
Abstract
'Bottom-up' method is a powerful approach to design nanomaterials with desired properties. The bottle neck of being oxidized of phosphorous structures may be conquered by cluster assembling method. Here, we used P8 and N2 as assembling units to construct one-dimensional (1D) nanowire (NW) and two-dimensional (2D) nanosheet (NS), the stability, electronic and magnetic properties of these assembled nanomaterials are investigated using density functional theory (DFT) calculations. The assembled 1D-P8N2 NW and 2D-P8N4 NS are identified to possess good stability, as demonstrated by their high cohesive energies, positive phonon dispersions, and structural integrity through molecular dynamics simulations at 300 and 500 K. Moreover, they also exhibit good anti-oxidization property. The 2D-P8N4 NS is a direct bandgap semiconductor with the HSE06 gap of 2.61 eV, and shows appropriate band-edge aliments and moderate carrier mobility for photocatalyzing water splitting. The 1D-P8N2 NW is an indirect bandgap semiconductor, and Mn doping could convert it into a dilute magnetic semiconductor (DMS) with one Dirac cone in the spin-up channel, while the vdW-type sheet composed of Mn1@1D-P8N2 NWs is a ferromagnetic metal. Our theoretical study is helpful to design stable phosphorus-based nanomaterials with diverse properties and potential applications.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
The successful experimental preparation [1] and theoretical exploration of phosphorene in 2014 [2] confirmed its direct-band-gap semiconducting characteristics, layer thickness-dependent bandgap adjustment, high carrier mobility, and anisotropic mechanical and transport properties [3–5]. It has potential applications in communications, energy and electronic devices. In addition, phosphorene has many allotropes, such as α-P, β-P, δ-P, ε-P, ζ-P, η-P, and θ-P [6–10]. Although phosphorene has many excellent properties, the shortcomings of being easily oxidized and decomposed in the ambient condition will make the phosphorene-based electronic devices rapidly invalidation, which extremely limits its practical application [11–15]. Typically, doping and functionalization were used to improve the stability of P-based structures [16–19]. Furthermore, assembling phosphorus clusters into nanomaterials might be an effective and promising procedure to overcome the bottle neck of the instability against oxygen in phosphorene [20].
Nowadays, various nanomaterials were designed based on 'bottom-up' method, and cluster assembly is a powerful and promising scheme to design nanomaterials, mainly due to the multi-dimensionality regulation of the properties and functions of the assembled material [21–23]. For instance, the In12As12 cage structure is proved an ideal building block for the synthesis of 1D NW, 2D graphene-like sheet and three-dimensional (3D) crystal materials, which could be used for heterogeneous catalysis, molecular transport, and gas storage [24]. The assembled 1D V1@Si12-NW-rI is reported to have good stabilities and exhibit feature of antiferromagnetic semiconductor [25], which is different to the ferromagnetic metallic character of the assembled 2D sheets [26]. Liu et al used the novel cage structures of M12N12 (M = Al and Ga) clusters as building blocks to construct new 2D and 3D crystal materials, enriching the properties and applications of the corresponding MN compounds [27]. Recently, based on the medium-sized phosphorus clusters containing P8 and P2 moieties, we successfully assembled 1D P10 nanowire (1D-P10 NW) with these two subunits [20]. We found that 1D-P10 NW is very stable with exposure to O2, and the semiconducting feature can be converted into various magnetic and electronic characters upon transition metal adsorption.
Considering that N has the same valence electrons as P, and the main group V-V binary compounds are stable and possess many interesting physical and chemical properties, as well as excellent photoelectronic and nanoelectronic properties [28, 29], we chose N atoms as the linker to connect the experimentally synthesized P8 clusters [30] into one dimensional nanowire (1D-P8N2 NW) and two dimensional nanosheet (2D-P8N4 NS). Their stabilities were verified by thermodynamic (high cohesive energies), dynamical, thermal and chemical aspects. The assembled 1D-P8N2 NW is a nonmagnetic semiconductor with an indirect bandgap of 0.76 eV, and Mn doping converts it into a dilute magnetic semiconductor. The assembled 2D-P8N4 NS is a direct-bandgap semiconductor, and it has suitable bandgap (2.61 eV) and bandedge aliments for water splitting, as well as comparable carrier mobility for electrons and holes; moreover, it also has good optical absorption covering the visible light and UV-light region. Our comprehensive studies provide a novel approach to design stable low-dimensional P-based nanomaterials with various magnetic and electronic properties, which are expected to serve as photocatalyst for water splitting, nanoelectronic devices and spintronics.
2. Computational methods
Spin-polarized density functional theory (DFT) calculations were carried out using the Vienna ab initio simulation package (VASP) [31–36]. The projector augmented wave (PAW) method [37] was used to describe the electron-ion interactions, and generalized gradient approximations (GGA) with the Perdew–Burke–Ernzerhof (PBE) functional [38] was adopted for electron-electron exchange correlations. In order to obtain precise electronic structures, we used the Heyd-Scuseria-Ernzerhof (HSE06) [39] screened hybrid density functional in this work. The energy cutoff for the plane-wave basis set was set to be 600 eV. The k-point mesh was set to be 9 × 1 × 1 and 9 × 9 × 1 for 1D NW and 2D sheet, respectively. The criteria for terminating electronic or ionic iterations was 1 × 10−6 eV and remaining force on each atom was less than 0.01 eV Å−1. A vacuum space of about 20 Å was added. The PBE + U (U = 4 eV) approach was used for both geometry optimizations and magnetic/electronic properties of Mn doped 1D-P8N2 NW, due to the strong correlations between d electrons of transition metals [40]. Spin–orbit coupling (SOC) was included to determine the ground state of magnetic order and the magnetic anisotropic energy (MAE), as well as the easy axis of the Mn doped nanowire. Thermal stability was assessed at 300 and 500 K based on first-principles molecular dynamics (FPMD) simulations conducted at the DFT level using a canonical ensemble having a constant number of atoms, volume, and temperature (NVT) with 0.5 fs time steps for a total simulated time duration of 5 ps, with the temperature controlled by the Nosé-Hoover thermostat [41]. The D2 method [42] was used to account for the van der Waals (vdW) interaction. The barriers of O2 dissociation were determined by the climbing image nudged elastic-band (CI-NEB) method [43]. The cohesive energy (Ecoh ) is defined as follows:
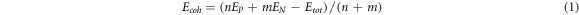
where EP , EN and Etot are the energy of an isolated P atom, N atom and the total energy of the unit cell of assembled materials, respectively; n and m is the number of P atoms and N atoms in the unit cell.
The work function of a material is a critical parameter commonly used as an intrinsic reference for band alignment [44, 45]. Work function is defined as follows:

where Evac the energy of a stationary electron in the vacuum nearby the surface, EF is the Fermi energy.
The carrier mobility of 2D material is given by the following expression [46, 47]:
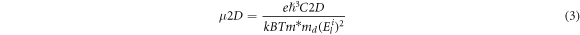
where e, ħ, C2D, kB, T, m*, md
and  are the charge of the electron, Plank's constant, elastic modulus, Boltzmann constant, temperature, the effective mass along the transport direction, average effective mass and deformation potential constant, respectively.
are the charge of the electron, Plank's constant, elastic modulus, Boltzmann constant, temperature, the effective mass along the transport direction, average effective mass and deformation potential constant, respectively.
3. Result and discussion
3.1. Geometries, stabilities and electronic properties of the assembled 1D NW and 2D sheet
3.1.1. Geometries
After replacing the P2 dimer in 1D-P10 with N2 dimer, it was found that the P8 unit remains its original cage-like structure in either 1D-P8N2 NW or 2D-P8N4 NS, while the N-N bond was broken (the N...N distance is 2.79 Å) after geometry relaxation, forming bridging N moiety, which is similar to the structural pattern of N in the global minimum of tetra-MoN2 [48] and S-SN2 monolayers [49]. The configurations of the two assembled 1D-P8N2 NW and 2D-P8N4 NS are presented in figure 1, and the key geometry parameters are summarized in table 1. For comparison, the parameters of black phosphorene (BP) and 1D-P10 NW are also given in table 1. The lattice a of 1D-P8N2 NW is 5.92 Å, a and b are both 5.99 Å for 2D-P8N4 NS. The P–N (P–P) bond lengths are slightly shorter (longer) in the 2D-P8N4 NS than the 1D-P8N2 NW, and the ∠P–N–P is also smaller in the assembled 2D structure.

Figure 1. Structures of the assembled (a) 1D-P8N2 (b) 2D-P8N4. The pink and blue balls denote phosphorus and nitrogen atoms, respectively. The area in dash lines represents the unit cell of each system.
Download figure:
Standard image High-resolution imageTable 1. The lattice parameters (a, b, in Å), bond lengths (dP–P, dP–N, in Å) and bond angels (∠P–P–P, ∠P–N–P, in °) of different materials.
| 1D-P8N2 | 2D-P8N4 | BP1 | 1D-P10 [20] | |
|---|---|---|---|---|
| a | 5.92 | 5.99 | 4.58 | 6.45 |
| b | — | 5.99 | 3.22 | 3.37 |
| dP–P | 2.24, 2.26 | 2.26, 2.28 | 2.22, 2.25 | 2.19, 2.27 |
| dP–N | 1.62 | 1.60 | — | — |
| ∠P–P–P | 79.59, 93.07, 103.33 | 88.07, 101.05, 102.31 | 96, 103.51 | 95.2, 100.35, 103.47 |
| ∠P–N–P | 137.12 | 125.62 | — | — |
3.1.2. Thermodynamic stability
Firstly, we examined the thermodynamic stabilities of two assembled materials by calculating their cohesive energies (Ecoh ). A more positive value of Ecoh means a more stable structure according to equation (1). The cohesive energy of the 1D-P8N2 NW is 3.79 eV/atom, as expected lower than the Ecoh of the 2D-P8N4 NS (4.17 eV/atom). By comparing the cohesive energies of black phosphene/1D-P10 NW (3.60/3.58 eV/atom) and PN monolayers with buckled or puckered hexagons [28] (4.08 ∼ 4.40 eV/atom) calculated at the same level of theory (table 2), we found that the high N content leads to high Ecoh value. The reasonably high cohesive energies of the assembled 1D-P8N2 NW or 2D-P8N4 NS suggest their appreciable thermodynamic stability.
Table 2. The Ecoh (eV/atom) of the 1D-P8N2, 2D-P8N4, α-PN, β-PN, and γ-PN.
| 1D-P8N2 | 2D-P8N4 | α-PN | β-PN | γ-PN | |
|---|---|---|---|---|---|
| Ecoh | 3.79 | 4.17 | 4.40 | 4.36 | 4.08 |
3.1.3. Dynamic stability
Then we examined the dynamic stability of the two assembled materials by calculating their phonon dispersion curves (figure 2). There is no imaginary frequency in Brillouin Zone (BZ) for 1D-P8N2 NW, however, a tiny U-shape negative frequencies (most imaginary value −9 cm−1) appear in the Г region for 2D-P8N4. Note that such U-shape characteristic in the 2D phonon spectra near the Г point is a signature of the flexural acoustic mode but not instability, and is a common phenomenon in first-principles calculations of 2D materials [50, 51]. Therefore, both of the two assembled materials are considered to be dynamically stable.

Figure 2. The calculated phonon dispersion curves of the assembled 1D-P8N2 (a) and 2D-P8N4 (b).
Download figure:
Standard image High-resolution image3.1.4. Thermal stability
We performed first-principles molecular dynamics (FPMD) simulations to examine the thermal stability of the two assembled materials at 300 (figure 3) and 500 K (figure S1 is available online at stacks.iop.org/NANOX/2/010004/mmedia), respectively. At the end of a 5 ps FPMD simulation, the structural integrity of 1D-P8N2 NW (a 5 × 1 × 1 supercell) and 2D-P8N4 NS (a 5 × 5 × 1 supercell) were retained at both 300 and 500 K. Thus, the result of FPMD simulations confirms that two assembled materials possess good thermal stability.

Figure 3. The evolution of total energies of 1D-P8N2 (a) and 2D-P8N4 (b) during FPMD simulation at 300 K and the corresponding structural snapshots at the end of 5 ps.
Download figure:
Standard image High-resolution image3.1.5. Chemical stability
The shortcoming of being easily oxidized inhibits the wide application of the 2D phosphorenes [52, 53]. Therefore, we investigated the oxygen resistance of the 1D-P8N2 NW and 2D-P8N4. The O2 dissociation on the two assembled materials are exothermic, releasing the reaction heat of 4.88 and 4.66 eV on the 1D-P8N2 NW and 2D-P8N4, respectively, and the corresponding activation energy barriers are as high as 1.51 and 1.24 eV, respectively (figure S2), which are comparable to the barrier on the 1D-P10 NW (1.55 eV) [20], but much higher than the result of α-P (0.56 eV) [51]. Furthermore, the FPMD simulations at 300 K were carried out for the 2 × 1 × 1 supercell of 1D-P8N2 NW and 2 × 2 × 1 supercell of 2D-P8N4 NS with one O2 molecule adsorption. During the 5 ps FPMD simulation, dissociation of the O2 was not observed on these two systems, and the O–O bond at the end of 5 ps was not elongated compared to the initial value (figure 4). Moreover, the O2 dissociation was not observed either at 500 K (figure S3). The above results demonstrate that the assembled 1D-P8N2 NW and 2D-P8N4 NS exhibit excellent chemical stability against O2.

Figure 4. The energy evolution of an O2 molecule adsorbed on the assembled 1D-P8N2 NW (a) and 2D-P8N4 (b) through a 5 ps FPMD simulation at 300 K, as well as the corresponding structural snapshot at the end of 5 ps.
Download figure:
Standard image High-resolution image3.1.6. Electronic properties
Typically, the PBE method underestimates bandgaps of materials. Therefore, we used the HSE06 method to calculate electronic band structures of the two assembled materials. The HSE06 results reveal that the 1D-P8N2 NW is an indirect semiconductor with a bandgap (Eg) of 0.76 eV, and the 2D-P8N4 NS is a direct semiconductor with Eg = 2.61 eV (figures 5(a) and (b)). In comparison with the conduction band minimum (CBM) and valance band maximum (VBM) both located on P atoms for the 1D-P8N2 NW (figure 5(c)), the CBM is distributed on the P8 unit, while the VBM is located on the N atoms for 2D-P8N4 NS (figure 5(d)). The different spatial distributions of CBM and VBM will reduce the probability of electron hole recombination, which is beneficial for the high efficiency of photocatalysis.

Figure 5. The HSE06 band structure of the assembled 1D-P8N2 NW (a) 2D-P8N4 NS (b), as well as their corresponding CBM (upper panel) and VBM (down panel) distributions (c), (d). The Fermi level is set at 0 eV, and the isosurface value is 0.01 a.u.
Download figure:
Standard image High-resolution image3.2. The assembled 2D-P8N4 NS as photocatalyst for water splitting
3.2.1. Band alignment
For photocatalytic water splitting, the bandgap of the photocatalyst is required to be in the range of 1.23 ∼ 3 eV. Both the reduction potential ( = 4.44 eV) for H+ to H2 and the oxidation potential (
= 4.44 eV) for H+ to H2 and the oxidation potential ( = 5.67 eV) of H2O to O2 should be located inside the bandgap for good photocatalysts [54]. The small bandgap (0.76 eV) of the 1D-P8N2 NW rules it out as a photocatalyst. The band edge of the 2D-P8N4 NS (figure 6(a)) was calculated to be −3.64 eV for CBM (higher than −4.44 eV) and −6.25 eV for VBM (lower than −5.67 eV), perfectly satisfying the band alignment requirement, i.e., the band positions of the 2D-P8N4 NS are favorable for water splitting. The energy difference (ΔE1) between CBM level and water reduction potential is 0.80 eV and the energy difference (ΔE2) between VBM level and water oxidation potential is 0.58 eV; the comparable energy differences provides comparable driving force for both water oxidation and water reduction.
= 5.67 eV) of H2O to O2 should be located inside the bandgap for good photocatalysts [54]. The small bandgap (0.76 eV) of the 1D-P8N2 NW rules it out as a photocatalyst. The band edge of the 2D-P8N4 NS (figure 6(a)) was calculated to be −3.64 eV for CBM (higher than −4.44 eV) and −6.25 eV for VBM (lower than −5.67 eV), perfectly satisfying the band alignment requirement, i.e., the band positions of the 2D-P8N4 NS are favorable for water splitting. The energy difference (ΔE1) between CBM level and water reduction potential is 0.80 eV and the energy difference (ΔE2) between VBM level and water oxidation potential is 0.58 eV; the comparable energy differences provides comparable driving force for both water oxidation and water reduction.

Figure 6. The band position (a) and optical adsorption spectrum (b) of 2D-P8N4 obtained by HSE06.
Download figure:
Standard image High-resolution image3.2.2. Optical property
The ability to absorb light is also an important factor for photocatalyzing water splitting. The optical absorptions of the 2D-P8N4 NS is illustrated in figure 6(b), where the wavelength of visible light is labeled at 400 ∼ 800 nm (1.55 ∼ 3.1 eV). It is observed that the 2D-P8N4 NS exhibits adsorption starting at ∼1.5 eV, and the adsorption coefficient increases as the wavelength decreases. The peak of the absorption (7.43 × 105 cm−1) appears in the UV-light region at the wavelength of ∼167 nm. The peak of the absorption in visible light rang is 1.42 × 105 cm−1 at the wavelength of 400 nm. These results indicate that the 2D-P8N4 NS provide good optical absorption from the visible light to UV-light region.
3.2.3. Carrier mobility
The carrier mobility of the 2D-P8N4 NS was investigated along the transport directions (x = y), and the results are shown in table 2. The 2D-P8N4 NS is isotropic, so we only examined carrier mobility in x direction. The elastic modulus of 2D materials is calculated by changed energy under applied strain (figure S4(a)). The deformation potential constants of the 2D-P8N4 NS are obtained by the CBM and VBM positions under external strain (figure S4(b)). In table 3, at 300 K, the calculated mobility of the electrons for the 2D-P8N4 NS is 207.94 cm2V−1s−1 along transport direction, which is higher than the theoretical value (80 cm2V−1s−1) of monolayer BP along zigzag direction and lower than theoretical value (1140 cm2V−1s−1) along armchair direction [2]. The holes mobility of the 2D-P8N4 NS is 207.66 cm2V−1s−1, which is almost identical to the value of its electron mobility, but smaller than monolayer BP along armchair and zigzag direction (700 and 2600 cm2V−1s−1, respectively). In addition, the carrier mobility of the electrons for the 2D-P8N4 NS is higher than some 2D water-splitting photocatalysts, such as MoS2 (72.16/60.32 cm2V−1s−1 along armchair/zigzag direction) [55] and WS2 (104.46/142.30 cm2V−1s−1 along armchair/zigzag direction) [56].
Table 3. The calculated carrier effective masses m* (m0), deformation potential E1 (eV), in-plane stiffness C2D (N/m) and mobility μ (cm2V−1s−1) for electron (e) and hole (h) along the x and y directions in 2D-P8N4, monolayer BP [2], MoS2 [54], and WS2 [55] at 300 K.
| Carrier | mx * | my * | Elx | Ely | C2D_x | C2D_y | μx | μy | |
|---|---|---|---|---|---|---|---|---|---|
| e | 2D-P8N4 | 1.51 | 1.51 | 2.56 | 2.56 | 147.56 | 147.56 | 207.94 | 207.94 |
| BP | 0.17 | 1.13 | 5.02 | 7.35 | 57.48 | 194.62 | 1140 | 80 | |
| MoS2 | 0.46 | 0.48 | −10.88 | −11.36 | 127.44 | 128.16 | 72.16 | 60.32 | |
| WS2 | 0.33 | 0.31 | 13.10 | 11.90 | 136.14 | 144.44 | 104.46 | 142.30 | |
| h | 2D-P8N4 | 0.63 | 0.63 | 6.14 | 6.14 | 147.56 | 147.56 | 207.66 | 207.66 |
| BP | 0.15 | 6.35 | 2.50 | 0.15 | 28.94 | 101.60 | 700 | 26 000 | |
| MoS2 | 0.57 | 0.60 | −5.29 | −5.77 | 127.44 | 128.16 | 200.52 | 152.18 | |
| WS2 | 0.45 | 0.42 | 5.18 | 4.60 | 136.14 | 144.44 | 373.88 | 542.29 |
Note that, to characterize a suitable material for photocatalytic water splitting, the reaction barrier of hydrogen evolution reaction (HER) and oxygen evolution reaction (OER), and good resistance to photoinduced corrosion are also important [57–59], our identification of the carrier separation, optical adsorption and band alignment suggest that the assembled 2D-P8N4 NS may serve as a photocatalyst candidate for water splitting.
3.3. Transition metal atom Mn doped the assembled 1D-P8N2 NW
The magnetic and electronic properties of materials can be engineered by transition metal atom doping. We examined the magnetic and electronic properties of Mn doped 1D-P8N2 NW by adding one Mn atom per unitcell of 1D-P8N2 NW. Considering the structural symmetry in the 1D-P8N2 NW, we examined six adsorption sites: the middle site of two adjacent P8 (A, figure S5(a)), the hollow site of the pentagon formed between the N2 and P8 (B, figure S5(b)), the hollow site of the pentagon of the P8 (C, figure S5(c)), the bridge site of N2 (D, figure S5(d)), and two bridge sites of two P atoms (E, figure S5(e); F, figure S5(f)).
After geometry optimization (figure S6), one P–P bond was broken (elongated to be 4.34, 4.74 and 3.63 Å, respectively) at the A, E and F sites, the formed Mn–P bonds range from 2.32 to 2.73 Å. Unlike the case of the 1D-P10 NW doped by one Mn atom with no magnetism introduced [20], adding one Mn atom to the 1D-P8N2 NW (Mn1@1D-P8N2 NW) introduces a magnetic moment at each doping site (table S1). Doping Mn at D site has the highest adsorption energy (Eads
= 2.32 eV, table 4), very close to the Eads
(2.12 eV) of Mn at B site, due to the very similar optimized structures (figure S6). Here the adsorption energy is given by  where
where  and
and  are the energy of an isolated Mn atom and 1D-P8N2 NW unitcell, respectively,
are the energy of an isolated Mn atom and 1D-P8N2 NW unitcell, respectively,  is the total energy of one Mn adsorbed on 1D-P8N2 NW. Furthermore, the stability of the Mn1@1D-P8N2 NW was verified by a FPMD simulation at 300 K (figure S7): the P8 and N2 moieties were kept well at the end of 5 ps.
is the total energy of one Mn adsorbed on 1D-P8N2 NW. Furthermore, the stability of the Mn1@1D-P8N2 NW was verified by a FPMD simulation at 300 K (figure S7): the P8 and N2 moieties were kept well at the end of 5 ps.
Table 4. The Eads (eV) and magnetic configuration (MC: FM, AFM and NM denote ferromagnetic, antiferromagnetic and nonmagnetic properties, respectively) of Mn1@1D-P8N2 NW with Mn at the considered six sites, as well as the Ecoh (eV) of the three Mn bulk phases.
| A | B | C | D | E | F | |
|---|---|---|---|---|---|---|
| Eads | 1.07 | 2.12 | 0.56 | 2.32 | 0.71 | 0.39 |
| MC | AFM | FM | AFM | FM | AFM | FM |
We then studied the magnetic properties and band structure of Mn1@1D-P8N2 NW with Mn at D site. The Mn1@1D-P8N2 NW is ferromagnetic (FM) with the magnetic momentum of 4.92 μB on each Mn atom. The ferromagnetic state is 0.06 eV lower in energy than the antiferromagnetic (AFM) configuration (table S1), and the Curie temperature (TC) was estimated to be as high as 521 K according to the equation of  [60]. The easy axis (EA) of Mn1@1D-P8N2 NW is along the direction of the nanowire (010), and the magnetic anisotropy energy (MAE) is as large as 0.211 meV per Mn atom (table S2), much larger than those of traditional FM bulk metals, such as Fe (0.0014 meV/Fe), Co (0.065 meV/Co), and Ni (0.0027 meV/Ni) [61]. The band structure features with one Dirac cone existing between Г and X points in spin-up channel, and a direct bandgap (0.79 eV) in spin-down channel, as illustrated in figure 7(a). Both P-p and N-p orbitals contribute to the Dirac cone of spin-up channel and the VBM of spin-down channel, while CBM of spin-down channel is dominated by P-p orbitals according to figure 7(b). Thus, the Mn doped 1D-P8N2 NW is good diluted magnetic semiconductor (DMS) with the spin-flip bandgap of 0.38 eV calculated at HSE06 method. We obtain the Fermi velocity (
[60]. The easy axis (EA) of Mn1@1D-P8N2 NW is along the direction of the nanowire (010), and the magnetic anisotropy energy (MAE) is as large as 0.211 meV per Mn atom (table S2), much larger than those of traditional FM bulk metals, such as Fe (0.0014 meV/Fe), Co (0.065 meV/Co), and Ni (0.0027 meV/Ni) [61]. The band structure features with one Dirac cone existing between Г and X points in spin-up channel, and a direct bandgap (0.79 eV) in spin-down channel, as illustrated in figure 7(a). Both P-p and N-p orbitals contribute to the Dirac cone of spin-up channel and the VBM of spin-down channel, while CBM of spin-down channel is dominated by P-p orbitals according to figure 7(b). Thus, the Mn doped 1D-P8N2 NW is good diluted magnetic semiconductor (DMS) with the spin-flip bandgap of 0.38 eV calculated at HSE06 method. We obtain the Fermi velocity ( ) of the Dirac Fermions in Mn1@1D-P8N2 NW via linear fitting the band. The Fermi velocity is about 5.86 × 105 m s−1 along k+
direction and 3.96 × 105 m s−1 along k-
direction, of the same order to Vf
of graphene (8.0 × 105 m s−1) [62]. The direction-dependent and high Fermi velocity indicates its great potential for direction-dependent quantum information devices.
) of the Dirac Fermions in Mn1@1D-P8N2 NW via linear fitting the band. The Fermi velocity is about 5.86 × 105 m s−1 along k+
direction and 3.96 × 105 m s−1 along k-
direction, of the same order to Vf
of graphene (8.0 × 105 m s−1) [62]. The direction-dependent and high Fermi velocity indicates its great potential for direction-dependent quantum information devices.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 7. The HSE06 band structure (a) and density of states (b) of Mn1@1D-P8N2 NW. The Fermi level is set at 0 eV, and the band curves of spin-up and spin-down channels were highlighted in red and blue, respectively.
Download figure:
Standard image High-resolution image{kind=link}
In order to investigate the inter-chain magnetic coupling of Mn1@1D-P8N2 NWs, we designed two species of van der Waals (vdW)-type 2D sheets (figure S8), namely, 2D-Mn1@P8N2-I and 2D-Mn1@P8N2-II, by placing Mn1@1D-P8N2 NWs repeatedly with the separation of 3.72 Å and alternatively with N 'connecting' to P and the separation of 3.28 Å, respectively. The 2D-Mn1@P8N2-II is slightly energy-preferred by 0.11 eV per Mn1@P8N2 unit, and the ferromagnetic ordering is also energetically favored by 0.05 eV per unitcell with each Mn atom carrying magnetic momentum of 4.98 μB, and the TC is estimated to be 426 K. Compared with the isolated Mn1@1D-P8N2 NW, the EA of 2D-Mn1@P8N2-II sheet remains the direction of the nanowire (010), while the MAE is lowered (0.135 versus 0.211 meV per Mn atom, table S2). To save computational cost, we employed PBE + U (U = 4 eV) method to evaluate the electronic band structure of 2D-Mn1@P8N2-II sheet, and our computations showed that the vdW-type 2D sheet is a metal with one Dirac cone remaining in spin-up channel, which is also contributed by the P-p and N-p orbitals (figure S9). The Fermi velocity is estimated to be 1.46 × 105 m s−1 along kx direction and 0.41 × 105 m s−1 along ky direction, smaller than the Vf values (1.58 × 105 and 1.06 × 105 m s−1 calculated at PBE + U level, figure S10) of isolated Mn1@1D-P8N2 chain.
4. Conclusions
In summary, based on cluster assembling method, we successfully designed 1D-P8N2 NW and 2D-P8N4 NS by using P8 and N2 as building blocks, both the two assembled nanomaterials exhibit extraordinary stability according to our first principles calculations: high cohesive energies (higher than that of black phosphene), almost no imaginary frequencies, structural integrity through a 5 ps molecular dynamics simulation at 500 K, high O2 dissociation energy barriers (1.51 and 1.24 eV on 1D-P8N2 NW and 2D-P8N4 NS, respectively) and O–O bond integrity upon adsorption through a 5 ps molecular dynamics simulation at 500 K. Both materials are semiconductors. The bandgap (2.61 eV) and band edge of 2D-P8N4 NS perfectly satisfy the requirement of a good photocatalyst for water splitting, the different spatial distributions of CBM and VBM, and the comparable ΔE1 and ΔE2, the equivalent electron and hole mobility, and the optical adsorption in visible and UV light region, together endow the 2D-P8N4 NS a potential photocatalyst candidate for water splitting with high efficiency. The indirect-bandgap semiconducting 1D-P8N2 NW (0.76 eV) was converted into a ferromagnetic half-metal by Mn doping, the Curie temperature of the Mn1@1D-P8N2 NW is estimated to be 521 K, the magnetic anisotropy energy (MAE) is as high as 0.211 meV per Mn atom, and its Fermi velocity at the Dirac cone in spin-up channel has the same order of graphene's. The vdW-type 2D-Mn1@P8N2-II sheet is a ferromagnetic metal also with one Dirac cone in spin-up channel.
Our comprehensive studies provide a novel approach to obtain stable low-dimensional phosphorus-based nanomaterials with various electronic and magnetic properties, which are expected to serve as high efficient photocatalyst for water splitting, nanoelectronic devices and spintronics.
Acknowledgments
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (11964022, 11964024, 91961204) and the Startup Project of Inner Mongolia University (21200-5175101). We also acknowledge the helpful discussion from Ms. Haihong Meng and Miss Bing Han.
Data availability statement
The data that support the findings of this study are available upon reasonable request from the authors.
Notes
The authors declare no competing financial interest.