.; van den Brand, M.A.; Hendriksfranssen, M.G.; van den Heuvel, L.P.; Smeitink, J.A.; Huynen, M.A.; Rodenburg, R.J. A mutation in C2orf64 causes impaired cytochrome c oxidase assembly and mitochondrial cardiomyopathy. Am. J. Hum. Genet. 2011, 88, 488–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, Z.N.; Cobine, P.A.; Leary, S.C. The mitochondrion: A central architect of copper homeostasis. Metallomics 2017, 9, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Leary, S.C.; Sasarman, F.; Nishimura, T.; Shoubridge, E.A. Human SCO2 is required for the synthesis of CO II and as a thiol-disulphide oxidoreductase for SCO1. Hum. Mol. Genet. 2009, 18, 2230–2240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.; Trivedi, P.P.; Timbalia, S.A.; Griffin, A.T.; Rahn, J.J.; Chan, S.S.; Gohil, V.M. Copper supplementation restores cytochrome c oxidase assembly defect in a mitochondrial disease model of COA6 deficiency. Hum. Mol. Genet. 2014, 23, 3596–3606. [Google Scholar] [CrossRef]
- Soma, S.; Latimer, A.J.; Chun, H.; Vicary, A.C.; Timbalia, S.A.; Boulet, A.; Rahn, J.J.; Chan, S.S.L.; Leary, S.C.; Kim, B.E.; et al. Elesclomol restores mitochondrial function in genetic models of copper deficiency. Proc. Natl. Acad. Sci. USA 2018, 115, 8161–8166. [Google Scholar] [CrossRef] [Green Version]
- Pacheu-Grau, D.; Bareth, B.; Dudek, J.; Juris, L.; Vogtle, F.N.; Wissel, M.; Leary, S.C.; Dennerlein, S.; Rehling, P.; Deckers, M. Cooperation between COA6 and SCO2 in COX2 maturation during cytochrome c oxidase assembly links two mitochondrial cardiomyopathies. Cell Metab. 2015, 21, 823–833. [Google Scholar] [CrossRef] [Green Version]
- Stroud, D.A.; Maher, M.J.; Lindau, C.; Vogtle, F.N.; Frazier, A.E.; Surgenor, E.; Mountford, H.; Singh, A.P.; Bonas, M.; Oeljeklaus, S.; et al. COA6 is a mitochondrial complex IV assembly factor critical for biogenesis of mtDNA-encoded COX2. Hum. Mol. Genet. 2015, 24, 5404–5415. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.; Pratt, A.T.; Soma, S.; Theriault, S.G.; Griffin, A.T.; Trivedi, P.P.; Gohil, V.M. Mitochondrial disease genes COA6, COX6B and SCO2 have overlapping roles in COX2 biogenesis. Hum. Mol. Genet. 2016, 25, 660–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soma, S.; Morgada, M.N.; Naik, M.T.; Boulet, A.; Roesler, A.A.; Dziuba, N.; Ghosh, A.; Yu, Q.; Lindahl, P.A.; Ames, J.B.; et al. COA6 is Structurally tuned to function as a thiol-disulfide oxidoreductase in copper delivery to mitochondrial cytochrome c oxidase. Cell Rep. 2019, 29, 4114–4126.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maghool, S.; Cooray, N.D.G.; Stroud, D.A.; Aragao, D.; Ryan, M.T.; Maher, M.J. Structural and functional characterization of the mitochondrial complex IV assembly factor Coa6. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef] [PubMed]
- Pacheu-Grau, D.; Wasilewski, M.; Oeljeklaus, S.; Gibhardt, C.S.; Aich, A.; Chudenkova, M.; Dennerlein, S.; Deckers, M.; Bogeski, I.; Warscheid, B.; et al. COA6 facilitates cytochrome c oxidase biogenesis as thiol-reductase for copper metallochaperones in mitochondria. J. Mol. Biol. 2020, 432, 2067–2079. [Google Scholar] [CrossRef] [PubMed]
- Calvo, S.E.; Compton, A.G.; Hershman, S.G.; Lim, S.C.; Lieber, D.S.; Tucker, E.J.; Laskowski, A.; Garone, C.; Liu, S.; Jaffe, D.B.; et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci. Transl. Med. 2012, 4, 118ra110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baertling, F.; M, A.M.v.d.B.; Hertecant, J.L.; Al-Shamsi, A.; L, P.v.d.H.; Distelmaier, F.; Mayatepek, E.; Smeitink, J.A.; Nijtmans, L.G.; Rodenburg, R.J. Mutations in COA6 cause cytochrome c oxidase deficiency and neonatal hypertrophic cardiomyopathy. Hum. Mutat. 2015, 36, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Vempati, U.D.; Han, X.; Moraes, C.T. Lack of cytochrome c in mouse fibroblasts disrupts assembly/stability of respiratory complexes I and IV. J. Biol. Chem. 2009, 284, 4383–4391. [Google Scholar] [CrossRef] [Green Version]
- Barrientos, A.; Pierre, D.; Lee, J.; Tzagoloff, A. Cytochrome oxidase assembly does not require catalytically active cytochrome c. J. Biol. Chem. 2003, 278, 8881–8887. [Google Scholar] [CrossRef] [Green Version]
- Bode, M.; Longen, S.; Morgan, B.; Peleh, V.; Dick, T.P.; Bihlmaier, K.; Herrmann, J.M. Inaccurately assembled cytochrome c oxidase can lead to oxidative stress-induced growth arrest. Antioxid. Redox Signal. 2013, 18, 1597–1612. [Google Scholar] [CrossRef] [Green Version]
- LaMarche, A.E.; Abate, M.I.; Chan, S.H.; Trumpower, B.L. Isolation and characterization of COX12, the nuclear gene for a previously unrecognized subunit of Saccharomyces cerevisiae cytochrome c oxidase. J. Biol. Chem. 1992, 267, 22473–22480. [Google Scholar] [CrossRef]
- Carrero-Valenzuela, R.D.; Quan, F.; Lightowlers, R.; Kennaway, N.G.; Litt, M.; Forte, M. Human cytochrome c oxidase subunit VIb: Characterization and mapping of a multigene family. Gene 1991, 102, 229–236. [Google Scholar] [CrossRef]
- Taanman, J.W.; Schrage, C.; Reuvekamp, P.; Bijl, J.; Hartog, M.; de Vries, H.; Agsteribbe, E. Identification of three human pseudogenes for subunit VIb of cytochrome c oxidase: A molecular record of gene evolution. Gene 1991, 102, 237–244. [Google Scholar] [CrossRef]
- Hüttemann, M.; Jaradat, S.; Grossman, L.I. Cytochrome c oxidase of mammals contains a testes-specific isoform of subunit VIb--the counterpart to testes-specific cytochrome c? Mol. Reprod. Dev. 2003, 66, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Roberts, V.A.; Pique, M.E. Definition of the interaction domain for cytochrome c on cytochrome c oxidase. III. Prediction of the docked complex by a complete, systematic search. J. Biol. Chem. 1999, 274, 38051–38060. [Google Scholar] [CrossRef] [Green Version]
- Massa, V.; Fernandez-Vizarra, E.; Alshahwan, S.; Bakhsh, E.; Goffrini, P.; Ferrero, I.; Mereghetti, P.; D’Adamo, P.; Gasparini, P.; Zeviani, M. Severe infantile encephalomyopathy caused by a mutation in COX6B1, a nucleus-encoded subunit of cytochrome c oxidase. Am. J. Hum. Genet. 2008, 82, 1281–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdulhag, U.N.; Soiferman, D.; Schueler-Furman, O.; Miller, C.; Shaag, A.; Elpeleg, O.; Edvardson, S.; Saada, A. Mitochondrial complex IV deficiency, caused by mutated COX6B1, is associated with encephalomyopathy, hydrocephalus and cardiomyopathy. Eur. J. Hum. Genet. 2015, 23, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Huh, W.K.; Falvo, J.V.; Gerke, L.C.; Carroll, A.S.; Howson, R.W.; Weissman, J.S.; O’Shea, E.K. Global analysis of protein localization in budding yeast. Nature 2003, 425, 686–691. [Google Scholar] [CrossRef]
- Gabriel, K.; Milenkovic, D.; Chacinska, A.; Muller, J.; Guiard, B.; Pfanner, N.; Meisinger, C. Novel mitochondrial intermembrane space proteins as substrates of the MIA import pathway. J. Mol. Biol. 2007, 365, 612–620. [Google Scholar] [CrossRef]
- Tkach, J.M.; Yimit, A.; Lee, A.Y.; Riffle, M.; Costanzo, M.; Jaschob, D.; Hendry, J.A.; Ou, J.; Moffat, J.; Boone, C.; et al. Dissecting DNA damage response pathways by analysing protein localization and abundance changes during DNA replication stress. Nat. Cell Biol. 2012, 14, 966–976. [Google Scholar] [CrossRef] [Green Version]
- Baughman, J.M.; Nilsson, R.; Gohil, V.M.; Arlow, D.H.; Gauhar, Z.; Mootha, V.K. A computational screen for regulators of oxidative phosphorylation implicates SLIRP in mitochondrial RNA homeostasis. PloS Genet. 2009, 5, e1000590. [Google Scholar] [CrossRef] [Green Version]
- Aras, S.; Pak, O.; Sommer, N.; Finley, R., Jr.; Hüttemann, M.; Weissmann, N.; Grossman, L.I. Oxygen-dependent expression of cytochrome c oxidase subunit 4–2 gene expression is mediated by transcription factors RBPJ, CXXC5 and CHCHD2. Nucleic. Acids. Res. 2013, 41, 2255–2266. [Google Scholar] [CrossRef] [Green Version]
- Aras, S.; Bai, M.; Lee, I.; Springett, R.; Hüttemann, M.; Grossman, L.I. MNRR1 (formerly CHCHD2) is a bi-organellar regulator of mitochondrial metabolism. Mitochondrion 2015, 20, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Aras, S.; Arrabi, H.; Purandare, N.; Hüttemann, M.; Kamholz, J.; Züchner, S.; Grossman, L.I. Abl2 kinase phosphorylates Bi-organellar regulator MNRR1 in mitochondria, stimulating respiration. Biochim. Biophys. Acta BBA Mol. Cell Res. 2017, 1864, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhou, F.; Zhang, Z.; Xing, D. Mitochondrial oxidative stress causes mitochondrial fragmentation via differential modulation of mitochondrial fission-fusion proteins. FEBS J. 2011, 278, 941–954. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [Green Version]
- MacVicar, T.; Langer, T. OPA1 processing in cell death and disease—The long and short of it. J. Cell Sci. 2016, 129, 2297–2306. [Google Scholar] [CrossRef] [Green Version]
- Floyd, B.J.; Wilkerson, E.M.; Veling, M.T.; Minogue, C.E.; Xia, C.; Beebe, E.T.; Wrobel, R.L.; Cho, H.; Kremer, L.S.; Alston, C.L.; et al. Mitochondrial protein interaction mapping identifies regulators of respiratory chain function. Mol. Cell 2016, 63, 621–632. [Google Scholar] [CrossRef] [Green Version]
- Aras, S.; Purandare, N.; Gladyck, S.; Somayajulu-Nitu, M.; Zhang, K.; Wallace, D.C.; Grossman, L.I. Mitochondrial Nuclear Retrograde Regulator 1 (MNNR1) rescues the cellular phenotype of the MELAS mutation by inductin stress-responsive homeostatic mechanisms. Proc. Natl. Acad. Sci. USA. in Press.
- Aras, S.; Maroun, M.C.; Song, Y.; Bandyopadhyay, S.; Stark, A.; Yang, Z.Q.; Long, M.P.; Grossman, L.I.; Fernandez-Madrid, F. Mitochondrial autoimmunity and MNRR1 in breast carcinogenesis. BMC Cancer 2019, 19, 411. [Google Scholar] [CrossRef]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Vellanki, R.N.; Coyaud, E.; Ignatchenko, V.; Li, L.; Krieger, J.R.; Taylor, P.; Tong, J.; Pham, N.A.; Liu, G.; et al. CHCHD2 Is coamplified with EGFR in NSCLC and regulates mitochondrial function and cell migration. Mol. Cancer Res. 2015, 13, 1119–1129. [Google Scholar] [CrossRef] [Green Version]
- Song, R.; Yang, B.; Gao, X.; Zhang, J.; Sun, L.; Wang, P.; Meng, Y.; Wang, Q.; Liu, S.; Cheng, J. Cyclic adenosine monophosphate response element-binding protein transcriptionally regulates CHCHD2 associated with the molecular pathogenesis of hepatocellular carcinoma. Mol. Med.Rep. 2015, 11, 4053–4062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funayama, M.; Ohe, K.; Amo, T.; Furuya, N.; Yamaguchi, J.; Saiki, S.; Li, Y.; Ogaki, K.; Ando, M.; Yoshino, H.; et al. CHCHD2 mutations in autosomal dominant late-onset Parkinson’s disease: A genome-wide linkage and sequencing study. Lancet Neurol. 2015, 14, 274–282. [Google Scholar] [CrossRef]
- Feyeux, M.; Bourgois-Rocha, F.; Redfern, A.; Giles, P.; Lefort, N.; Aubert, S.; Bonnefond, C.; Bugi, A.; Ruiz, M.; Deglon, N.; et al. Early transcriptional changes linked to naturally occurring Huntington’s disease mutations in neural derivatives of human embryonic stem cells. Hum. Mol. Genet. 2012, 21, 3883–3895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimojima, K.; Okumura, A.; Hayashi, M.; Kondo, T.; Inoue, H.; Yamamoto, T. CHCHD2 is down-regulated in neuronal cells differentiated from iPS cells derived from patients with lissencephaly. Genomics 2015, 106, 196–203. [Google Scholar] [CrossRef]
- Seo, M.; Lee, W.H.; Suk, K. Identification of novel cell migration-promoting genes by a functional genetic screen. FASEB J. 2010, 24, 464–478. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Wu, B.P.; Nguyen, D.; Liu, Y.T.; Marani, M.; Hench, J.; Benit, P.; Kozjak-Pavlovic, V.; Rustin, P.; Frank, S.; et al. CHCHD2 accumulates in distressed mitochondria and facilitates oligomerization of CHCHD10. Hum. Mol. Genet. 2018, 27, 3881–3900. [Google Scholar] [CrossRef]
- Hüttemann, M.; Lee, I.; Liu, J.; Grossman, L.I. Transcription of mammalian cytochrome c oxidase subunit IV-2 is controlled by a novel conserved oxygen responsive element. FEBS J. 2007, 274, 5737–5748. [Google Scholar] [CrossRef]
- Hüttemann, M.; Kadenbach, B.; Grossman, L.I. Mammalian subunit IV isoforms of cytochrome c oxidase. Gene 2001, 267, 111–123. [Google Scholar] [CrossRef]
- Moreno-Dominguez, A.; Ortega-Saenz, P.; Gao, L.; Colinas, O.; Garcia-Flores, P.; Bonilla-Henao, V.; Aragones, J.; Hüttemann, M.; Grossman, L.I.; Weissmann, N.; et al. Acute O2 sensing through HIF2alpha-dependent expression of atypical cytochrome oxidase subunits in arterial chemoreceptors. Sci. Signal. 2020, 13. [Google Scholar] [CrossRef] [Green Version]
- Hüttemann, M.; Lee, I.; Gao, X.; Pecina, P.; Pecinova, A.; Liu, J.; Aras, S.; Sommer, N.; Sanderson, T.H.; Tost, M.; et al. Cytochrome c oxidase subunit 4 isoform 2-knockout mice show reduced enzyme activity, airway hyporeactivity, and lung pathology. FASEB J. 2012, 26, 3916–3930. [Google Scholar] [CrossRef] [Green Version]
- Martherus, R.S.; Sluiter, W.; Timmer, E.D.; VanHerle, S.J.; Smeets, H.J.; Ayoubi, T.A. Functional annotation of heart enriched mitochondrial genes GBAS and CHCHD10 through guilt by association. Biochem. Biophys. Res. Commun. 2010, 402, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Ajroud-Driss, S.; Fecto, F.; Ajroud, K.; Lalani, I.; Calvo, S.E.; Mootha, V.K.; Deng, H.X.; Siddique, N.; Tahmoush, A.J.; Heiman-Patterson, T.D.; et al. Mutation in the novel nuclear-encoded mitochondrial protein CHCHD10 in a family with autosomal dominant mitochondrial myopathy. Neurogenetics 2015, 16, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Purandare, N.; Somayajulu, M.; Hüttemann, M.; Grossman, L.I.; Aras, S. The cellular stress proteins CHCHD10 and MNRR1 (CHCHD2): Partners in mitochondrial and nuclear function and dysfunction. J. Biol. Chem. 2018, 293, 6517–6529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannwarth, S.; Ait-El-Mkadem, S.; Chaussenot, A.; Genin, E.C.; Lacas-Gervais, S.; Fragaki, K.; Berg-Alonso, L.; Kageyama, Y.; Serre, V.; Moore, D.G.; et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 2014, 137, 2329–2345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penttila, S.; Jokela, M.; Bouquin, H.; Saukkonen, A.M.; Toivanen, J.; Udd, B. Late onset spinal motor neuronopathy is caused by mutation in CHCHD10. Ann. Neurol. 2015, 77, 163–172. [Google Scholar] [CrossRef]
- Chaussenot, A.; Le Ber, I.; Ait-El-Mkadem, S.; Camuzat, A.; de Septenville, A.; Bannwarth, S.; Genin, E.C.; Serre, V.; Auge, G.; French research network on, F.T.D.; et al. Screening of CHCHD10 in a French cohort confirms the involvement of this gene in frontotemporal dementia with amyotrophic lateral sclerosis patients. Neurobiol. Aging 2014, 35, 2884.e1–2884.e4. [Google Scholar] [CrossRef]
- Muller, K.; Andersen, P.M.; Hubers, A.; Marroquin, N.; Volk, A.E.; Danzer, K.M.; Meitinger, T.; Ludolph, A.C.; Strom, T.M.; Weishaupt, J.H. Two novel mutations in conserved codons indicate that CHCHD10 is a gene associated with motor neuron disease. Brain 2014, 137, e309. [Google Scholar] [CrossRef] [Green Version]
- Auranen, M.; Ylikallio, E.; Shcherbii, M.; Paetau, A.; Kiuru-Enari, S.; Toppila, J.P.; Tyynismaa, H. CHCHD10 variant p.(Gly66Val) causes axonal Charcot-Marie-Tooth disease. Neurol. Genet. 2015, 1, e1. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Salokas, K.; Tamene, F.; Jiu, Y.; Weldatsadik, R.G.; Ohman, T.; Varjosalo, M. An AP-MS- and BioID-compatible MAC-tag enables comprehensive mapping of protein interactions and subcellular localizations. Nat. Commun. 2018, 9, 1188. [Google Scholar] [CrossRef] [Green Version]
- Madani, A.; Choukroun, V.; Soulier, J.; Cacheux, V.; Claisse, J.F.; Valensi, F.; Daliphard, S.; Cazin, B.; Levy, V.; Leblond, V.; et al. Expression of p13MTCP1 is restricted to mature T-cell proliferations with t(X;14) translocations. Blood 1996, 87, 1923–1927. [Google Scholar] [CrossRef] [Green Version]
- Madani, A.; Soulier, J.; Schmid, M.; Plichtova, R.; Lerme, F.; Gateau-Roesch, O.; Garnier, J.P.; Pla, M.; Sigaux, F.; Stern, M.H. The 8 kD product of the putative oncogene MTCP-1 is a mitochondrial protein. Oncogene 1995, 10, 2259–2262. [Google Scholar] [PubMed]
- Soulier, J.; Madani, A.; Cacheux, V.; Rosenzwajg, M.; Sigaux, F.; Stern, M.H. The MTCP-1/c6.1B gene encodes for a cytoplasmic 8 kD protein overexpressed in T cell leukemia bearing a t(X;14) translocation. Oncogene 1994, 9, 3565–3570. [Google Scholar] [PubMed]
- Stern, M.H.; Soulier, J.; Rosenzwajg, M.; Nakahara, K.; Canki-Klain, N.; Aurias, A.; Sigaux, F.; Kirsch, I.R. MTCP-1: A novel gene on the human chromosome Xq28 translocated to the T cell receptor alpha/delta locus in mature T cell proliferations. Oncogene 1993, 8, 2475–2483. [Google Scholar] [PubMed]
- Fisch, P.; Forster, A.; Sherrington, P.D.; Dyer, M.J.; Rabbitts, T.H. The chromosomal translocation t(X;14)(q28;q11) in T-cell pro-lymphocytic leukaemia breaks within one gene and activates another. Oncogene 1993, 8, 3271–3276. [Google Scholar] [PubMed]
- Schoenfeld, R.A.; Napoli, E.; Wong, A.; Zhan, S.; Reutenauer, L.; Morin, D.; Buckpitt, A.R.; Taroni, F.; Lonnerdal, B.; Ristow, M.; et al. Frataxin deficiency alters heme pathway transcripts and decreases mitochondrial heme metabolites in mammalian cells. Hum. Mol. Genet. 2005, 14, 3787–3799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Vizarra, E.; Tiranti, V.; Zeviani, M. Assembly of the oxidative phosphorylation system in humans: What we have learned by studying its defects. Biochim. Biophys. Acta. 2009, 1793, 200–211. [Google Scholar] [CrossRef] [Green Version]
- Pecina, P.; Houstkova, H.; Hansikova, H.; Zeman, J.; Houstek, J. Genetic defects of cytochrome c oxidase assembly. Physiol. Res. 2004, 53 (Suppl 1), S213–S223. [Google Scholar]
- Barrientos, A.; Gouget, K.; Horn, D.; Soto, I.C.; Fontanesi, F. Suppression mechanisms of COX assembly defects in yeast and human: Insights into the COX assembly process. Biochim. Biophys. Acta. 2009, 1793, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Soto, I.C.; Fontanesi, F.; Liu, J.; Barrientos, A. Biogenesis and assembly of eukaryotic cytochrome c oxidase catalytic core. Biochim. Biophys. Acta. 2012, 1817, 883–897. [Google Scholar] [CrossRef] [Green Version]
- Valnot, I.; Osmond, S.; Gigarel, N.; Mehaye, B.; Amiel, J.; Cormier-Daire, V.; Munnich, A.; Bonnefont, J.P.; Rustin, P.; Rotig, A. Mutations of the SCO1 gene in mitochondrial cytochrome c oxidase deficiency with neonatal-onset hepatic failure and encephalopathy. Am. J. Hum. Genet. 2000, 67, 1104–1109. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulou, L.C.; Sue, C.M.; Davidson, M.M.; Tanji, K.; Nishino, I.; Sadlock, J.E.; Krishna, S.; Walker, W.; Selby, J.; Glerum, D.M.; et al. Fatal infantile cardioencephalomyopathy with COX deficiency and mutations in SCO2, a COX assembly gene. Nat. Genet. 1999, 23, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Sue, C.M.; Karadimas, C.; Checcarelli, N.; Tanji, K.; Papadopoulou, L.C.; Pallotti, F.; Guo, F.L.; Shanske, S.; Hirano, M.; De Vivo, D.C.; et al. Differential features of patients with mutations in two COX assembly genes, SURF-1 and SCO2. Ann. Neurol. 2000, 47, 589–595. [Google Scholar] [CrossRef]
- Salviati, L.; Sacconi, S.; Rasalan, M.M.; Kronn, D.F.; Braun, A.; Canoll, P.; Davidson, M.; Shanske, S.; Bonilla, E.; Hays, A.P.; et al. Cytochrome c oxidase deficiency due to a novel SCO2 mutation mimics Werdnig-Hoffmann disease. Arch. Neurol. 2002, 59, 862–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valnot, I.; von Kleist-Retzow, J.C.; Barrientos, A.; Gorbatyuk, M.; Taanman, J.W.; Mehaye, B.; Rustin, P.; Tzagoloff, A.; Munnich, A.; Rotig, A. A mutation in the human heme A:farnesyltransferase gene (COX10 ) causes cytochrome c oxidase deficiency. Hum. Mol. Genet. 2000, 9, 1245–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonicka, H.; Leary, S.C.; Guercin, G.H.; Agar, J.N.; Horvath, R.; Kennaway, N.G.; Harding, C.O.; Jaksch, M.; Shoubridge, E.A. Mutations in COX10 result in a defect in mitochondrial heme A biosynthesis and account for multiple, early-onset clinical phenotypes associated with isolated COX deficiency. Hum. Mol. Genet. 2003, 12, 2693–2702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonicka, H.; Mattman, A.; Carlson, C.G.; Glerum, D.M.; Hoffbuhr, K.C.; Leary, S.C.; Kennaway, N.G.; Shoubridge, E.A. Mutations in COX15 produce a defect in the mitochondrial heme biosynthetic pathway, causing early-onset fatal hypertrophic cardiomyopathy. Am. J. Hum. Genet. 2003, 72, 101–114. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Yao, J.; Johns, T.; Fu, K.; De Bie, I.; Macmillan, C.; Cuthbert, A.P.; Newbold, R.F.; Wang, J.; Chevrette, M.; et al. SURF1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat. Genet. 1998, 20, 337–343. [Google Scholar] [CrossRef]
- Tiranti, V.; Hoertnagel, K.; Carrozzo, R.; Galimberti, C.; Munaro, M.; Granatiero, M.; Zelante, L.; Gasparini, P.; Marzella, R.; Rocchi, M.; et al. Mutations of SURF-1 in Leigh disease associated with cytochrome c oxidase deficiency. Am. J. Hum. Genet. 1998, 63, 1609–1621. [Google Scholar] [CrossRef] [Green Version]
- Naoe, M.; Ohwa, Y.; Ishikawa, D.; Ohshima, C.; Nishikawa, S.; Yamamoto, H.; Endo, T. Identification of Tim40 that mediates protein sorting to the mitochondrial intermembrane space. J. Biol. Chem. 2004, 279, 47815–47821. [Google Scholar] [CrossRef] [Green Version]
- Mesecke, N.; Terziyska, N.; Kozany, C.; Baumann, F.; Neupert, W.; Hell, K.; Herrmann, J.M. A disulfide relay system in the intermembrane space of mitochondria that mediates protein import. Cell 2005, 121, 1059–1069. [Google Scholar] [CrossRef] [Green Version]
- Allen, S.; Balabanidou, V.; Sideris, D.P.; Lisowsky, T.; Tokatlidis, K. Erv1 mediates the Mia40-dependent protein import pathway and provides a functional link to the respiratory chain by shuttling electrons to cytochrome c. J. Mol. Biol. 2005, 353, 937–944. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic. Acids. Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bak, D.W.; Weerapana, E. Cysteine-mediated redox signalling in the mitochondria. Mol. Biosyst. 2015, 11, 678–697. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Jedrychowski, M.P.; Schweppe, D.K.; Huttlin, E.L.; Yu, Q.; Heppner, D.E.; Li, J.; Long, J.; Mills, E.L.; Szpyt, J.; et al. A quantitative tissue-specific landscape of protein redox regulation during aging. Cell 2020, 180, 968–983. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Assembly of cytochrome c oxidase. The assembly of COX is thought to be both modular and linear. Dark blue, mitochondrial encoded COX core subunits; light blue, nuclear encoded COX subunit; purple, assembly factors; gray, unknown function. *, Twin CX9C protein. Gray dashed lines represent assembly factor interactions. Black dashed lines represent subunit assembly.
Figure 1.
Assembly of cytochrome c oxidase. The assembly of COX is thought to be both modular and linear. Dark blue, mitochondrial encoded COX core subunits; light blue, nuclear encoded COX subunit; purple, assembly factors; gray, unknown function. *, Twin CX9C protein. Gray dashed lines represent assembly factor interactions. Black dashed lines represent subunit assembly.

Figure 2.
COX17. Alignment of human and yeast COX17 protein sequences. Human sequence annotation is as follows: Black boxes indicate conserved residues. Blue box indicates the CHCH domain. Purple boxes indicate a helix. Solid black lines indicate the twin CX9C motif. Dashed lines indicate the cysteine pairs that form structural disulfide bonds. Arrows indicate copper binding cysteines.
Figure 2.
COX17. Alignment of human and yeast COX17 protein sequences. Human sequence annotation is as follows: Black boxes indicate conserved residues. Blue box indicates the CHCH domain. Purple boxes indicate a helix. Solid black lines indicate the twin CX9C motif. Dashed lines indicate the cysteine pairs that form structural disulfide bonds. Arrows indicate copper binding cysteines.

Figure 3.
COX19. Alignment of human and yeast COX19 protein sequences. Black boxes indicate conserved residues. Blue box indicates the CHCH domain. Purple boxes indicate a helix. Solid black lines indicate the twin CX9C motif. Dashed lines indicate the cysteine pairs that form structural disulfide bonds. Arrows indicate copper binding cysteines.
Figure 3.
COX19. Alignment of human and yeast COX19 protein sequences. Black boxes indicate conserved residues. Blue box indicates the CHCH domain. Purple boxes indicate a helix. Solid black lines indicate the twin CX9C motif. Dashed lines indicate the cysteine pairs that form structural disulfide bonds. Arrows indicate copper binding cysteines.

Figure 4.
CHCHD7/Cox23p. Alignment of human and yeast CHCHD7/COX23 protein sequences. Black boxes indicate conserved residues. Human sequence annotation is as follows: Blue box indicates the CHCH domain. Purple boxes indicate a helix. Green boxes indicate a turn. Solid black lines indicate the twin CX9C motif. Dashed lines indicate the cysteine pairs that form structural disulfide bonds.
Figure 4.
CHCHD7/Cox23p. Alignment of human and yeast CHCHD7/COX23 protein sequences. Black boxes indicate conserved residues. Human sequence annotation is as follows: Blue box indicates the CHCH domain. Purple boxes indicate a helix. Green boxes indicate a turn. Solid black lines indicate the twin CX9C motif. Dashed lines indicate the cysteine pairs that form structural disulfide bonds.

Figure 5.
CMC1. Alignment of human and yeast CMC1 protein sequences. Black boxes indicate conserved residues. Human sequence annotation is as follows: Blue box indicates the CHCH domain. Solid black lines indicate the twin CX9C motif. Dashed lines indicate the cysteine pairs that form structural disulfide bonds.
Figure 5.
CMC1. Alignment of human and yeast CMC1 protein sequences. Black boxes indicate conserved residues. Human sequence annotation is as follows: Blue box indicates the CHCH domain. Solid black lines indicate the twin CX9C motif. Dashed lines indicate the cysteine pairs that form structural disulfide bonds.

Figure 6.
CMC2. Alignment of human and yeast CMC2 protein sequences. Black boxes indicate conserved residues. Human sequence annotation is as follows: Blue box indicates the CHCH domain. Solid black lines indicate the twin CX9C motif. Dashed lines indicate the cysteine pairs that form structural disulfide bonds.
Figure 6.
CMC2. Alignment of human and yeast CMC2 protein sequences. Black boxes indicate conserved residues. Human sequence annotation is as follows: Blue box indicates the CHCH domain. Solid black lines indicate the twin CX9C motif. Dashed lines indicate the cysteine pairs that form structural disulfide bonds.

Figure 7.
COA5/Pet191p. Alignment of human COA5 and yeast Pet191p protein sequences. Black boxes indicate conserved residues. Human sequence annotation is as follows: Blue box indicates the CHCH domain. Solid black lines indicate the CX9C-CX10C motif. Dashed lines indicate the cysteine pairs that form structural disulfide bonds.
Figure 7.
COA5/Pet191p. Alignment of human COA5 and yeast Pet191p protein sequences. Black boxes indicate conserved residues. Human sequence annotation is as follows: Blue box indicates the CHCH domain. Solid black lines indicate the CX9C-CX10C motif. Dashed lines indicate the cysteine pairs that form structural disulfide bonds.

Figure 8.
COA6. Alignment of human and yeast COA6 protein sequences. Purple boxes indicate a helix. Black boxes indicate conserved residues. Human sequence annotation is as follows: Blue box indicates the CHCH domain. Green boxes indicate a turn. Solid black lines indicate the CX9C-CX10C motif. Dashed lines indicate the cysteine pairs that form structural disulfide bonds.
Figure 8.
COA6. Alignment of human and yeast COA6 protein sequences. Purple boxes indicate a helix. Black boxes indicate conserved residues. Human sequence annotation is as follows: Blue box indicates the CHCH domain. Green boxes indicate a turn. Solid black lines indicate the CX9C-CX10C motif. Dashed lines indicate the cysteine pairs that form structural disulfide bonds.

Figure 9.
COA4/CHCHD8. Alignment of human and yeast COA4 protein sequences. Black boxes indicate conserved residues. Human sequence annotation is as follows: Blue box indicates the CHCH domain. Solid black lines indicate the twin CX9C motif. Dashed lines indicate the cysteine pairs that form structural disulfide bonds.
Figure 9.
COA4/CHCHD8. Alignment of human and yeast COA4 protein sequences. Black boxes indicate conserved residues. Human sequence annotation is as follows: Blue box indicates the CHCH domain. Solid black lines indicate the twin CX9C motif. Dashed lines indicate the cysteine pairs that form structural disulfide bonds.

Figure 10.
COX VIb1/Cox12p. Alignment of human and yeast COX VIb1/Cox12p protein sequences. Black boxes indicate conserved residues. Human sequence annotation is as follows: Blue box indicates the CHCH domain. Solid black lines indicate the CX9C-CX10C motif. Dashed lines indicate the cysteine pairs that form structural disulfide bonds.
Figure 10.
COX VIb1/Cox12p. Alignment of human and yeast COX VIb1/Cox12p protein sequences. Black boxes indicate conserved residues. Human sequence annotation is as follows: Blue box indicates the CHCH domain. Solid black lines indicate the CX9C-CX10C motif. Dashed lines indicate the cysteine pairs that form structural disulfide bonds.

Figure 11.
MNRR1, CHCHD10, and Mix17. Alignment of human MNRR1 and CHCHD10 (D10) and yeast Mix17 protein sequences. Black boxes indicate conserved residues. Human sequence annotation is as follows: Blue box indicates the CHCH domain. Solid black lines indicate the twin CX9C motif. Dashed lines indicate the cysteine pairs that form structural disulfide bonds. The arrow indicates a residue of interest discussed in this review.
Figure 11.
MNRR1, CHCHD10, and Mix17. Alignment of human MNRR1 and CHCHD10 (D10) and yeast Mix17 protein sequences. Black boxes indicate conserved residues. Human sequence annotation is as follows: Blue box indicates the CHCH domain. Solid black lines indicate the twin CX9C motif. Dashed lines indicate the cysteine pairs that form structural disulfide bonds. The arrow indicates a residue of interest discussed in this review.

Figure 12.
Sequence similarity of twin CX9C proteins. Human and yeast protein sequences were aligned using MUSCLE, phylogenetic tree created with PhyML and TreeDeign (“One Click” MAB LIRMM). Homologs/orthologs between species are colored the same. Human proteins are underlined.
Figure 12.
Sequence similarity of twin CX9C proteins. Human and yeast protein sequences were aligned using MUSCLE, phylogenetic tree created with PhyML and TreeDeign (“One Click” MAB LIRMM). Homologs/orthologs between species are colored the same. Human proteins are underlined.

Figure 13.
CMC4. Alignment of human and yeast CMC4 protein sequences. Black boxes indicate conserved residues. Human sequence annotation is as follows: Purple boxes indicate a helix. Green boxes indicate a turn. Blue box indicates the CHCH domain. Solid black lines indicate the twin CX9C motif. Dashed lines indicate the cysteine pairs that form structural disulfide bonds.
Figure 13.
CMC4. Alignment of human and yeast CMC4 protein sequences. Black boxes indicate conserved residues. Human sequence annotation is as follows: Purple boxes indicate a helix. Green boxes indicate a turn. Blue box indicates the CHCH domain. Solid black lines indicate the twin CX9C motif. Dashed lines indicate the cysteine pairs that form structural disulfide bonds.


{kind=link}
{kind=link}
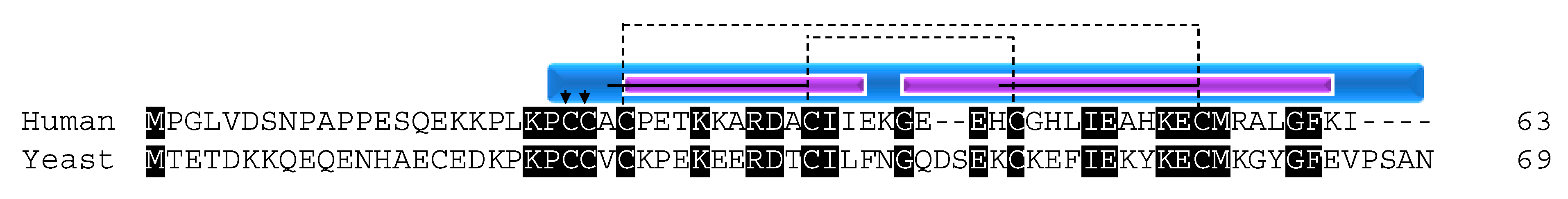{kind=link}
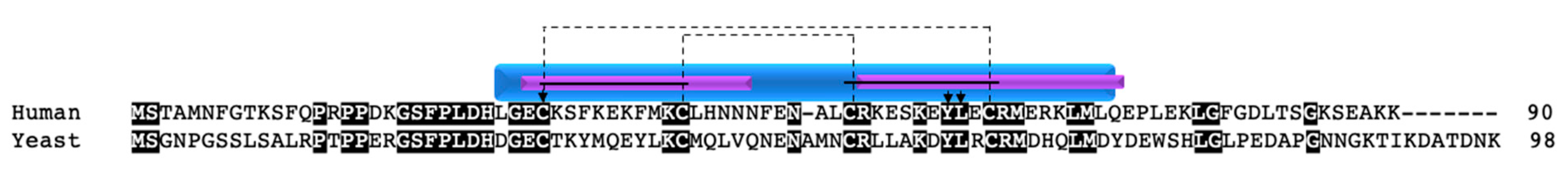{kind=link}
{kind=link}
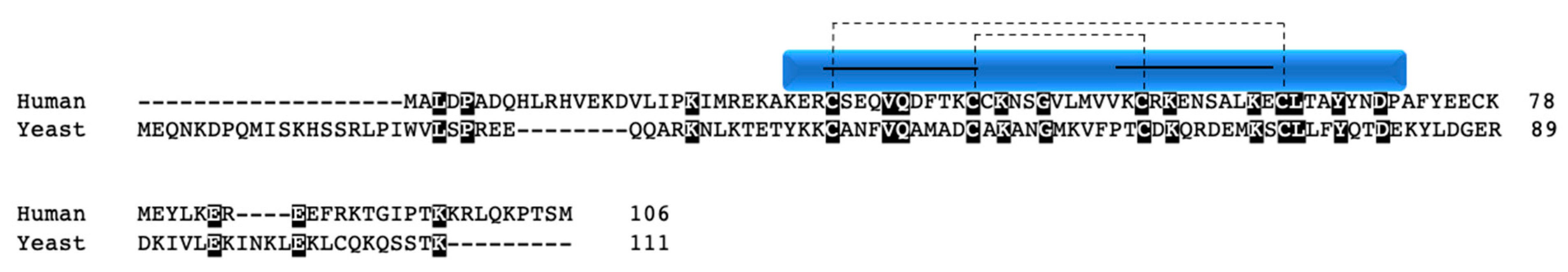{kind=link}
{kind=link}
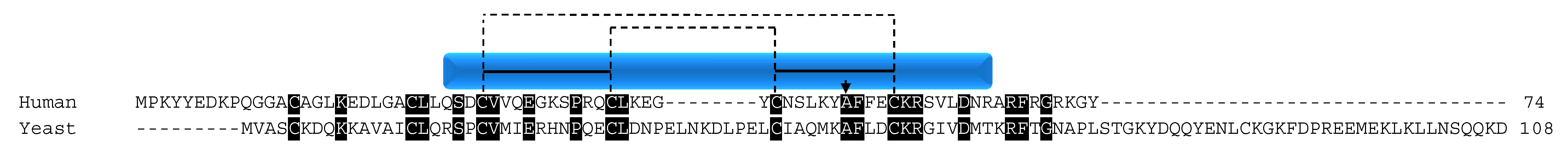{kind=link}
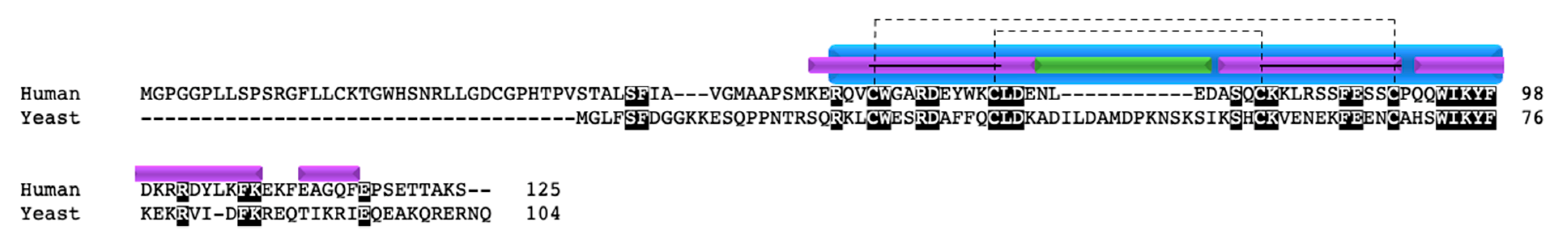{kind=link}
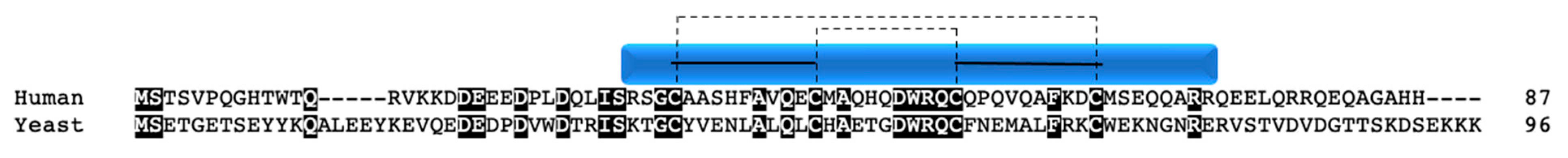{kind=link}
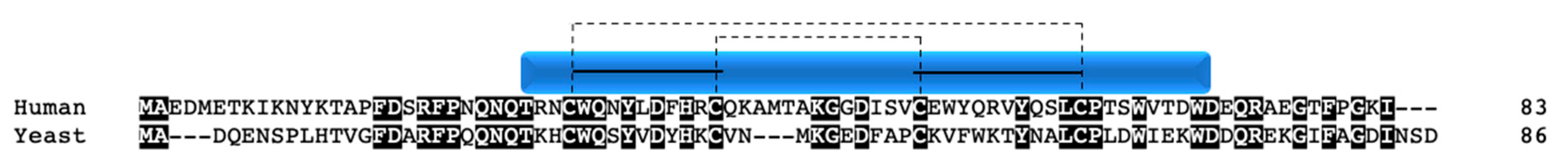{kind=link}
{kind=link}
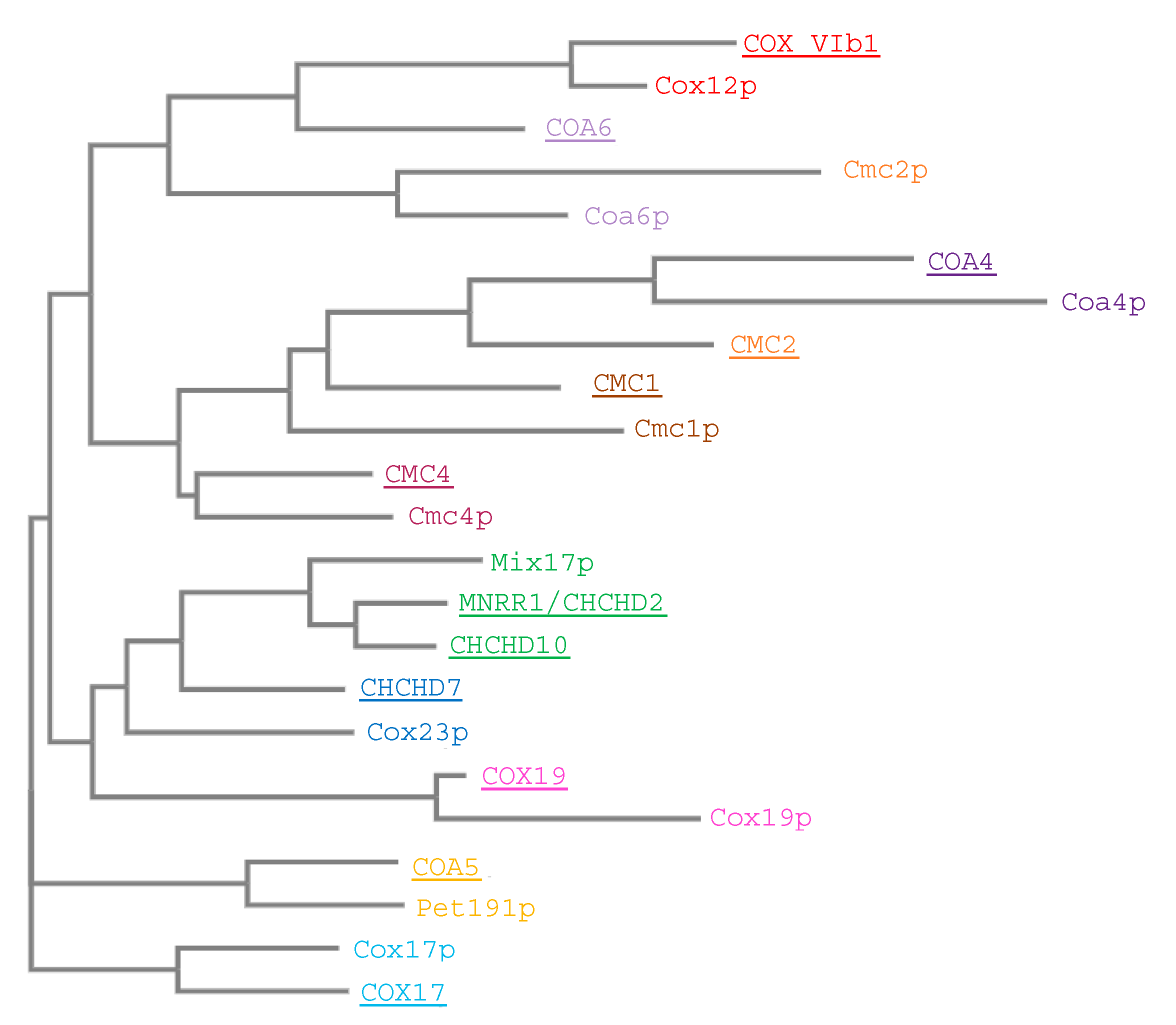{kind=link}
{kind=link}
Table 1.
COX regulation.
| Types of Regulation of COX |
|---|
| Expression of tissue-, developmental-, and/or species-specific isoforms of subunits |
| Interaction with small molecules |
| Reversible phosphorylation of subunits |
| Protein–protein interactions |
| Supercomplex formation |
Table 2.
Human and yeast proteins, nomenclature, and functions of twin CX9C proteins with COX [9].
Table 2.
Human and yeast proteins, nomenclature, and functions of twin CX9C proteins with COX [9].
| Human Protein | Yeast Protein | Function |
|---|---|---|
| CX9C Proteins | ||
| COX17 | Cox17p | COX copper chaperone |
| COX19 | Cox19p | COX assembly |
| CMC1 | Cmc1p | COX assembly |
| CMC2 | Cmc2p | COX assembly |
| COA5 | Pet191p | COX assembly |
| COA6 | Coa6p | COX assembly |
| CHCHD7 | Cox23p | COX assembly |
| CHCHD8 | Coa4p | COX/complex III assembly/function |
| MNRR1/CHCHD2 | Mix17p | Activity regulation |
| CHCHD10 | Mix17p | Activity regulation |
| CMC4 | Cmc4p | Unknown |
| COX VIb1 | Cox12p | Subunit |
| Cytochrome c Oxidase | ||
| COX I | Cox1p | Subunit |
| COX II | Cox2p | |
| COX III | Cox3p | |
| COX IV | Cox4p | |
| COX Va | Cox5Ap | |
| COX Vb | Cox5Bp | |
| COX VI | Cox6p | |
| COX VII | Cox7p | |
| COX VIII | COX8p | |
| COX IX | Cox9p | |
| COX XIII | Cox13p | |
Table 3.
COX-associated twin CX9C proteins and diseases.
| Protein | Disease Association | Reference |
|---|---|---|
| COX17 | COX17-null embryonic lethal in mice | [43] |
| COX19 | None | |
| CHCHD7 | Salivary gland pleiomorphic adenomas (benign) | [65] |
| Lung adenocarcinoma | [66] | |
| Acute myeloid leukemia | [66] | |
| CMC1 | None | |
| CMC2 | None | |
| COA5 | Hypertrophic cardiomyopathy | [67] |
| COA6 | Hypertrophic cardiomyopathy | [68,69] |
| CHCHD8 | None | |
| MNRR1/CHCHD2 | Parkinson’s disease | [70] |
| Huntington’s disease | [71] | |
| Lissencephaly | [72] | |
| Breast cancer | [73] | |
| EGFR-positive non-small cell lung cancer | [74] | |
| Hepatitis B or C virus associated hepatocellular carcinoma | [75] | |
| CHCHD10 | Spinal muscular atrophy, Jokela type | [76] |
| ALS and/or front temporal dementia | [77,78] | |
| Autosomal dominant mitochondrial myopathy | [79] | |
| Lower motor neuron syndrome | [80] | |
| Charcot-Marie-Tooth disease type 2 | [81] | |
| CMC4 | T cell leukemia with translocation (X:14) | [82,83,84,85,86] |
| Frataxin mRNA deficiency | [87] | |
| COXVIb1 | COX deficiency/mitochondrial encephalomyopathy | [88,89] |
Table 4.
COX subunit and assembly genes containing oxygen responsive element (ORE) within 500 bp of transcription start site. TCCCA is the minimal ORE element required for R1 mediated transcription. Bold and italicized bases match the classical ORE motif found in the promoter of COX4I2. Shaded sequences represent the non-coding strand.
Table 4.
COX subunit and assembly genes containing oxygen responsive element (ORE) within 500 bp of transcription start site. TCCCA is the minimal ORE element required for R1 mediated transcription. Bold and italicized bases match the classical ORE motif found in the promoter of COX4I2. Shaded sequences represent the non-coding strand.
| ID/Sequence Window Parameters | Gene | ORE |
|---|---|---|
| Published ORE | COXIVI2 | GGACGT TCCCA CGCTGGGGCGG |
| NC_000015.10:74937913-74938533 | COXVa | TCGGCC TCCCA AAAGTGCTGGG |
| CTGTAA TCCCA GCACTTTTGGG | ||
| NC_000002.12:97645525-97646190 | COXVb | TCCGCC TCCCA CACACAGTACA |
| AGACTT TCCCA CCTCCCAGCGT | ||
| CCCACC TCCCA GCGTTATTAAA | ||
| GCCCAC TCCCA AGACTGTGGCG | ||
| GCTTCC TCCCA GCCCTCGAGCC | ||
| NC_000012.12:120437600-120438231 | COXVIa | TTCTGA TCCCA GACCGCCCTAT |
| CCGCCC TCCCA CTGGCATTAAC | ||
| NC_000029.10:3560717-35651348 | COXVIb1 | GATGAA TCCCA GAAACCTCTCG |
| NC_000008.11:99891909-99892550 | COXVIc | CCGGAA TCCCA GCACTTTGGGA |
| TCGTCC TCCCA AAGTGCTGGGA | ||
| NC_000019.10:36152390-36153000 | COXVIIa | TTTTAC TCCCA AAGACTTTAGC |
| GCTTGG TCCCA CCGGAGCCCAA | ||
| NC_000023.11:77899014-77899593 | COXVIIb | TACATT TCCCA AAAGGCCTTTA |
| NC_000005.10:86617391-86618131 | COXVIIc | TTCGAT TCCCA TTATCTTTAGT |
| TACATT TCCCA CAATCCTTCGG | ||
| TACAAT TCCCA CAATCCAGGGC | ||
| CCCATT TCCCA TCTTTCTTTTC | ||
| TAACCT TCCCA GGGCCCTCCTC | ||
| NC_000011.10:63974100-63974795 | COXVIIIa | CCGGCC TCCCA AAGTGCTGGGA |
| CTCTAA TCCCA GCACTTTGGGA | ||
| ACGGCC TCCCA AAAGTTCTGAG | ||
| CACACC TCCCA CAGCCCTTGGC | ||
| CCATGA TCCCA AGCTTCCCCTC | ||
| NC_000017.11:14068795-14069649 | COX10 | CTGGGC TCCCA TTTGATAGAGA |
| TCGACC TCCCA GGCTCAAGCGA | ||
| TCGAGT TCCCA GGATCAAGCGA | ||
| CTGCAA TCCCA GTGCTTAGGGA | ||
| TGCAGC TCCCA GAAGTTGACAG | ||
| NC_000017.11:54968279-54969164 | COX11 | TGTTAC TCCCA CTTCGCTTCTC |
| NC_000012.12:50119498-50120218 | COX14 | TCAGCC TCCCA AGTAGCTGACG |
| CCACCC TCCCA CCTCAGCCTCC | ||
| NC_000010.11:99731958-99732640 | COX15 | None |
| NC_000003.12:119677203-119677899 | COX17 | None |
| NC_000004.12:73069629-73030150 | COX18 | CGCCTT TCCCA CGCGGCACTGG |
| TGACCT TCCCA GTCAAGCCGGT | ||
| NC_000001.11:244834975-244835753 | COX20 | CGGGGC TCCCA CGCCCACCCCA |
| NC_000007.14:43647530-4348140 | COA1 | TCCTCA TCCCA AGGGTGGTGCA |
| TAGGAT TCCCA GTTTTGTGCCA | ||
| CCTTGC TCCCA GAGGCATTGAC | ||
| NC_000011.10:73873321-73874201 | COA4 | TCAGTC TCCCA AAGTGCTGGGA |
| CTATAA TCCCA GCACTTTGGGA | ||
| TCAGCC TCCCA TAATGCTAGGA | ||
| CTGTAA TCCCA GCATTTTGGGT | ||
| CTGTAG TCCCA GCTACTCTGGC | ||
| NC_000001.11:234372861-234372961 | COA6 | TTATTT TCCCA ACTCCCCTGCC |
| GTGACG TCCCA GAACCCATGGC | ||
| CTGTAG TCCCA GCTACCTGGGA | ||
| CCTGCC TCCCA GGTAGCTGGGA | ||
| AGATCC TCCCA CCTCAGCCTGC | ||
| NC_000003.12:28241299-28241909 | CMC1 | GCACAG TCCCA AAGTCCTGCAG |
| GCACAG TCCCA AAGTCCTGCAG | ||
| CTTCCT TCCCA GGCTCCTACTT | ||
| CACCTT TCCCA GGTCGGCATCC | ||
| NC_000016.10:80997314-80997940 | CMC2 | None |
| NC_000023.11:155063965-155064531 | CMC4 | TATTAT TCCCA AGACTTTTTTT |
| TATGAT TCCCA TTTTATATATG | ||
| TACTCT TCCCA AACATAGGATG | ||
| NC_000009.12:133356399-133357002 | SURF1 | CCCTCA TCCCA ACTGCGCCCTT |
| CACCCG TCCCA GCCCCGCCGCC | ||
| NC_000003.12:42804144-42804699 | HIGD1A | CAGCCG TCCCA GCCAATCAGAG |
| ACGGAC TCCCA GCCCCCACCCC | ||
| NC_000007.14:10940000-10940611 | NDUFA4 | CTCGGC TCCCA GAGGCGCCGCG |
| CTGCCC TCCCA GCCAAGGGTCC | ||
| NC_000007.14:56106405-56106909 | CHCHD2/MNRR1 | CCCGCC TCCCA TCTTCCGGTCT |
| TGGTTG TCCCA CGTCCGGAGGC | ||
| NC_000008.11:56212000-56212924 | CHCHD7 | CTGATT TCCCA TCCAGTGGTTC |
| NC_000022.11:23767863-237687376 | CHCHD10 | TCCCCT TCCCA GCTGGCCCCAT |
| GCAGGG TCCCA TTTCCAGCCGA | ||
| CCCGGG TCCCA CCGCCGCCACC | ||
| NC_000020.11:18137355-18138051 | PET117 | CGGGGT TCCCA TCCGTGGAGTT |
| CCCCTC TCCCA CAGACGTCCCT | ||
| NC_000017.11:10697489-10697998 | SCO1 | None |
| NC_000022:11:50524401-50524931 | SCO2 | CCACAC TCCCA GCAGAAAACCT |
| TGGTGA TCCCA GGTAGAGGACA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Gladyck, S.; Aras, S.; Hüttemann, M.; Grossman, L.I. Regulation of COX Assembly and Function by Twin CX9C Proteins—Implications for Human Disease. Cells 2021, 10, 197. https://doi.org/10.3390/cells10020197
AMA Style
Gladyck S, Aras S, Hüttemann M, Grossman LI. Regulation of COX Assembly and Function by Twin CX9C Proteins—Implications for Human Disease. Cells. 2021; 10(2):197. https://doi.org/10.3390/cells10020197
Chicago/Turabian StyleGladyck, Stephanie, Siddhesh Aras, Maik Hüttemann, and Lawrence I. Grossman. 2021. "Regulation of COX Assembly and Function by Twin CX9C Proteins—Implications for Human Disease" Cells 10, no. 2: 197. https://doi.org/10.3390/cells10020197
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.