
Beyond the Warburg Effect: Oxidative and Glycolytic Phenotypes Coexist within the Metabolic Heterogeneity of Glioblastoma

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Culture of GSCs from Human GBM Samples, U87MG and Mesenchymal Stem Cells
2.2. Reagents and Metabolic Inhibitors
2.3. MTS Assays and Drug Combination Studies using the Chou-Talalay Method
2.4. Real-Time Quantitative Reverse Transcription PCR (RT-qRT-PCR) Analysis
2.5. Antibodies
2.6. Protein Isolation/Quantification and Western Blotting
2.7. Seahorse XFp Protocol for Real-Time Metabolic Evaluation of U87MG Adherent Cells and GSCs Neurospheres
2.8. TCGA Gene-Set Variation Analysis
2.9. Statistical Analysis
3. Results
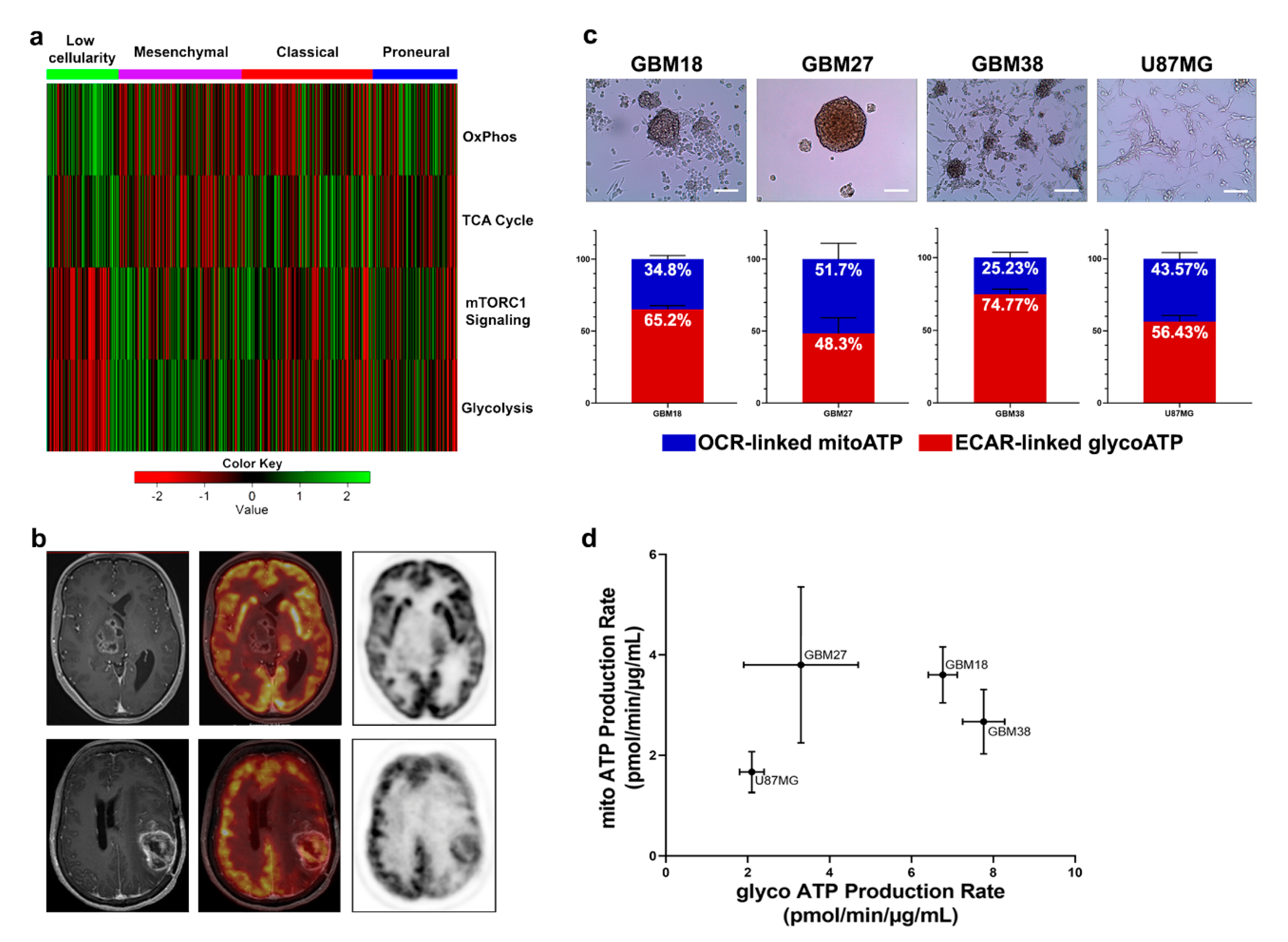
3.1. GBM can be Stratified into Glycolytic and Oxidative Phenotypes
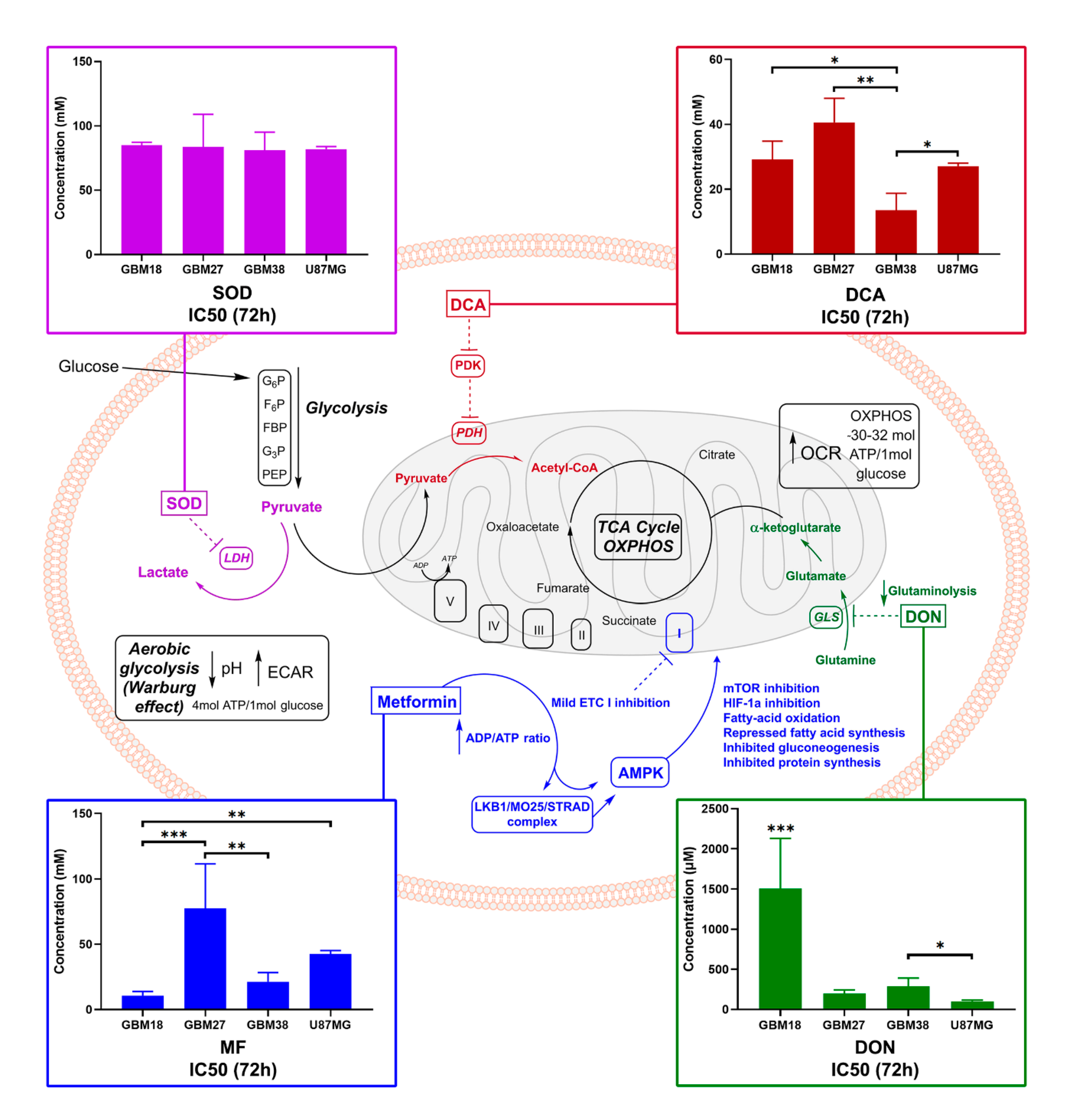
3.2. GSCs Display a Heterogeneous Pattern of Resistance to Metabolic Inhibitors
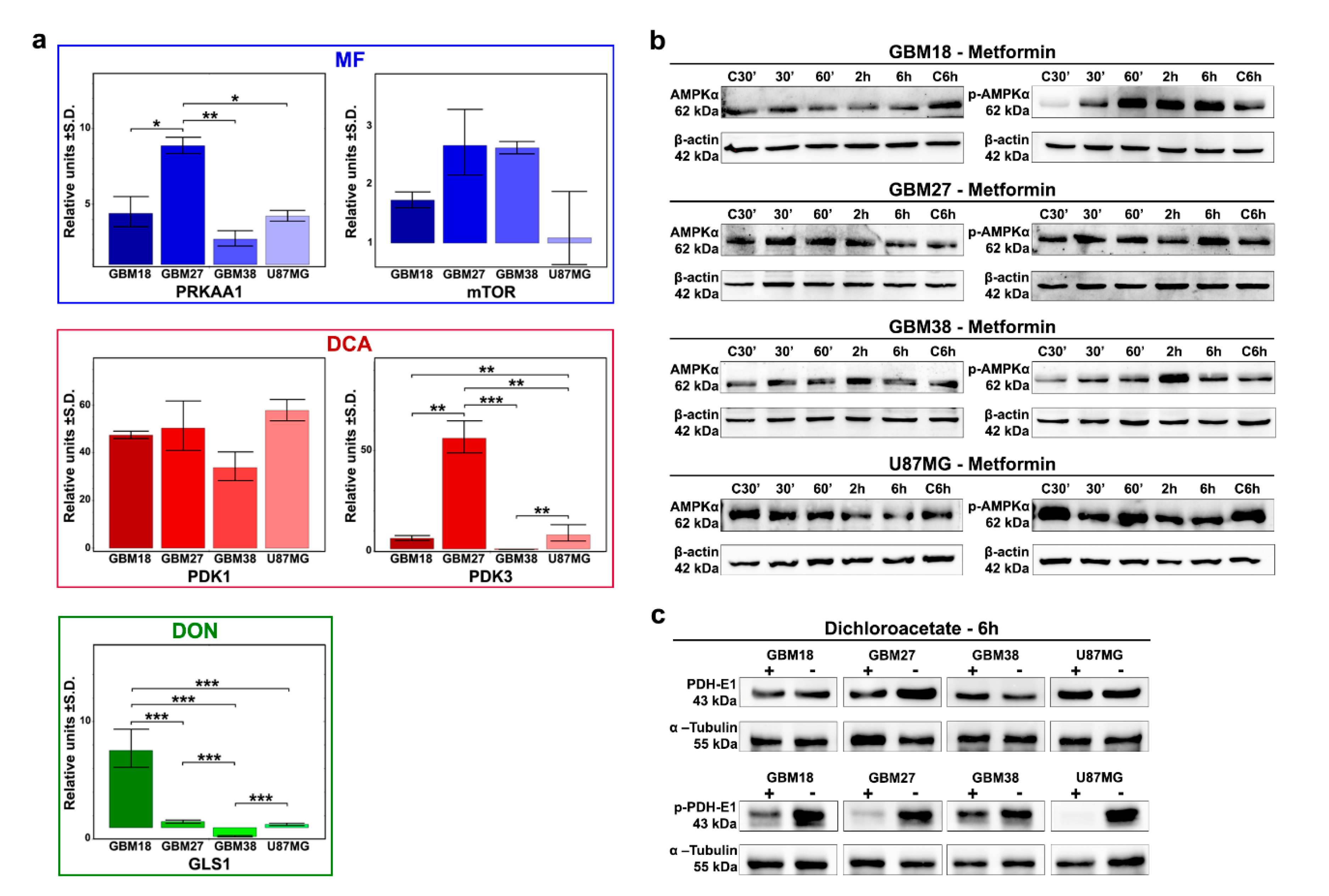
3.3. Differences of Target Enzymes across Cell Lines Predicts Responses to Metabolic Inhibitors
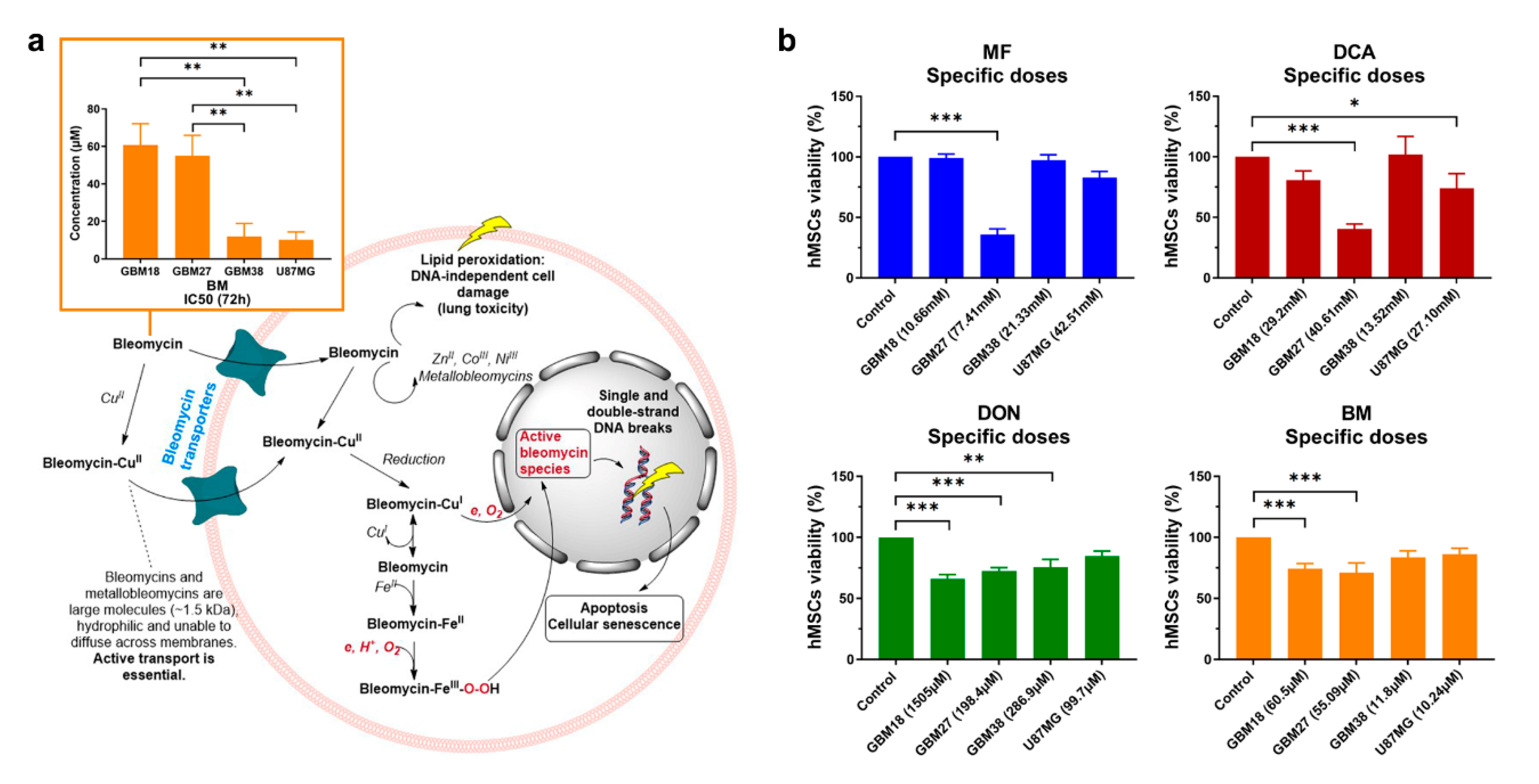
3.4. Doses of Metabolic Inhibitors and Radiomimetic Bleomycin Corresponding to Warburg-Like Phenotypes Spare Viability of Non-Tumoral hMSCs
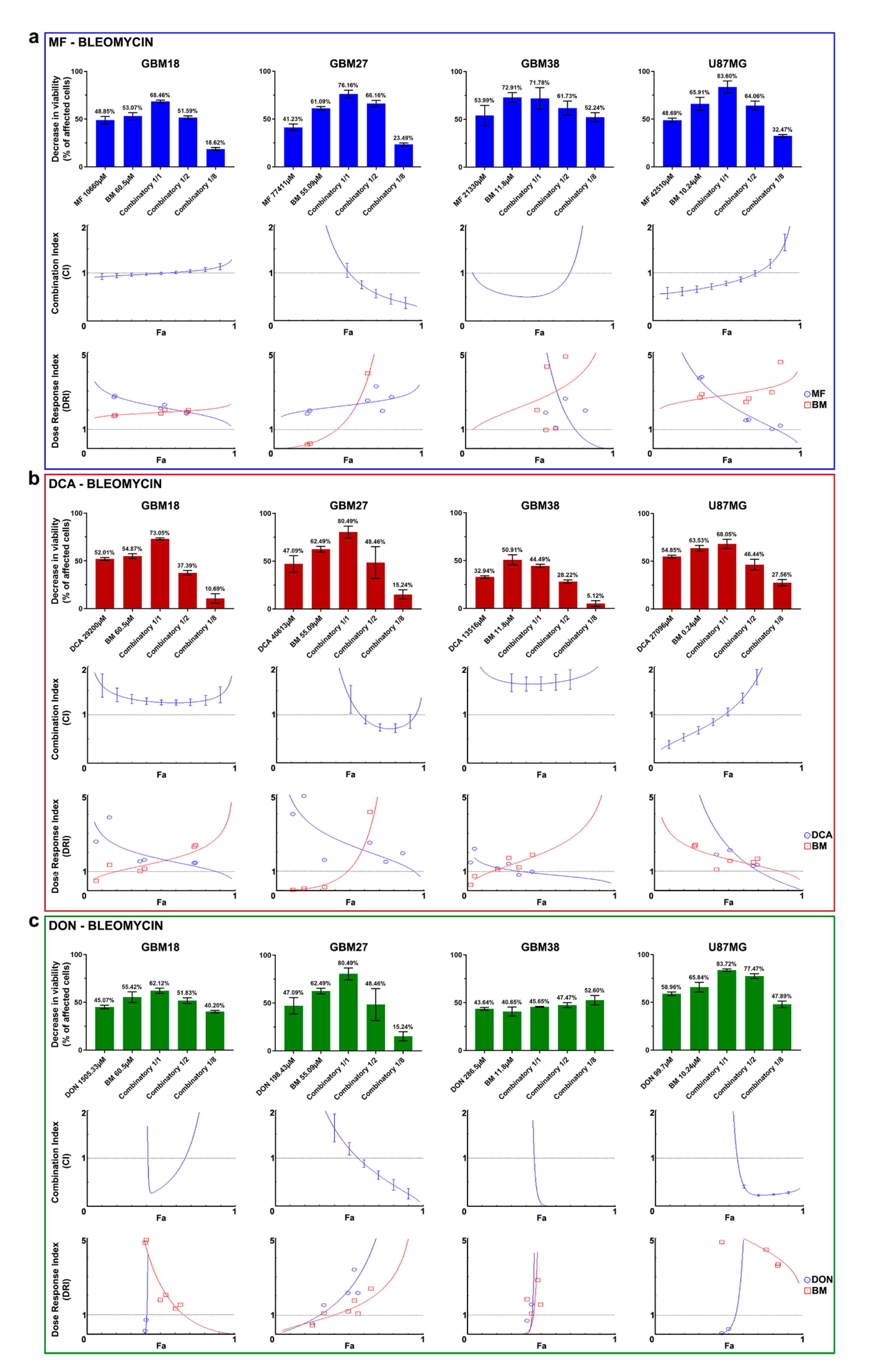
3.5. Synergy between Bleomycin and Metabolic Inhibitors Helps to Overcome Dose-Limitng Toxicity in Predominantly Oxidative Metabolic Phenotypes
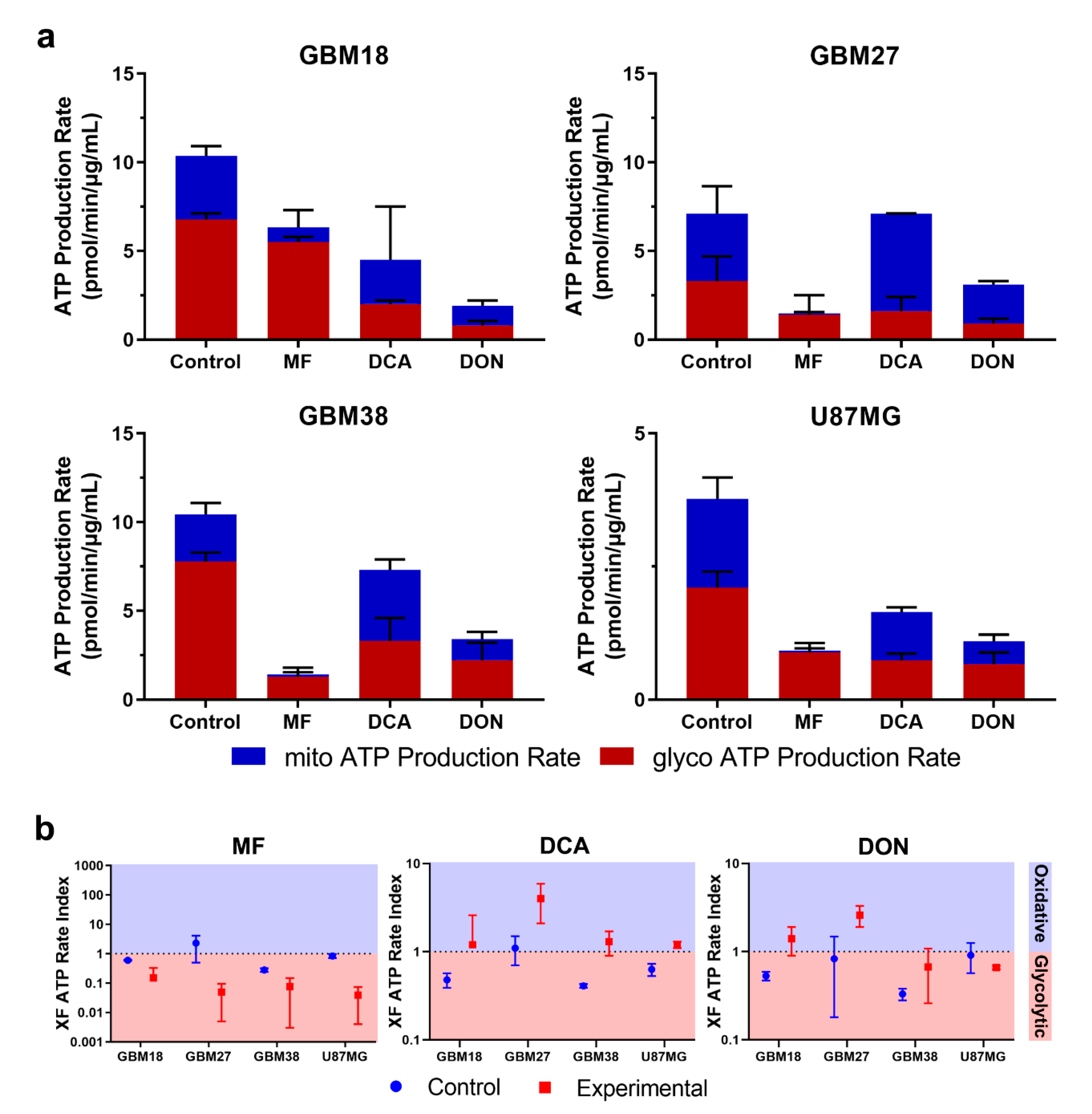
3.6. Bioenergetic Profiling after Metabolic Treatment Reveals Opportunities for Metabolic Priming in Surviving Cell Populations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Farah, P.; Ondracek, A.; Chen, Y.; Wolinsky, Y.; Stroup, N.E.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2006–2010. Neuro-Oncology 2013, 15, ii1–ii56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koshy, M.; Villano, J.L.; Dolecek, T.A.; Howard, A.; Mahmood, U.; Chmura, S.J.; Weichselbaum, R.R.; McCarthy, B.J. Improved survival time trends for glioblastoma using the SEER 17 population-based registries. J. Neuro-Oncol. 2012, 107, 207–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamimi, A.F.; Juweid, M. Epidemiology and Outcome of Glioblastoma. In Glioblastoma [Internet]; 201921345W; Codon Publications: Singapore, 2017. [Google Scholar]
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teo, W.-Y.; Sekar, K.; Seshachalam, P.; Shen, J.; Chow, W.-Y.; Lau, C.C.; Yang, H.; Park, J.; Kang, S.-G.; Li, X.; et al. Relevance of a TCGA-derived Glioblastoma Subtype Gene-Classifier among Patient Populations. Sci. Rep. 2019, 9, 7442. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.J.J. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [Green Version]
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M.; et al. The epidemiology of glioma in adults: A “state of the science” review. Neuro-Oncology 2014, 16, 896–913. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.R.; O’Neill, B.P. Glioblastoma survival in the United States before and during the temozolomide era. J. Neuro-Oncol. 2012, 107, 359–364. [Google Scholar] [CrossRef]
- Han, X.; Xue, X.; Zhou, H.; Zhang, G. A molecular view of the radioresistance of gliomas. Oncotarget 2017, 8, 100931–100941. [Google Scholar] [CrossRef] [Green Version]
- Seyfried, T.N.; Shelton, L.M. Cancer as a metabolic disease. Nutr. Metab. 2010, 7, 7. [Google Scholar] [CrossRef] [Green Version]
- Jose, C.; Bellance, N.; Rossignol, R. Choosing between glycolysis and oxidative phosphorylation: A tumor’s dilemma? Biochim. Biophys. Acta 2011, 1807, 552–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potter, M.; Newport, E.; Morten, K.J. The Warburg effect: 80 years on. Biochem. Soc. Trans. 2016, 44, 1499–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, P.P.; Sabatini, D.M. Cancer Cell Metabolism: Warburg and Beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seyfried, T.N.; Flores, R.E.; Poff, A.M.; D’Agostino, D.P. Cancer as a metabolic disease: Implications for novel therapeutics. Carcinogenesis 2014, 35, 515–527. [Google Scholar] [CrossRef]
- Ristow, M. Oxidative metabolism in cancer growth. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 339–345. [Google Scholar] [CrossRef]
- Semenza, G.L.; Artemov, D.; Bedi, A.; Bhujwalla, Z.; Chiles, K.; Feldser, D.; Laughner, E.; Ravi, R.; Simons, J.; Taghavi, P.; et al. ‘The Metabolism of Tumours’: 70 Years Later. In Proceedings of the Novartis Foundation Symposium, Singapore, 30 August–1 September 2004; pp. 251–259. [Google Scholar]
- Timm, K.N.; Kennedy, B.W.; Brindle, K.M. Imaging Tumor Metabolism to Assess Disease Progression and Treatment Response. Clin. Cancer Res. 2016, 22, 5196–5203. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.M.; Parolia, A.; Dunphy, M.P.; Venneti, S. Non-invasive metabolic imaging of brain tumours in the era of precision medicine. Nat. Rev. Clin. Oncol. 2016, 13, 725–739. [Google Scholar] [CrossRef] [Green Version]
- Paech, D.; Nagel, A.M.; Schultheiss, M.N.; Umathum, R.; Regnery, S.; Scherer, M.; Wick, A.; Platt, T.; Wick, W.; Bendszus, M.; et al. Quantitative Dynamic Oxygen 17 MRI at 7.0 T for the Cerebral Oxygen Metabolism in Glioma. Radiology 2020, 295, 181–189. [Google Scholar] [CrossRef]
- Grasso, D.; Zampieri, L.X.; Capelôa, T.; van de Velde, J.A.; Sonveaux, P. Mitochondria in Cancer. Cell Stress 2020, 4, 114–146. [Google Scholar] [CrossRef]
- Xu, X.D.; Shao, S.X.; Jiang, H.P.; Cao, Y.W.; Wang, Y.H.; Yang, X.C.; Wang, Y.L.; Wang, X.S.; Niu, H.T. Warburg Effect or Reverse Warburg Effect? A Review of Cancer Metabolism. Oncol. Res. Treat. 2015, 38, 117–122. [Google Scholar] [CrossRef]
- Vidali, S.; Aminzadeh, S.; Lambert, B.; Rutherford, T.; Sperl, W.; Kofler, B.; Feichtinger, R.G. Mitochondria: The ketogenic diet—A metabolism-based therapy. Int. J. Biochem. Cell Biol. 2015, 63, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Patel, S.; Affleck, V.S.; Wilson, I.; Turnbull, D.M.; Joshi, A.R.; Maxwell, R.; Stoll, E.A. Fatty acid oxidation is required for the respiration and proliferation of malignant glioma cells. Neuro-Oncology 2017, 19, 43–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinopoulos, C.; Seyfried, T.N. Mitochondrial substrate-level phosphorylation as energy source for glioblastoma: Review and hypothesis. ASN Neuro 2018, 10, 1759091418818261. [Google Scholar] [CrossRef]
- Hinohara, K.; Polyak, K. Intratumoral heterogeneity: More than just mutations. Trends Cell Biol. 2019, 29, 569–579. [Google Scholar] [CrossRef]
- Saga, I.; Shibao, S.; Okubo, J.; Osuka, S.; Kobayashi, Y.; Yamada, S.; Fujita, S.; Urakami, K.; Kusuhara, M.; Yoshida, K.; et al. Integrated analysis identifies different metabolic signatures for tumor-initiating cells in a murine glioblastoma model. Neuro-Oncology 2014, 16, 1048–1056. [Google Scholar] [CrossRef] [Green Version]
- Shibao, S.; Minami, N.; Koike, N.; Fukui, N.; Yoshida, K.; Saya, H.; Sampetrean, O. Metabolic heterogeneity and plasticity of glioma stem cells in a mouse glioblastoma model. Neuro-Oncology 2018, 20, 343–354. [Google Scholar] [CrossRef] [Green Version]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Matsutani, T.; Masui, K.; Poulou, M.; Popescu, R.; Della Donna, L.; Evers, P.; Dekmezian, C.; et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16062–16067. [Google Scholar] [CrossRef] [Green Version]
- Lemberg, K.M.; Vornov, J.J.; Rais, R.; Slusher, B.S. We’re not “DON” yet: Optimal dosing and prodrug delivery of 6-Diazo-5-oxo-L-norleucine. Mol. Cancer Ther. 2018, 17, 1824–1832. [Google Scholar] [CrossRef] [Green Version]
- Tataranni, T.; Piccoli, C. Dichloroacetate (DCA) and Cancer: An Overview towards Clinical Applications. Oxidative Med. Cell. Longev. 2019, 2019, 8201079. [Google Scholar] [CrossRef]
- Kasznicki, J.; Sliwinska, A.; Drzewoski, J. Metformin in cancer prevention and therapy. Ann. Transl. Med. 2014, 2, 57. [Google Scholar]
- Granchi, C.; Paterni, I.; Rani, R.; Minutolo, F. Small-molecule inhibitors of human LDH5. Future Med. Chem. 2013, 5, 1967–1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Hajj, M.; Clarke, M.F. Self-renewal and solid-tumor stem cells. Oncogene 2005, 11, 14–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattabiraman, D.R.; Weinberg, R.A. Tackling the cancer stem cells—What challenges do they pose? Nat. Rev. Drug Discov. 2014, 13, 497–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-Inducible Factors Regulate Tumorigenic Capacity of Glioma Stem Cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A Perivascular Niche for Brain Tumor Stem Cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Peiris-Pagès, M.; Martinez-Outschoorn, U.E.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer stem cell metabolism. Breast Cancer Res. 2016, 18, 1–10. [Google Scholar] [CrossRef]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- Behnan, J.; Finocchiaro, G.; Hanna, G. The landscape of the mesenchymal signature in brain tumours. Brain 2019, 142, 847–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Povirk, L.F.; Mutagenesis, M. DNA damage and mutagenesis by radiomimetic DNA-cleaving agents: Bleomycin, neocarzinostatin and other enediynes. Mutat. Res. 1996, 355, 71–89. [Google Scholar] [CrossRef]
- Chen, J.; Stubbe, J. Bleomycins: Towards better therapeutics. Nat. Rev. Cancer 2005, 5, 102–112. [Google Scholar] [CrossRef] [PubMed]
- García-Romero, N.; González-Tejedo, C.; Carrión-Navarro, J.; Esteban-Rubio, S.; Rackov, G.; Rodríguez-Fanjul, V.; Oliver-De La Cruz, J.; Prat-Acín, R.; Peris-Celda, M.; Blesa, D.; et al. Cancer stem cells from human glioblastoma resemble but do not mimic original tumors after in vitro passaging in serum-free media. Oncotarget 2016, 7, 65888–65901. [Google Scholar] [CrossRef]
- Chou, T.-C. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.-C. Theoretical Basis, Experimental Design, and Computerized Simulation of Synergism and Antagonism in Drug Combination Studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Mahmood, T.; Yang, P.-C. Western blot: Technique, theory, and trouble shooting. N. Am. J. Med. Sci. 2012, 4, 429–434. [Google Scholar] [CrossRef]
- Divakaruni, A.S.; Paradyse, A.; Ferrick, D.A.; Murphy, A.N.; Jastroch, M. Analysis and Interpretation of Microplate-Based Oxygen Consumption and pH Data. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2014; Volume 547, pp. 309–354. [Google Scholar]
- Yépez, V.A.; Kremer, L.S.; Iuso, A.; Gusic, M.; Kopajtich, R.; Koňaříková, E.; Nadel, A.; Wachutka, L.; Prokisch, H.; Gagneur, J. OCR-Stats: Robust estimation and statistical testing of mitochondrial respiration activities using Seahorse XF Analyzer. PLoS ONE 2018, 13, e0199938. [Google Scholar] [CrossRef] [Green Version]
- Bowman, R.L.; Wang, Q.; Carro, A.; Verhaak, R.G.; Squatrito, M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro-Oncology 2017, 19, 139–141. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Wahl, D.R. Metabolic Abnormalities in Glioblastoma and Metabolic Strategies to Overcome Treatment Resistance. Cancers 2019, 11, 1231. [Google Scholar] [CrossRef] [Green Version]
- Madurga, R.; García-Romero, N.; Jiménez, B.; Collazo, A.; Pérez-Rodríguez, F.; Hernández-Laín, A.; Fernández-Carballal, C.; Prat-Acín, R.; Zanin, M.; Menasalvas, E.; et al. Normal tissue content impact on the GBM molecular classification. Brief. Bioinform. 2020, bbaa129. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, T.; Sun, G.; Sun, X.; Zhao, L.; Zhong, R.; Peng, Y. Tumor Energy Metabolism and Potential of 3-Bromopyruvate as an Inhibitor of Aerobic Glycolysis: Implications in Tumor Treatment. Cancers 2019, 11, 317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aljofan, M.; Riethmacher, D. Anticancer activity of metformin: A systematic review of the literature. Future Sci. OA 2019, 5, FSO410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeaman, S.J.; Hutcheson, E.T.; Roche, T.E.; Pettit, F.H.; Brown, J.R.; Reed, L.J.; Watson, D.C.; Dixon, G.H. Sites of phosphorylation on pyruvate dehydrogenase from bovine kidney and heart. Biochemistry 1978, 17, 2364–2370. [Google Scholar] [CrossRef]
- Korotchkina, L.G.; Patel, M.S. Site specificity of four pyruvate dehydrogenase kinase isoenzymes toward the three phosphorylation sites of human pyruvate dehydrogenase. J. Biol. Chem. 2001, 276, 37223–37229. [Google Scholar] [CrossRef] [Green Version]
- Sesen, J.; Dahan, P.; Scotland, S.J.; Saland, E.; Dang, V.-T.; Lemarié, A.; Tyler, B.M.; Brem, H.; Toulas, C.; Moyal, E.C.-J.; et al. Metformin Inhibits Growth of Human Glioblastoma Cells and Enhances Therapeutic Response. PLoS ONE 2015, 10, e0123721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velpula, K.K.; Guda, M.R.; Sahu, K.; Tuszynski, J.; Asuthkar, S.; Bach, S.E.; Lathia, J.D.; Tsung, A.J. Metabolic targeting of EGFRvIII/PDK1 axis in temozolomide resistant glioblastoma. Oncotarget 2017, 8, 35639–35655. [Google Scholar] [CrossRef] [Green Version]
- Prabhu, A.; Sarcar, B.; Miller, C.R.; Kim, S.-H.; Nakano, I.; Forsyth, P.; Chinnaiyan, P. Ras-mediated modulation of pyruvate dehydrogenase activity regulates mitochondrial reserve capacity and contributes to glioblastoma tumorigenesis. Neuro-Oncology 2015, 17, 1220–1230. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.V.; Stylli, S.S.; Kaye, A.H.; Mantamadiotis, T. Multilayered Heterogeneity of Glioblastoma Stem Cells: Biological and Clinical Significance. In Cannabinoids and Neuropsychiatric Disorders; Springer: Berlin/Heidelberg, Germany, 2019; pp. 1–21. [Google Scholar]
- Spehalski, E.I.; Lee, J.A.; Peters, C.; Tofilon, P.; Camphausen, K. The Quiescent Metabolic Phenotype of Glioma Stem Cells. J. Proteom. Bioinform. 2019, 12, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Zhou, Y.; Shingu, T.; Feng, L.; Chen, Z.; Ogasawara, M.; Keating, M.J.; Kondo, S.; Huang, P. Metabolic alterations in highly tumorigenic glioblastoma cells preference for hypoxia and high dependency on glycolysis. J. Biol. Chem. 2011, 286, 32843–32853. [Google Scholar] [CrossRef] [Green Version]
- Massalha, W.; Markovits, M.; Pichinuk, E.; Feinstein-Rotkopf, Y.; Tarshish, M.; Mishra, K.; Llado, V.; Weil, M.; Escriba, P.V.; Kakhlon, O. Minerval (2-hydroxyoleic acid) causes cancer cell selective toxicity by uncoupling oxidative phosphorylation and compromising bioenergetic compensation capacity. Biosci. Rep. 2019, 39, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cvrljevic, A.; Akhavan, D.; Wu, M.; Martinello, P.; Furnari, F.B.; Johnston, A.J.; Guo, D.; Pike, L.; Cavenee, W.K.; Scott, A.M.; et al. Activation of Src induces mitochondrial localisation of de2-7EGFR (EGFRvIII) in glioma cells: Implications for glucose metabolism. J. Cell Sci. 2011, 124, 2938–2950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, H.; Hau, E.; Joshi, S.; Dilda, P.J.; McDonald, K.L. Sensitization of Glioblastoma Cells to Irradiation by Modulating the Glucose Metabolism. Mol. Cancer Ther. 2015, 14, 1794–1804. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Han, J.; Jang, Y.; Kim, S.J.; Lee, M.J.; Ryu, M.J.; Kweon, G.R.; Heo, J.Y. High-capacity glycolytic and mitochondrial oxidative metabolisms mediate the growth ability of glioblastoma. Int. J. Oncol. 2015, 47, 1009–1016. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Chhipa, R.R.; Pooya, S.; Wortman, M.; Yachyshin, S.; Chow, L.M.L.; Kumar, A.; Zhou, X.; Sun, Y.; Quinn, B.; et al. Discrete mechanisms of mTOR and cell cycle regulation by AMPK agonists independent of AMPK. Proc. Natl. Acad. Sci. USA 2014, 111, E435–E444. [Google Scholar] [CrossRef] [Green Version]
- Baker, J.C.; Yan, X.; Peng, T.; Kasten, S.; Roche, T.E. Marked Differences between Two Isoforms of Human Pyruvate Dehydrogenase Kinase. J. Biol. Chem. 2000, 275, 15773–15781. [Google Scholar] [CrossRef] [Green Version]
- Bowker-Kinley, M.M.; Davis, I.W.; Wu, P.; Harris, A.R.; Popov, M.K. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem. J. 1998, 329, 191–196. [Google Scholar] [CrossRef]
- Goldberg, E.B.; Colowick, S.P. The role of glycolysis in the growth of tumor cells. 3. Lactic dehydrogenase as the site of action of oxamate on the growth of cultured cells. J. Biol. Chem. 1965, 240, 2786–2790. [Google Scholar] [CrossRef]
- Elwood, J.C. Effect of oxamate on glycolysis and respiration in sarcoma 37 ascites cells. Cancer Res. 1968, 28, 2056–2060. [Google Scholar] [PubMed]
- Fiume, L.; Manerba, M.; Vettraino, M.; Di Stefano, G. Impairment of Aerobic Glycolysis by Inhibitors of Lactic Dehydrogenase Hinders the Growth of Human Hepatocellular Carcinoma Cell Lines. Pharmacology 2010, 86, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.; Yang, Y.; Wan, J.; Zhu, R.; Wu, Y. Inhibition of LDH-A by oxamate induces G2/M arrest, apoptosis and increases radiosensitivity in nasopharyngeal carcinoma cells. Oncol. Rep. 2013, 30, 2983–2991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Yang, Z.; Chen, Z.; Chen, R.; Zhaofeng, C.; Zhou, Y.; Qiao, L. Effects of the suppression of lactate dehydrogenase A on the growth and invasion of human gastric cancer cells. Oncol. Rep. 2014, 33, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Still, E.R.; Yuneva, M.O. Hopefully devoted to Q: Targeting glutamine addiction in cancer. Br. J. Cancer 2017, 116, 1375–1381. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohba, S.; Hirose, Y. L-asparaginase and 6-diazo-5-oxo-L-norleucine synergistically inhibit the growth of glioblastoma cells. J. Neuro-Oncol. 2020, 146, 469–475. [Google Scholar] [CrossRef]
- Linnert, M.; Gehl, J. Bleomycin treatment of brain tumors: An evaluation. Anti-Cancer Drugs 2009, 20, 157–164. [Google Scholar] [CrossRef]
- Chou, T.-C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Shen, H.; Decollogne, S.; Dilda, P.J.; Hau, E.; Chung, S.A.; Luk, P.P.; Hogg, P.J.; McDonald, K.L. Dual-targeting of aberrant glucose metabolism in glioblastoma. J. Exp. Clin. Cancer Res. 2015, 34, 14. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Jeon, J.-H.; Min, B.-K.; Ha, C.-M.; Thoudam, T.; Park, B.-Y.; Lee, I. Role of the Pyruvate Dehydrogenase Complex in Metabolic Remodeling: Differential Pyruvate Dehydrogenase Complex Functions in Metabolism. Diabetes Metab. J. 2018, 42, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, S.-L.; Hu, X.; Tam, K.Y. Targeting Tumor Metabolism for Cancer Treatment: Is Pyruvate Dehydrogenase Kinases (PDKs) a Viable Anticancer Target? Int. J. Biol. Sci. 2015, 11, 1390–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oizel, K.; Chauvin, C.; Oliver, L.; Gratas, C.; Geraldo, F.; Jarry, U.; Scotet, E.; Rabe, M.; Alves-Guerra, M.-C.; Teusan, R.; et al. Efficient Mitochondrial Glutamine Targeting Prevails Over Glioblastoma Metabolic Plasticity. Clin. Cancer Res. 2017, 23, 6292–6304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shelton, L.M.; Huysentruyt, L.C.; Seyfried, T.N. Glutamine targeting inhibits systemic metastasis in the VM-M3 murine tumor model. Int. J. Cancer 2010, 127, 2478–2485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cervantes-Madrid, D.; Romero, Y.; Dueñas-González, A. Reviving Lonidamine and 6-Diazo-5-oxo-L-norleucine to Be Used in Combination for Metabolic Cancer Therapy. BioMed Res. Int. 2015, 2015, 690492. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Lukinius, A.; Bergström, M.; Eriksson, B.; Watanabe, Y.; Långström, B. A mechanism behind the antitumour effect of 6-diazo-5-oxo-L-norleucine (DON): Disruption of mitochondria. Eur. J. Cancer 1999, 35, 1155–1161. [Google Scholar] [CrossRef]
- Huber, K.R.; Mayer, E.P.; Mitchell, D.; Roberts, J.W. Cell cycle phase perturbations by 6-diazo-5-oxo-L-norleucine and acivicin in normal and neoplastic human cell lines. Br. J. Cancer 1987, 55, 653–656. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.S.; Hoyte, C. Review of biguanide (metformin) toxicity. J. Intensive Care Med. 2019, 34, 863–876. [Google Scholar] [CrossRef]
- Erices, R.; Bravo, M.L.; Gonzalez, P.; Oliva, B.; Racordon, D.; Garrido, M.; Ibañez, C.; Kato, S.; Brañes, J.; Pizarro, J.; et al. Metformin, at Concentrations Corresponding to the Treatment of Diabetes, Potentiates the Cytotoxic Effects of Carboplatin in Cultures of Ovarian Cancer Cells. Reprod. Sci. 2013, 20, 1433–1446. [Google Scholar] [CrossRef] [Green Version]
- Song, C.W.; Lee, H.; Dings, R.P.M.; Williams, B.; Powers, J.; Dos Santos, T.; Choi, B.-H.; Park, H.J. Metformin kills and radiosensitizes cancer cells and preferentially kills cancer stem cells. Sci. Rep. 2012, 2, 362. [Google Scholar] [CrossRef] [Green Version]
- Stacpoole, P.W.; Kurtz, T.L.; Han, Z.; Langaee, T.Y. Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Adv. Drug Deliv. Rev. 2008, 60, 1478–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandsma, D.; Dorlo, T.P.; Haanen, J.H.; Beijnen, J.H.; Boogerd, W. Severe encephalopathy and polyneuropathy induced by dichloroacetate. J. Neurol. 2010, 257, 2099–2100. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.B.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Bonnet, S.; et al. A Mitochondria-K+ Channel Axis Is Suppressed in Cancer and Its Normalization Promotes Apoptosis and Inhibits Cancer Growth. Cancer Cell 2007, 11, 37–51. [Google Scholar] [CrossRef] [Green Version]
- Sun, R.C.; Fadia, M.; Dahlstrom, J.E.; Parish, C.R.; Board, P.G.; Blackburn, A.C. Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast Cancer Res. Treat. 2009, 120, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Heshe, D.; Hoogestraat, S.; Brauckmann, C.; Karst, U.; Boos, J.; Lanvers-Kaminsky, C. Dichloroacetate metabolically targeted therapy defeats cytotoxicity of standard anticancer drugs. Cancer Chemother. Pharmacol. 2010, 67, 647–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madhok, B.M.; Yeluri, S.; Perry, S.L.; Hughes, T.A.; Jayne, D.G. Dichloroacetate induces apoptosis and cell-cycle arrest in colorectal cancer cells. Br. J. Cancer 2010, 102, 1746–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockwin, L.H.; Yu, S.X.; Borgel, S.; Hancock, C.; Wolfe, T.L.; Phillips, L.R.; Hollingshead, M.G.; Newton, D.L. Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC. Int. J. Cancer 2010, 127, 2510–2519. [Google Scholar] [CrossRef]
- Pinkus, L.M. [45] Glutamine binding sites. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1977; Volume 46, pp. 414–427. [Google Scholar]
- Alt, J.; Potter, M.C.; Rojas, C.; Slusher, B.S. Bioanalysis of 6-diazo-5-oxo-l-norleucine in plasma and brain by ultra-performance liquid chromatography mass spectrometry. Anal. Biochem. 2015, 474, 28–34. [Google Scholar] [CrossRef] [Green Version]
- Sarkaria, J.N.; Hu, L.S.; Parney, I.F.; Pafundi, D.H.; Brinkmann, D.H.; Laack, N.N.; Giannini, C.; Burns, T.C.; Kizilbash, S.H.; Laramy, J.K.; et al. Is the blood–brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro-Oncology 2018, 20, 184–191. [Google Scholar] [CrossRef]
- Song, M.; Kim, S.-H.; Im, C.Y.; Hwang, H.-J. Recent development of small molecule glutaminase inhibitors. Curr. Top. Med. Chem. 2018, 18, 432–443. [Google Scholar] [CrossRef]
- Strickland, M.; Stoll, E.A. Metabolic reprogramming in glioma. Front. Cell Dev. Biol. 2017, 5, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woolf, E.C.; Syed, N.; Scheck, A.C. Tumor metabolism, the ketogenic diet and β-hydroxybutyrate: Novel approaches to adjuvant brain tumor therapy. Front. Mol. Neurosci. 2016, 9, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akins, N.S.; Nielson, T.C.; Le, H.V. Inhibition of glycolysis and glutaminolysis: An emerging drug discovery approach to combat cancer. Curr. Top. Med. Chem. 2018, 18, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, A.F.; Mahmoud, W.; Al-Harizy, R.M. Targeting glucose metabolism to suppress cancer progression: Prospective of anti-glycolytic cancer therapy. Pharmacol. Res. 2019, 150, 104511. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Alesi, G.; Kang, S. Glutaminolysis as a target for cancer therapy. Oncogene 2016, 35, 3619–3625. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Shi, Y. Mitochondria as a target in cancer treatment. MedComm 2020, 1, 129–139. [Google Scholar] [CrossRef]
- Ma, Y.; Temkin, S.M.; Hawkridge, A.M.; Guo, C.; Wang, W.; Wang, X.-Y.; Fang, X. Fatty acid oxidation: An emerging facet of metabolic transformation in cancer. Cancer Lett. 2018, 435, 92–100. [Google Scholar] [CrossRef]
- Xia, S.; Lin, R.; Jin, L.; Zhao, L.; Kang, H.-B.; Pan, Y.; Liu, S.; Qian, G.; Qian, Z.; Konstantakou, E. Prevention of dietary-fat-fueled ketogenesis attenuates BRAF V600E tumor growth. Cell Metab. 2017, 25, 358–373. [Google Scholar] [CrossRef] [Green Version]
- De Feyter, H.M.; Behar, K.L.; Rao, J.U.; Madden-Hennessey, K.; Ip, K.L.; Hyder, F.; Drewes, L.R.; Geschwind, J.-F.; De Graaf, R.A.; Rothman, D.L. A ketogenic diet increases transport and oxidation of ketone bodies in RG2 and 9L gliomas without affecting tumor growth. Neuro-Oncology 2016, 18, 1079–1087. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | 5′-Sequence-3′ |
|---|---|
| β-actin FW | TTCTACAATGAGCTGCGTGTG |
| β-actin RV | GGGGTGTTGAAGGTCTCAAA |
| GAPDH FW | TCCTCCACCTTTGACGCTG |
| GAPDH RV | ACCACCCTGTTGCTGTAGCC |
| GLS1 FW | GCCCGCTTTGTGTGACTAAA |
| GLS1 RV | CAGGGGTAAATAACGGCACA |
| GLS2 FW | GCACTAAAGGCCACTGGAC |
| GLS2 RV | CCAAGAGGCCACCACTACTG |
| MTOR FW | CTGACCGCTAGTAGGGAGGT |
| MTOR RV | AACATCCCAGAACCCTGCTG |
| PDK1 FW | ATCCTCCTGCCTGAGTCTCT |
| PDK1 RV | CAAATGCCAAGGACTGCTGT |
| PDK2 FW | TGCCTACGACATGGCTAAGCTC |
| PDK2 RV | GACGTAGACCATGTGAATCGGC |
| PDK3 FW | TGGAAGGAGTGGGTACTGATGC |
| PDK3 RV | GGATTGCTCCAATCATCGGCTTC |
| PDK4 FW | AACTCGGGATGTTGGGGATT |
| PDK4 RV | AGAGAAAAGCCCTTCCTACTGA |
| PRKAA1 FW | GTCCAGGGCTTGTTCTATTCA |
| PRKAA1 RV | ATGCTGCACTTAGAGACCCT |
| PRKAA2 FW | TGGAACATTGTTACAGCAGGC |
| PRKAA2 RV | AGCTCTTCTCCCGTGTCTTC |
| MF + Bleomycin | DCA + Bleomycin | DON + Bleomycin | ||||
|---|---|---|---|---|---|---|
| Cell Line | Effect at Fa = 0.6 | DRI at Fa = 0.6 | Effect at Fa = 0.6 | DRI at Fa = 0.6 | Effect at Fa = 0.6 | DRI at Fa = 0.6 |
| GBM18 | Additive | DRI > 1 for both | Antagonism | DRI > 1 for both | Synergism | DRI > 1 for both |
| GBM27 | Synergism | DRI > 1 for both | Synergism | DRI > 1 for both | Synergism | DRI > 1 for both |
| GBM38 | Synergism | DRI > 1 for both | Antagonism | DRI > 1 for bleomycin | Synergism | DRI > 1 for both |
| U87MG | Synergism | DRI > 1 for both | Antagonism | DRI > 1 for both | Synergism | DRI > 1 for both |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duraj, T.; García-Romero, N.; Carrión-Navarro, J.; Madurga, R.; Ortiz de Mendivil, A.; Prat-Acin, R.; Garcia-Cañamaque, L.; Ayuso-Sacido, A. Beyond the Warburg Effect: Oxidative and Glycolytic Phenotypes Coexist within the Metabolic Heterogeneity of Glioblastoma. Cells 2021, 10, 202. https://doi.org/10.3390/cells10020202
Duraj T, García-Romero N, Carrión-Navarro J, Madurga R, Ortiz de Mendivil A, Prat-Acin R, Garcia-Cañamaque L, Ayuso-Sacido A. Beyond the Warburg Effect: Oxidative and Glycolytic Phenotypes Coexist within the Metabolic Heterogeneity of Glioblastoma. Cells. 2021; 10(2):202. https://doi.org/10.3390/cells10020202
Chicago/Turabian StyleDuraj, Tomás, Noemí García-Romero, Josefa Carrión-Navarro, Rodrigo Madurga, Ana Ortiz de Mendivil, Ricardo Prat-Acin, Lina Garcia-Cañamaque, and Angel Ayuso-Sacido. 2021. "Beyond the Warburg Effect: Oxidative and Glycolytic Phenotypes Coexist within the Metabolic Heterogeneity of Glioblastoma" Cells 10, no. 2: 202. https://doi.org/10.3390/cells10020202