Whole-Genome DNA Methylation Analysis in Hydrogen Peroxide Overproducing Transgenic Tobacco Resistant to Biotic and Abiotic Stresses

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Phenotypic Characteristics
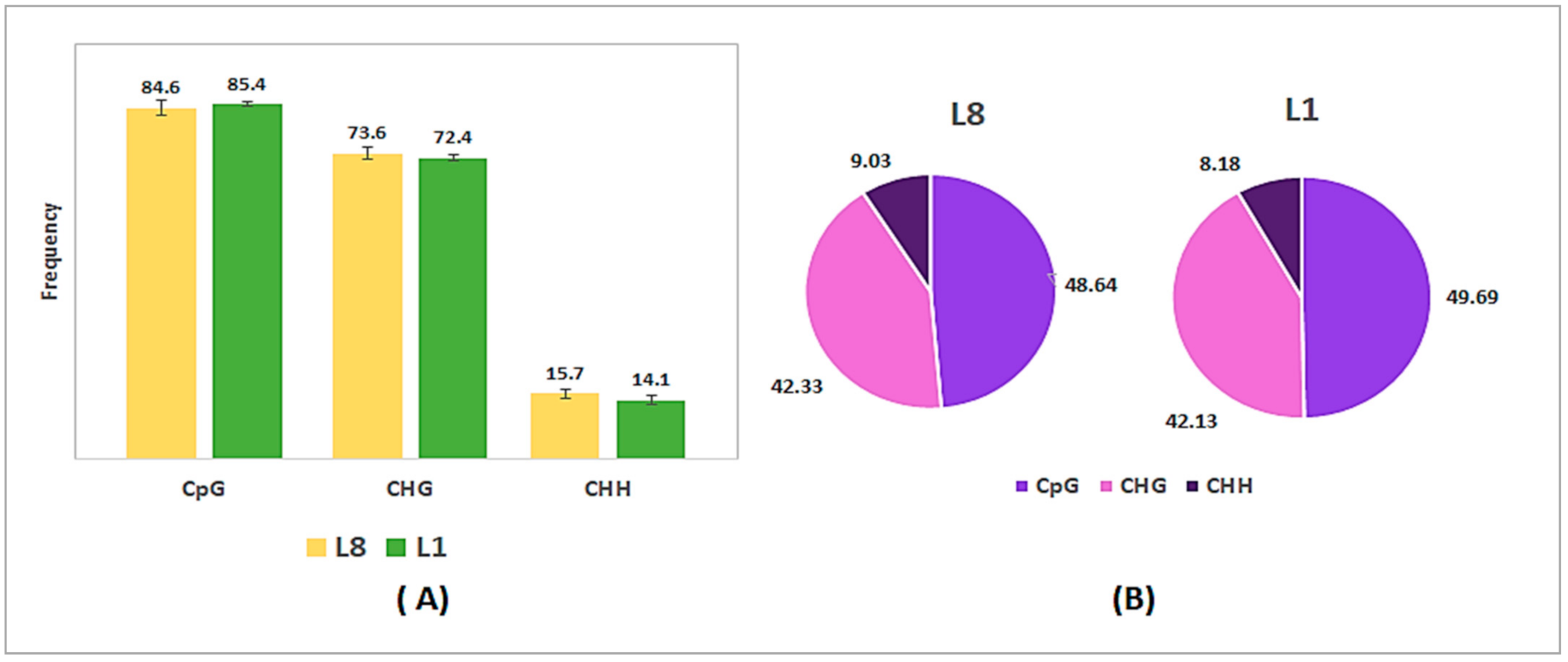
2.2. DNA Methylation Levels and Distribution
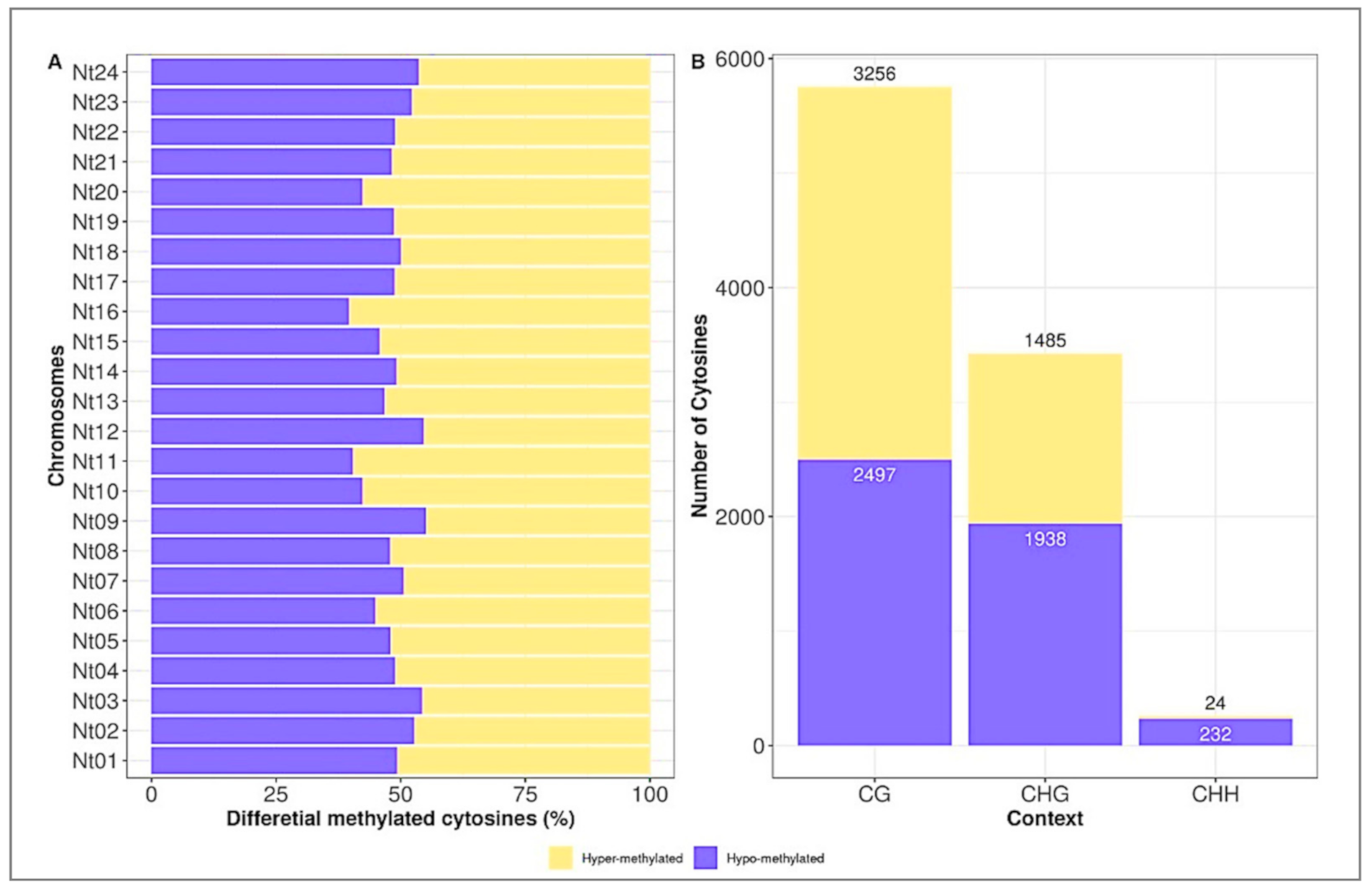
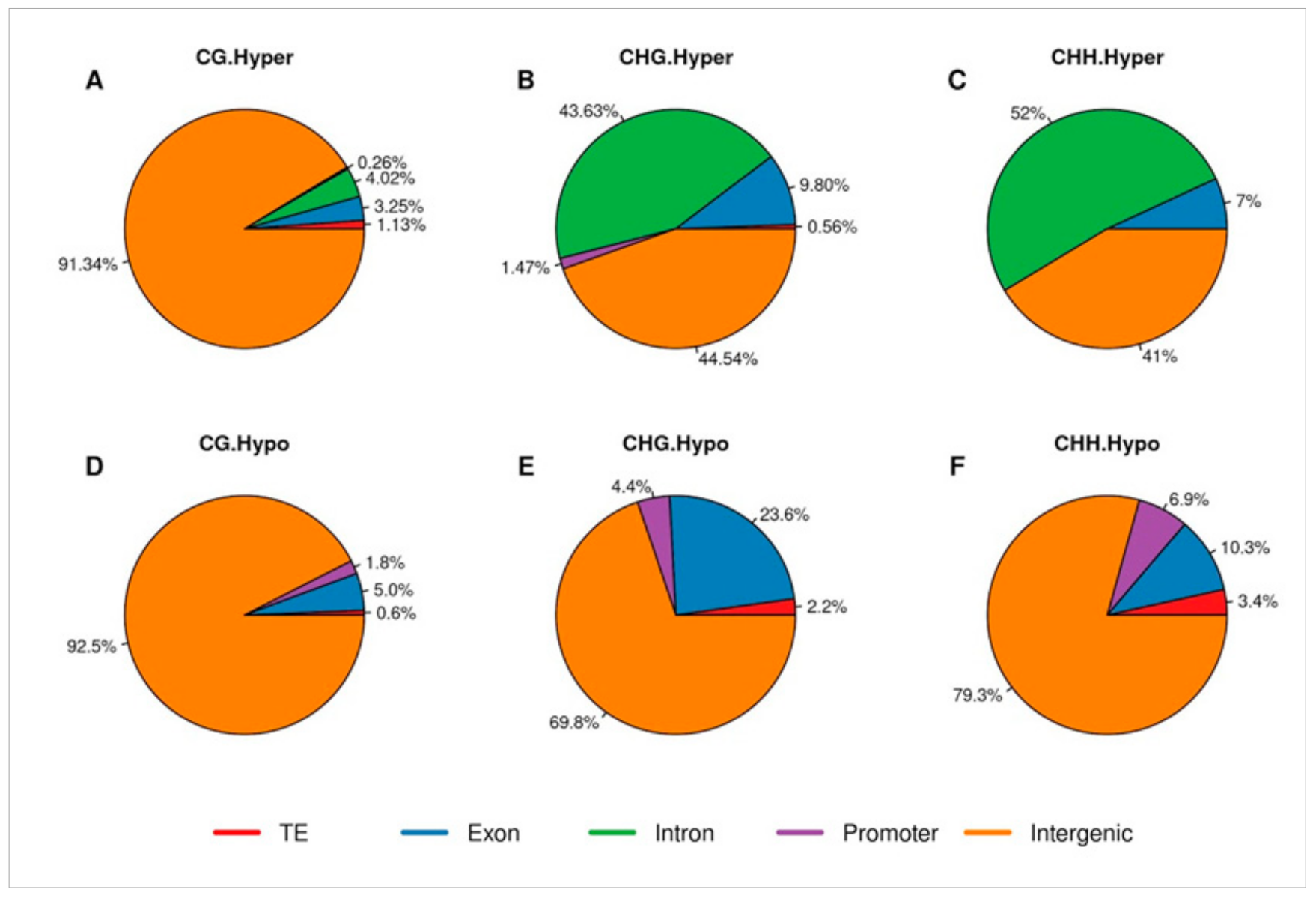
2.3. Distribution of Differentially Methylated Cytosines
2.4. Gene Ontology Analysis
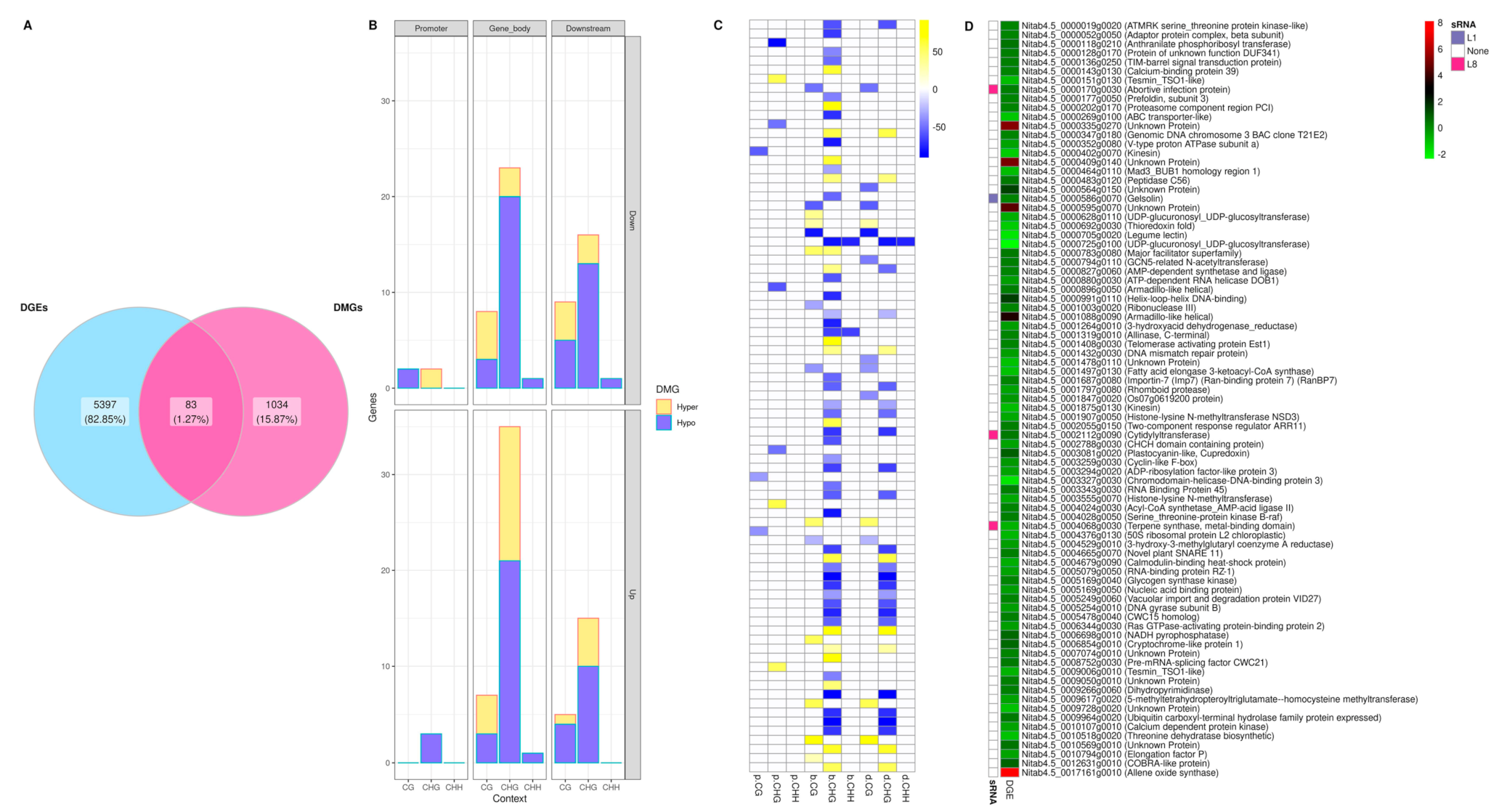
2.5. Correlation of Differentially Methylated Cytosines and Differential Gene Expression
3. Discussion
4. Materials and Methods
4.1. Transgenic Plants Growth Conditions
4.2. Tissue Sampling and Physiological Testing
4.3. DNA Extraction
4.4. WGBS
4.5. Data Processing
4.6. Correlation between DmC and DEGs
4.7. Statistics and Data Visualization
4.8. Data Availability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kapazoglou, A.; Ganopoulos, I.; Tani, E.; Tsaftaris, A. Epigenetics, Epigenomics and Crop Improvement. Adv. Bot. Res. 2018, 86, 287–324. [Google Scholar] [CrossRef]
- Lämke, J.; Bäurle, I. Epigenetic and chromatin-based mechanisms in environmental stress adaptation and stress memory in plants. Genome Biol. 2017, 18, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Balao, F.; Paun, O.; Alonso, C. Uncovering the contribution of epigenetics to plant phenotypic variation in Mediterranean ecosystems. Plant Biol. 2018, 20, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Avramova, Z. Transcriptional ‘memory’ of a stress: Transient chromatin and memory (epigenetic) marks at stress-response genes. Plant J. 2015, 83, 149–159. [Google Scholar] [CrossRef]
- Karanthamalai, J.; Chodon, A.; Chauhan, S.; Pandi, G. DNA N6-Methyladenine Modification in Plant Genomes—A Glimpse into Emerging Epigenetic Code. Plants 2020, 9, 247. [Google Scholar] [CrossRef] [Green Version]
- Manova, V.; Gruszka, D. DNA damage and repair in plants—From models to crops. Front. Plant Sci. 2015, 6, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Yaish, M.W.; Al-Lawati, A.; Al-Harrasi, I.; Patankar, H.V. Genome-wide DNA Methylation analysis in response to salinity in the model plant caliph medic (Medicago truncatula). BMC Genom. 2018, 19, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Asensi-Fabado, M.-A.; Amtmann, A.; Perrella, G. Plant responses to abiotic stress: The chromatin context of transcriptional regulation. Biochim. Biophys. Acta (BBA) Bioenerg. 2017, 1860, 106–122. [Google Scholar] [CrossRef] [Green Version]
- Holeski, L.M.; Jander, G.; Agrawal, A.A. Transgenerational defense induction and epigenetic inheritance in plants. Trends Ecol. Evol. 2012, 27, 618–626. [Google Scholar] [CrossRef]
- Pastor, V.; Luna, E.J.; Mauch-Mani, B.; Ton, J.; Flors, V. Primed plants do not forget. Environ. Exp. Bot. 2013, 94, 46–56. [Google Scholar] [CrossRef]
- Gallusci, P.; Dai, Z.; Génard, M.; Gauffretau, A.; Leblanc-Fournier, N.; Richard-Molard, C.; Vile, D.; Brunel-Muguet, S. Epigenetics for Plant Improvement: Current Knowledge and Modeling Avenues. Trends Plant Sci. 2017, 22, 610–623. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Grewal, A.; Sharma, P. Comparative analysis of DNA methylation changes in two contrasting wheat genotypes under water deficit. Biol. Plant 2018, 62, 471–478. [Google Scholar] [CrossRef]
- Bilichak, A.; Ilnystkyy, Y.; Hollunder, J.; Kovalchuk, I. The Progeny of Arabidopsis thaliana Plants Exposed to Salt Exhibit Changes in DNA Methylation, Histone Modifications and Gene Expression. PLoS ONE 2012, 7, e30515. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Wang, Q.; Huang, X. Chilling-induced DNA Demethylation is associated with the cold tolerance of Hevea brasili-ensis. BMC Plant Biol. 2018, 18, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, C.; Chi, J.; Zhao, Y.; He, Y.; Guo, J. Cadmium stress alters cytosine methylation status and expression of a select set of genes in Nicotiana benthamiana. Plant Sci. 2019, 284, 16–24. [Google Scholar] [CrossRef]
- Mager, S.; Ludewig, U. Massive Loss of DNA Methylation in Nitrogen-, but Not in Phosphorus-Deficient Zea mays Roots Is Poorly Correlated with Gene Expression Differences. Front. Plant Sci. 2018, 9, 497. [Google Scholar] [CrossRef] [Green Version]
- Kou, H.P.; Li, Y.; Song, X.X.; Ou, X.F.; Xing, S.C.; Ma, J.; von Wettstein, D.; Liu, B. Heritable alteration in DNA methylation induced by nitrogen-deficiency stress accompanies enhanced tolerance by progenies to the stress in rice (Oryza sativa L.). J. Plant Physiol. 2011, 168, 1685–1693. [Google Scholar] [CrossRef]
- Lewandowska-Gnatowska, E.; Polkowska-Kowalczyk, L.; Szczegielniak, J.; Barciszewska, M.; Barciszewski, J.; Muszyńska, G. Is DNA methylation modulated by wounding-induced oxidative burst in maize? Plant Physiol. Biochem. 2014, 82, 202–208. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Wang, C.; Xu, W.; Zou, J.; Qiu, Y.; Kong, J.; Yang, Y.; Zhang, B.; Zhu, S. Epigenetic Changes in the Regulation of Nicotiana tabacum Response to Cucumber Mosaic Virus Infection and Symptom Recovery through Single-Base Resolution Methylomes. Viruses 2018, 10, 402. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.-Y.; Pan, X.-L.; Peng, T.-C.; Chen, Y.-Y.; Zhao, H.; Mu, L.; Peng, Y.; He, R.; Tang, H. DNA methylation patterns of banana leaves in response to Fusarium oxysporum f. sp. cubense tropical race 4. J. Integr. Agric. 2016, 15, 2736. [Google Scholar] [CrossRef]
- Zhou, M.; Sng, N.J.; Le Frois, C.; Paul, A.-L.; Ferl, R.J. Epigenomics in an extraterrestrial environment: Organ-specific alteration of DNA methylation and gene expression elicited by spaceflight in Arabidopsis thaliana. BMC Genom. 2019, 20, 205. [Google Scholar] [CrossRef] [PubMed]
- Kuźnicki, D.; Meller, B.; Arasimowicz-Jelonek, M.; Brąszewska, A.; Drozda, A.; Floryszak-Wieczorek, J. BABA-Induced DNA Methylome Adjustment to Intergenerational Defense Priming in Potato to Phytophthora infestans. Front. Plant Sci. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Bertini, L.; Proietti, S.; Focaracci, F.; Sabatini, B.; Caruso, C. Epigenetic control of defense genes following MeJA-induced priming in rice. J. Plant Physiol. 2018, 228, 166–177. [Google Scholar]
- Smirnoff, N.; Arnaud, D. Hydrogen peroxide metabolism and functions in plants. New Phytol. 2018, 221, 1197–1214. [Google Scholar] [CrossRef]
- Mittler, R. ROS Are Good. Trends Plant Sci. 2017, 22, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Qi, J.; Wang, J.; Gong, Z.; Zhou, J.M. Apoplastic ROS signaling in plant immunity. Curr. Opin. Plant Biol. 2017, 38, 92–100. [Google Scholar] [CrossRef]
- Hossain, M.A.; Bhattacharjee, S.; Armin, S.-M.; Qian, P.; Xin, W.; Li, H.-Y.; Burritt, D.J.; Fujita, M.; Tran, L.P. Hydrogen peroxide priming modulates abiotic oxidative stress tolerance: Insights from ROS detoxification and scavenging. Front. Plant Sci. 2015, 6, 420. [Google Scholar] [CrossRef] [Green Version]
- Wahid, A.; Perveen, M.; Gelani, S.; Basra, S.M. Pretreatment of seed with H2O2 improves salt tolerance of wheat seedlings by alleviation of oxidative damage and expression of stress proteins. J. Plant Physiol. 2007, 164, 283–294. [Google Scholar] [CrossRef]
- Christou, A.; Manganaris, G.A.; Fotopoulos, V. Systemic mitigation of salt stress by hydrogen peroxide and sodium nitroprus-side in strawberry plants via transcriptional regulation of enzymatic and non-enzymatic antioxidants. Environ Exp Bot. 2014, 107, 46–54. [Google Scholar] [CrossRef]
- Ellouzi, H.; Sghayar, S.; Abdelly, C. H2O2 seed priming improves tolerance to salinity; drought and their combined effect more than mannitol in Cakile maritima when compared to Eutrema salsugineum. J. Plant Physiol. 2017, 210, 38–50. [Google Scholar] [CrossRef]
- Bagheri, M.; Gholami, M.; Baninasab, B. Hydrogen peroxide-induced salt tolerance in relation to antioxidant systems in pis-tachio seedlings. Sci. Hortic. 2019, 243, 207–213. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, H.; Liu, S.; Peng, X. Exogenous application of hydrogen peroxide alleviates drought stress in cucumber seedlings. S. Afr. J. Bot. 2016, 106, 23–28. [Google Scholar] [CrossRef]
- Ishibashi, Y.; Yamaguchi, H.; Yuasa, T.; Iwaya-Inoue, M.; Arima, S.; Zheng, S.-H. Hydrogen peroxide spraying alleviates drought stress in soybean plants. J. Plant Physiol. 2011, 168, 1562–1567. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.-Y.; Gao, Y.; Sun, W.-J.; Huang, Y.-W.; Zhang, J.; Bai, J.-G. Role of hydrogen peroxide pretreatment in heat-induced alteration of DNA methylation in cucumber leaves. Sci. Hortic. 2013, 151, 173–183. [Google Scholar] [CrossRef]
- Gao, Y.; Guo, Y.-K.; Lin, S.-H.; Fang, Y.-Y.; Bai, J.-G. Hydrogen peroxide pretreatment alters the activity of antioxidant enzymes and protects chloroplast ultrastructure in heat-stressed cucumber leaves. Sci. Hortic. 2010, 126, 20–26. [Google Scholar] [CrossRef]
- Andrade, C.A.; de Souza, K.R.D.; Santos, M.O.; Silva, D.M.; Alves, J.D. Hydrogen peroxide promotes the tolerance of soybeans to waterlogging. Sci. Hortic. 2018, 232, 40–45. [Google Scholar] [CrossRef]
- Mejía-Teniente, L.; Durán-Flores, B.A.; Torres-Pacheco, I.; Gonzalez-Chavira, M.M.; Rivera-Bustamante, R.; Feregrino-Perez, A.A.; Pérez-Ramírez, I.; Rocha-Guzmán, N.E.; Reynoso-Camacho, R.; Guevara-Gonzalez, R.G. Hydrogen peroxide protects pepper (Capsicum annuum L.) against pepper golden mosaic geminivirus (PepGMV) infections. Physiol. Mol. Plant Pathol. 2019, 106, 23–29. [Google Scholar] [CrossRef]
- Vargas-Hernández, M.; Torres-Pacheco, I.; Gautier, F.; Álvarez-Mayorga, B.; Cruz-Hernández, A.; García-Mier, L.; Jiménez-García, S.N.; Ocampo-Velázquez, R.V.; Feregrino-Perez, A.A.; Guevara-Gonzalez, R.G. Influence of hydrogen peroxide foliar applications on in vitro antimicrobial activity in Capsicum chinense Jacq. Plant Biosyst. Int. J. Deal. all Asp. Plant. Biol. 2016, 151, 269–275. [Google Scholar] [CrossRef]
- Zhang, X.-L.; Jia, X.-F.; Yu, B.; Gao, Y.; Bai, J.-G. Exogenous hydrogen peroxide influences antioxidant enzyme activity and lipid peroxidation in cucumber leaves at low light. Sci. Hortic. 2011, 129, 656–662. [Google Scholar] [CrossRef]
- Godínez-Hernández, Y.; Anaya-López, J.L.; Díaz-Plaza, R.; González-Chavira, M.; Torres-Pacheco, I.; Rivera-Bustamante, R.F.; Guevara-González, R.G. Characterization of resistance to pepper huasteco geminivirus in chili peppers from Yucatán, México. Hort. Sci. 2001, 36, 139–142. [Google Scholar]
- Guevara-Olvera, L.; Ruíz-Nito, M.; Rangel-Cano, R.; Torres-Pacheco, I.; Rivera-Bustamante, R.; Muñoz-Sánchez, C.; González-Chavira, M.; Cruz-Hernandez, A.; Guevara-Gonzalez, R.; Rivera-Bustamante, R. Expression of a germin-like protein gene (CchGLP) from a geminivirus-resistant pepper (Capsicum chinense Jacq.) enhances tolerance to geminivirus infection in transgenic tobacco. Physiol. Mol. Plant Pathol. 2012, 78, 45–50. [Google Scholar] [CrossRef]
- Cardenas-Manríquez, G.; Cruz-Hernandez, A.; Torres-Pacheco, I.; Caballero-Pérez, J.; González-Chavira, M.M.; García-Ortega, L.F.; Guevara-González, R.G. Transcriptome profiling of transgenic tobacco (Nicotiana tabacum cv. xanthi nc) expressing CchGLP gene from Capsicum chinense Jacq. reveals gene expression associated with stress tolerance. J. Hortic. Sci. Biotechnol. 2018, 93, 595–604. [Google Scholar] [CrossRef]
- Sáenz, O.D.; Cedillo-Jimenez, C.A.; García-Ortega, L.F.; Martínez-Reséndiz, M.; Arné-Robles, D.; Cruz-Hernandez, A.; Guevara-Gonzalez, R.G. Response of transgenic tobacco overexpressing the CchGLP gene to cadmium and aluminium: Phenotypic and microRNAs expression changes. Physiol. Mol. Biol. Plants 2020, 26, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Cárdenas-Manríquez, G.; Vega-Muñoz, I.; Villagómez-Aranda, A.; León-Galvan, M.; Cruz-Hernandez, A.; Torres-Pacheco, I.; Rangel-Cano, R.; Rivera-Bustamante, R.; Guevara-Gonzalez, R.G.; Rivera-Bustamante, R. Proteomic and metabolomic profiles in transgenic tobacco (N. tabacum xanthi nc) to CchGLP from Capsicum chinense BG-3821 resistant to biotic and abiotic stresses. Environ. Exp. Bot. 2016, 130, 33–41. [Google Scholar] [CrossRef]
- Cárdenas-Manríquez, G.; Robles-Bustos, D.A.; Vega-Muñoz, I.; Villagómez-Aranda, A.L.; Torres-Pacheco, I.; Guevara-Gonzalez, R.G. Determination of molecular communication network in transgenic tobbaco expressing CchGLP gene. In Proceedings of the 2017 XIII International Engineering Congress (CONIIN), Santiago de Queretaro, Mexico, 15–19 May 2017; pp. 1–9. [Google Scholar] [CrossRef]
- Locato, V.; Cimini, S.; De Gara, L. ROS and redox balance as multifaceted players of cross-tolerance: Epigenetic and retrograde control of gene expression. J. Exp. Bot. 2018, 69, 3373–3391. [Google Scholar] [CrossRef]
- Saravana Kumar, R.M.; Wang, Y.; Zhang, X.; Cheng, H.; Sun, L.; He, S.; Hao, F. Redox components: Key regulators of epigenetic modifications in plants. Int. J. Mol. Sci. 2020, 21, 1419. [Google Scholar]
- Li, R.; Hu, F.; Li, B.; Zhang, Y.; Chen, M.; Fan, T.; Wang, T. Whole genome bisulfite sequencing methylome analysis of mulberry (Morus alba) reveals epigenome modifications in response to drought stress. Sci. Rep. 2020, 10, 1–17. [Google Scholar] [CrossRef]
- Sun, Y.; Fan, M.; He, Y. DNA Methylation Analysis of the Citrullus lanatus Response to Cucumber Green Mottle Mosaic Virus Infection by Whole-Genome Bisulfite Sequencing. Genes 2019, 10, 344. [Google Scholar] [CrossRef] [Green Version]
- Fan, S.K.; Ye, J.Y.; Zhang, L.L.; Chen, H.S.; Zhang, H.H.; Zhu, Y.X.; Liu, X.X.; Jin, C.W. Inhibition of DNA demethylation enhances plant tolerance to cadmium toxicity by improving iron nutrition. Plant Cell Environ. 2019, 43, 275–291. [Google Scholar] [CrossRef]
- Ji, L.; Mathioni, S.M.; Johnson, S.; Tucker, D.; Bewick, A.J.; Kim, K.D.; Daron, J.; Slotkin, R.K.A.; Jackson, S.; Parrott, W.; et al. Genome-Wide Reinforcement of DNA Methylation Occurs during Somatic Embryogenesis in Soybean. Plant Cell 2019, 31, 2315–2331. [Google Scholar] [CrossRef]
- Zuo, J.; Wang, Y.; Zhu, B.; Luo, Y.; Wang, Q.; Gao, L. Comparative Analysis of DNA Methylation Reveals Specific Regulations on Ethylene Pathway in Tomato Fruit. Genes 2018, 9, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manoharlal, R.; Saiprasad, G.V.S.; Kaikala, V.; Kumar, R.S.; Kovarik, A. Analysis of DNA methylome and transcriptome profiling following Gibberellin A3 (GA3) foliar application in Nicotiana tabacum L. Indian J. Plant Physiol. 2018, 23, 543–556. [Google Scholar] [CrossRef]
- Konate, M.; Wilkinson, M.J.; Mayne, B.T.; Pederson, S.M.; Scott, E.S.; Berger, B.; Lopez, C.R. Salt Stress Induces Non-CG Methylation in Coding Regions of Barley Seedlings (Hordeum vulgare). Epigenomes 2018, 2, 12. [Google Scholar] [CrossRef] [Green Version]
- Gallego-Bartolomé, J. DNA methylation in plants: Mechanisms and tools for targeted manipulation. New Phytol. 2020, 227, 38–44. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Meng, X.; Dobrovolskaya, O.B.; Orlov, Y.L.; Chen, M. Non-coding RNAs and Their Roles in Stress Response in Plants. Genom. Proteom. Bioinform. 2017, 15, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Sano, H.; Choi, S.C. Abiotic-stress induces demethylation and transcriptional activation of a gene encoding a glycerophosphodiesterase-like protein in tobacco plants. Mol. Genet. Genom. 2007, 277, 589–600. [Google Scholar]
- Poborilova, Z.; Ohlsson, A.B.; Berglund, T.; Vildova, A.; Provaznik, I.; Babula, P. DNA hypomethylation concomitant with the overproduction of ROS induced by naphthoquinone juglone on tobacco BY-2 suspension cells. Environ. Exp. Bot. 2015, 113, 28–39. [Google Scholar] [CrossRef]
- Niu, L.; Liao, W. Hydrogen Peroxide Signaling in Plant Development and Abiotic Responses: Crosstalk with Nitric Oxide and Calcium. Front. Plant Sci. 2016, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Habibi, G. Hydrogen Peroxide (H2O2) Generation, Scavenging and Signaling in Plants. Oxid. Damage Plants 2014, 557–584. [Google Scholar]
- Smékalová, V.; Doskočilová, A.; Komis, G.; Šamaj, J. Crosstalk between secondary messengers, hormones and MAPK modules during abiotic stress signalling in plants. Biotechnol. Adv. 2014, 32, 2–11. [Google Scholar] [CrossRef]
- Atighi, M.R.; Verstraeten, B.; De Meyer, T.; Kyndt, T. Genome-wide DNA hypomethylation shapes nematode pattern-triggered immunity in plants. New Phytol. 2020, 227, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Project, G.; et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef] [Green Version]
- Ihaka, R.; Gentleman, R. R: A Language for Data Analysis and Graphics. J. Comput. Graph. Stat. 1996, 5, 299–314. [Google Scholar]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | L8 (6 Weeks) | L8 (3 Months) | L1 (6 Weeks) | L1 (3 Months) |
|---|---|---|---|---|
| Height (cm) | 6.4 ± 0.51 | 72 ± 3.46 | 7.1 ± 0.24 | 74 ± 1.0 |
| Fresh weight leaves (g) | 0.55 ± 0.03 | 41.6 ± 4.19 | 0.57 ± 0.02 | 56.9 ± 3.43 |
| Fresh weight root (g) | 0.12 ± 0.02 | 25.9± 5.23 | 0.13 ± 0.01 | 28.2 ± 2.68 |
| Stem width (mm) | 1.54 ± 0.11 | 11.3 ± 0.12 | 1.52 ± 0.14 | 10.8 ± 0.22 |
| Leaf area (cm2) | 47.5 ± 3.7 | 1775 ± 313 | 49.1 ± 2.9 | 2607 ± 134 |
| SPAD value | 39.4 ± 2.09 | 46.7 ± 2.24 | 38.2 ± 2.14 | 44.8 ± 2.55 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villagómez-Aranda, A.L.; García-Ortega, L.F.; Torres-Pacheco, I.; Guevara-González, R.G. Whole-Genome DNA Methylation Analysis in Hydrogen Peroxide Overproducing Transgenic Tobacco Resistant to Biotic and Abiotic Stresses. Plants 2021, 10, 178. https://doi.org/10.3390/plants10010178
Villagómez-Aranda AL, García-Ortega LF, Torres-Pacheco I, Guevara-González RG. Whole-Genome DNA Methylation Analysis in Hydrogen Peroxide Overproducing Transgenic Tobacco Resistant to Biotic and Abiotic Stresses. Plants. 2021; 10(1):178. https://doi.org/10.3390/plants10010178
Chicago/Turabian StyleVillagómez-Aranda, Ana L., Luis F. García-Ortega, Irineo Torres-Pacheco, and Ramón G. Guevara-González. 2021. "Whole-Genome DNA Methylation Analysis in Hydrogen Peroxide Overproducing Transgenic Tobacco Resistant to Biotic and Abiotic Stresses" Plants 10, no. 1: 178. https://doi.org/10.3390/plants10010178