Modeling the Influenza A NP-vRNA-Polymerase Complex in Atomic Detail

 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Modeling of the vRNA Phosphate-Ribose Backbone
2.2. Homology Modeling Using a Motif-Matching Fragment Assembly Method (MMFA)
2.3. Structure Alignment and Superposition
2.4. Modeling of Protein Loop Structure
2.5. Structural Refinement
2.6. Graphics
3. Results
3.1. Modeling of RNA and Nucleoprotein
3.1.1. All-Atom Model of a Single-Stranded RNA from a Low-Resolution Structure
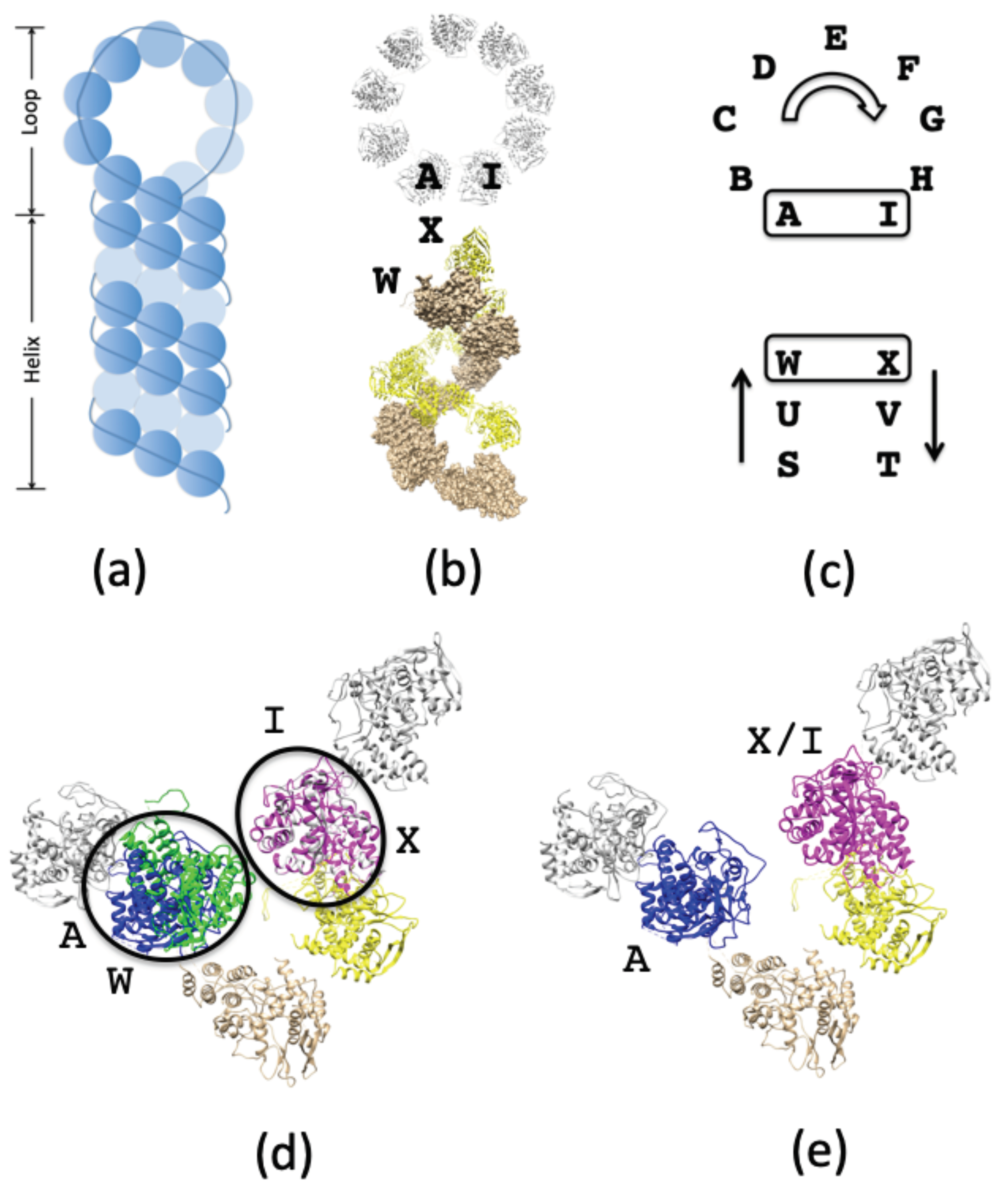
3.1.2. Connecting NPs between RNP Helical and Circular Regions
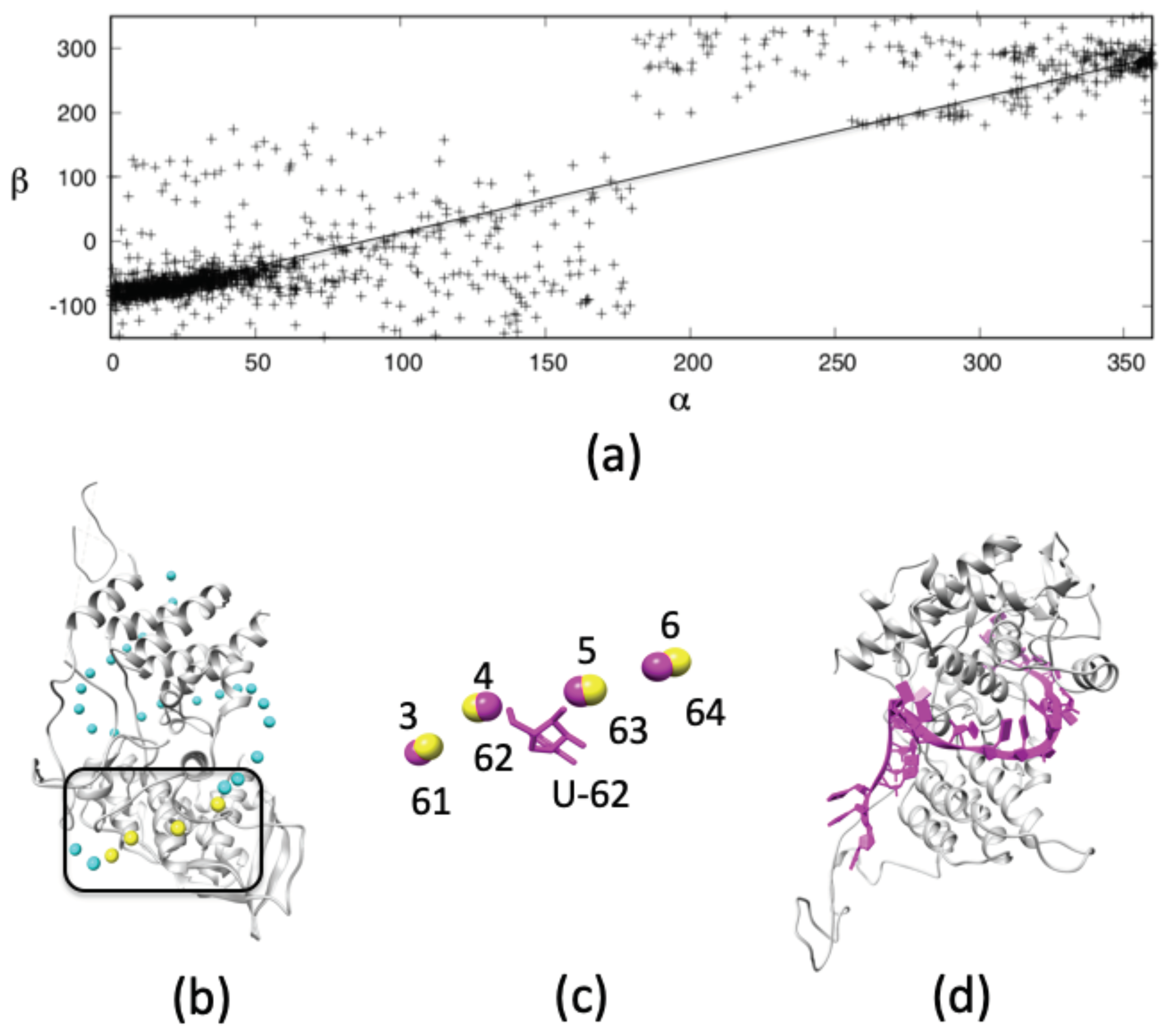
3.1.3. Structure of RNA Loops between NP-Bound vRNA
3.1.4. Model of NP-RNA Complex
3.2. Modeling the NP—vRNA—POL Complex
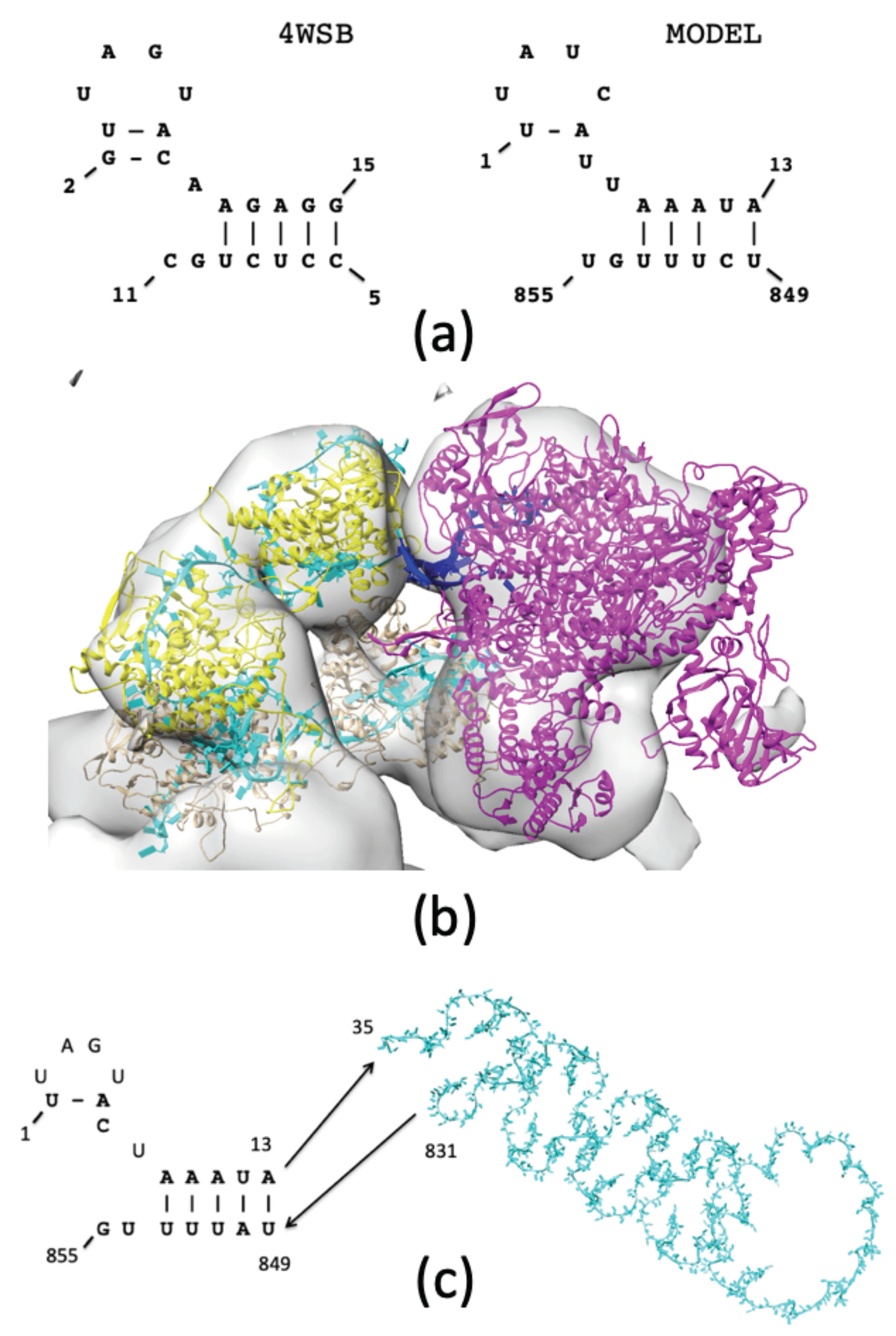
3.2.1. Selecting a Binding Mode for POL to the NP—RNA Complex
3.2.2. POL Binding to Double-Stranded RNA
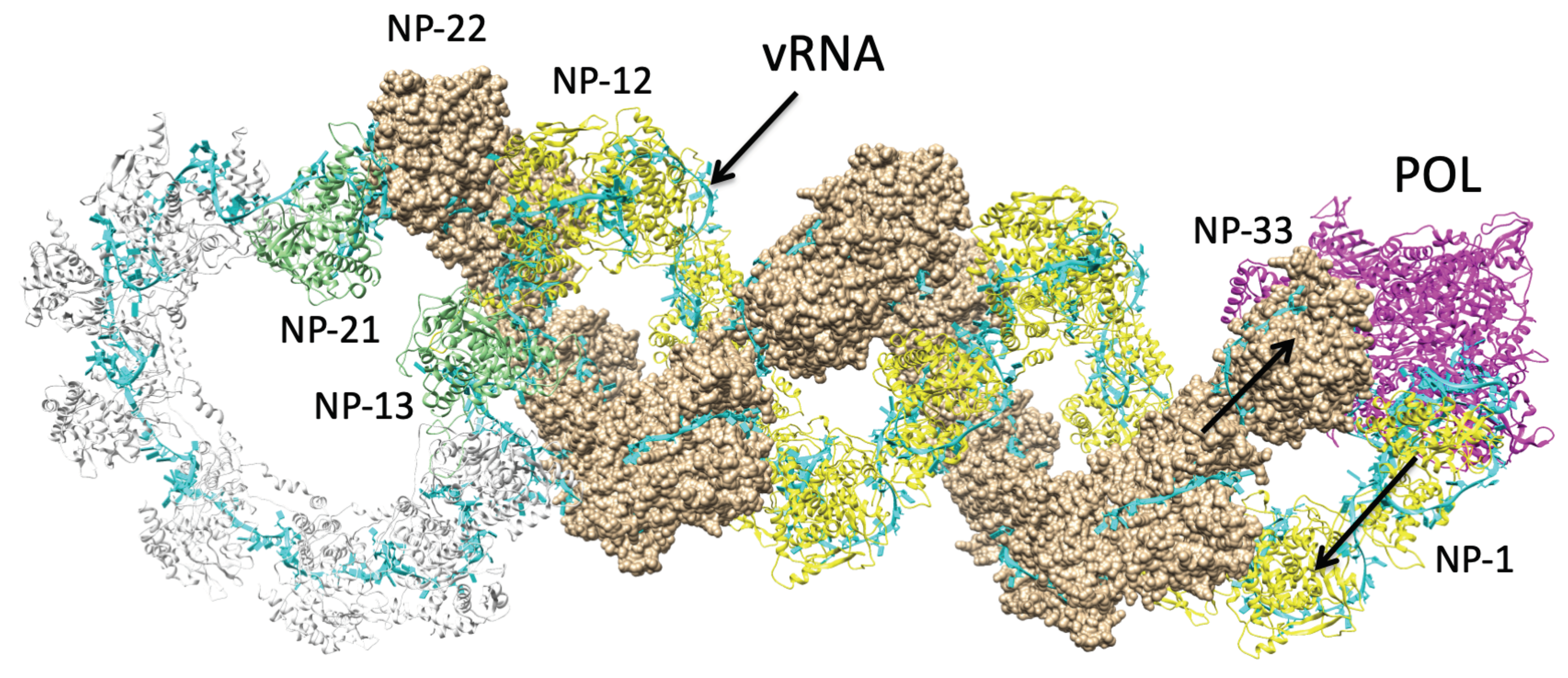
3.2.3. Structure of NP—vRNA—POL
3.3. Determining Structure-Function Relations of the Influenza A RNP Complex
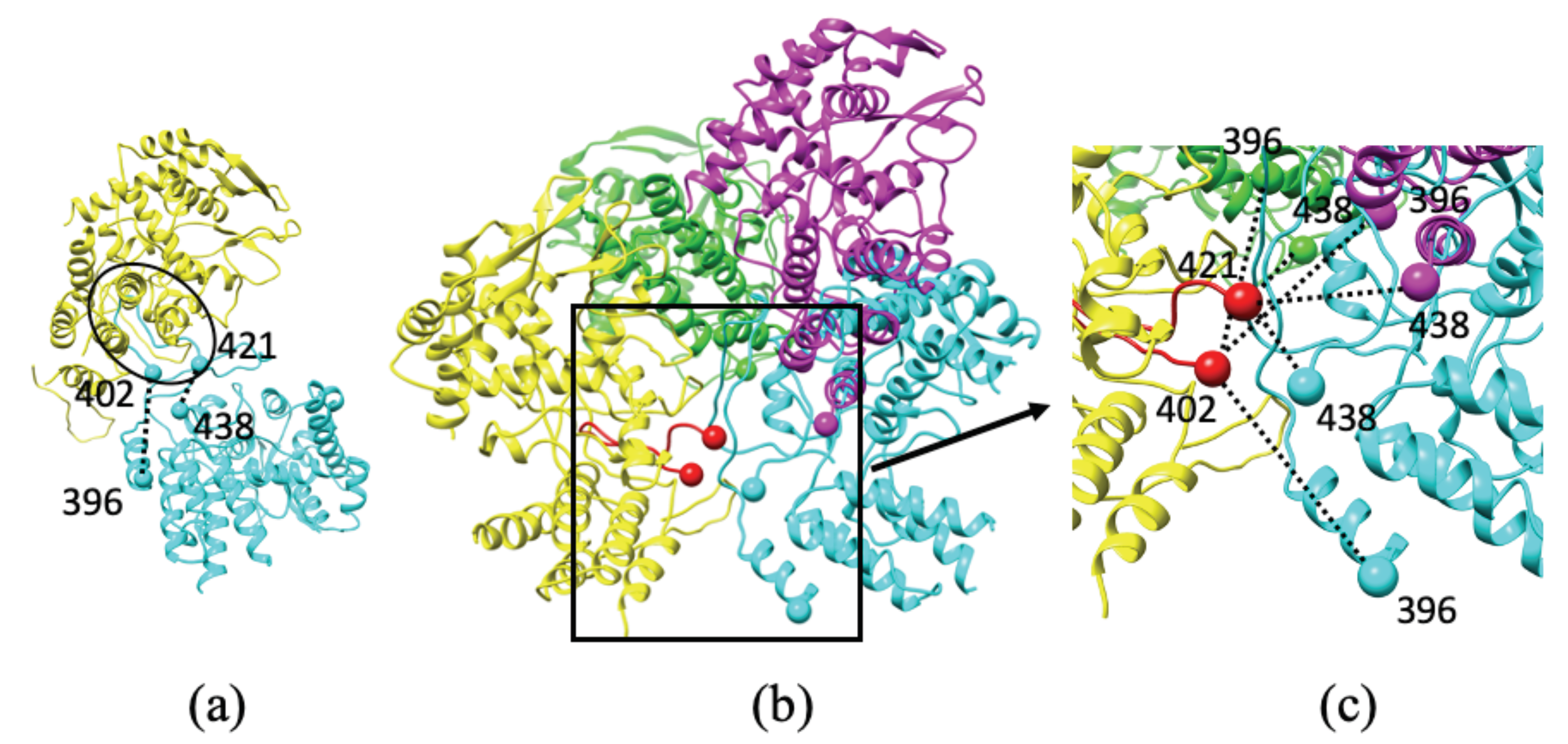
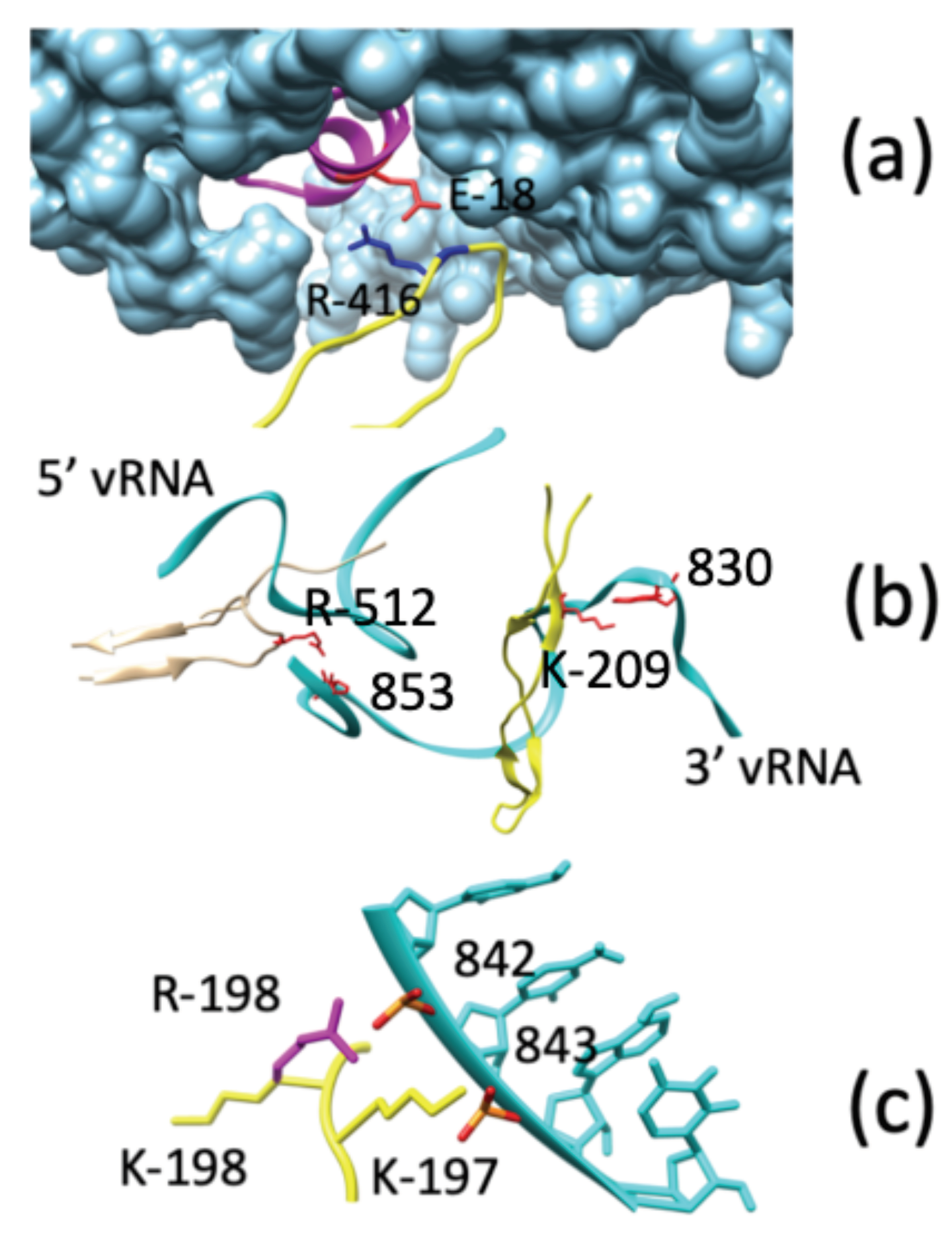
Direct Interactions between NP and POL
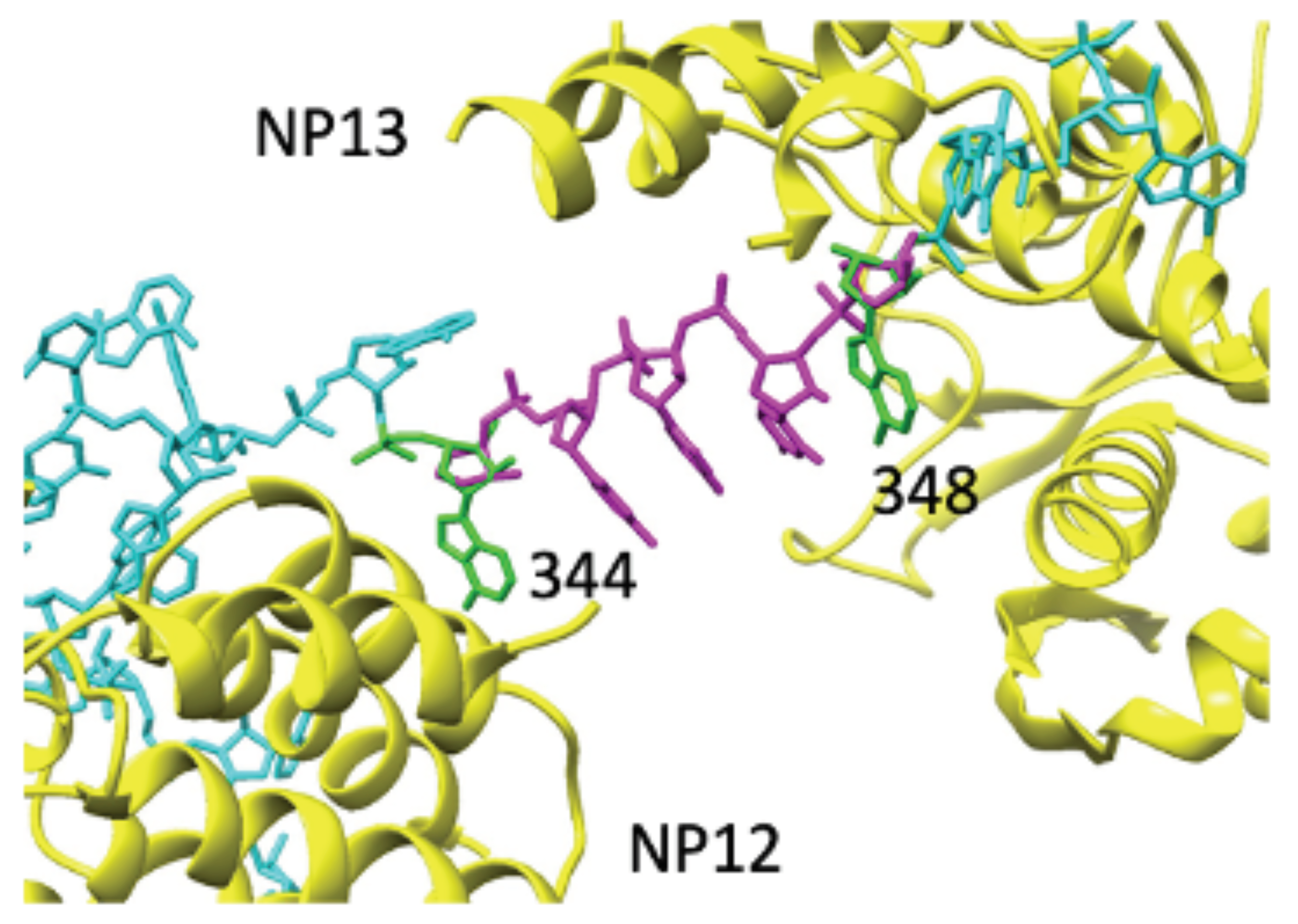
3.4. Direct Interactions between vRNA and POL
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary Digital Publishing Institute |
| DOAJ | Directory of open access journals |
| RNP | ribonucleoprotein |
| RNA | ribonucleic acid |
| POL | polymerase |
| MMFA | motif-matching fragment assembly |
References
- Bedford, T.; Riley, S.; Barr, I.G.; Broor, S.; Chadha, M.; Cox, N.J.; Daniels, R.S.; Gunasekaran, C.P.; Hurt, A.C.; Kelso, A.; et al. Global circulation patterns of seasonal influenza viruses vary with antigenic drift. Nature 2015, 523, 217–220. [Google Scholar] [CrossRef] [Green Version]
- Iuliano, A.D.; Roguski, K.M.; Chang, H.H.; Muscatello, D.J.; Palekar, R.; Tempia, S.; Cohen, C.; Gran, J.M.; Schanzer, D.; Cowling, B.J.; et al. Estimates of global seasonal influenza-associated respiratory mortality: A modelling study. Lancet 2018, 391, 1285–1300. [Google Scholar] [CrossRef]
- Pflug, A.; Lukarska, M.; Resa-Infante, P.; Reich, S.; Cusack, S. Structural insights into RNA synthesis by the influenza virus transcription-replication machine. Virus Res. 2017, 234, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Chaimayo, C.; Hayashi, T.; Underwood, A.; Hodges, E.; Takimoto, T. Selective incorporation of vRNP into influenza A virions determined by its specific interaction with M1 protein. Virology 2017, 505, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Tao, Y.J. Structure and assembly of the influenza A virus ribonucleoprotein complex. FEBS Lett. 2013, 587, 1206–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabriel, G.; Dauber, B.; Wolff, T.; Planz, O.; Klenk, H.D.; Stech, J. The viral polymerase mediates adaptation of an avian influenza virus to a mammalian host. Proc. Natl. Acad. Sci. USA 2005, 102, 18590–18595. [Google Scholar] [CrossRef] [Green Version]
- Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2015, 44, D67–D72. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- wwPDB consortium. Protein Data Bank: The single global archive for 3D macromolecular structure data. Nucleic Acids Res. 2018, 47, D520–D528. [Google Scholar]
- Lawson, C.L.; Baker, M.L.; Best, C.; Bi, C.; Dougherty, M.; Feng, P.; van Ginkel, G.; Devkota, B.; Lagerstedt, I.; Ludtke, S.J.; et al. EMDataBank.org: Unified data resource for CryoEM. Nucleic Acids Res. 2010, 39, D456–D464. [Google Scholar] [CrossRef] [Green Version]
- Tung, C.S.; Sanbonmatsu, K.Y. Atomic Model of the Thermus thermophilus 70S Ribosome Developed in Silico. Biophys. J. 2004, 87, 2714–2722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korostelev, A.; Trakhanov, S.; Laurberg, M.; Noller, H.F. Crystal Structure of a 70S Ribosome-tRNA Complex Reveals Functional Interactions and Rearrangements. Cell 2006, 126, 1065–1077. [Google Scholar] [CrossRef] [Green Version]
- Tung, C.S.; Joseph, S.; Sanbonmatsu, K.Y. All-atom homology model of the Escherichia coli 30S ribosomal subunit. Nat. Struct. Biol. 2002, 9, 750–755. [Google Scholar] [CrossRef] [PubMed]
- Lappala, A.; Nishima, W.; Miner, J.; Fenimore, P.; Fischer, W.; Hraber, P.; Zhang, M.; McMahon, B.; Tung, C.S. Structural Transition and Antibody Binding of EBOV GP and ZIKV E Proteins from Pre-Fusion to Fusion-Initiation State. Biomolecules 2018, 8, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tung, C.S.; McMahon, B.H. A structural model of the E. coli PhoB Dimer in the transcription initiation complex. BMC Struct. Biol. 2012, 12, 3. [Google Scholar] [CrossRef] [Green Version]
- Arranz, R.; Coloma, R.; Chichon, F.J.; Conesa, J.J.; Carrascosa, J.L.; Valpuesta, J.M.; Ortin, J.; Martin-Benito, J. The Structure of Native Influenza Virion Ribonucleoproteins. Science 2012, 338, 1634–1637. [Google Scholar] [CrossRef]
- Moeller, A.; Kirchdoerfer, R.N.; Potter, C.S.; Carragher, B.; Wilson, I.A. Organization of the Influenza Virus Replication Machinery. Science 2012, 338, 1631–1634. [Google Scholar] [CrossRef] [Green Version]
- Coloma, R.; Arranz, R.; de la Rosa-Trevín, J.M.; Sorzano, C.O.S.; Munier, S.; Carlero, D.; Naffakh, N.; Ortín, J.; Martín-Benito, J. Structural insights into influenza A virus ribonucleoproteins reveal a processive helical track as transcription mechanism. Nat. Microbiol. 2020, 5, 727–734. [Google Scholar] [CrossRef]
- Bao, Y.; Bolotov, P.; Dernovoy, D.; Kiryutin, B.; Zaslavsky, L.; Tatusova, T.; Ostell, J.; Lipman, D. The Influenza Virus Resource at the National Center for Biotechnology Information. J. Virol. 2007, 82, 596–601. [Google Scholar] [CrossRef] [Green Version]
- Ruigrok, R.W.H.; Baudin, F. Structure of influenza virus ribonucleoprotein particles. II. Purified RNA-free influenza virus ribonucleoprotein forms structures that are indistinguishable from the intact influenza virus ribonucleoprotein particles. J. Gen. Virol. 1995, 76, 1009–1014. [Google Scholar] [CrossRef]
- Ye, Q.; Krug, R.M.; Tao, Y.J. The mechanism by which influenza A virus nucleoprotein forms oligomers and binds RNA. Nature 2006, 444, 1078–1082. [Google Scholar] [CrossRef] [PubMed]
- Tarus, B.; Bakowiez, O.; Chenavas, S.; Duchemin, L.; Estrozi, L.; Bourdieu, C.; Lejal, N.; Bernard, J.; Moudjou, M.; Chevalier, C.; et al. Oligomerization paths of the nucleoprotein of influenza A virus. Biochimie 2012, 94, 776–785. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Guu, T.S.Y.; Mata, D.A.; Kuo, R.L.; Smith, B.; Krug, R.M.; Tao, Y.J. Biochemical and Structural Evidence in Support of a Coherent Model for the Formation of the Double-Helical Influenza A Virus Ribonucleoprotein. mBio 2012, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Compans, R.W.; Content, J.; Duesberg, P.H. Structure of the Ribonucleoprotein of Influenza Virus. J. Virol. 1972, 10, 795–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portela, A.; Digard, P. The influenza virus nucleoprotein: A multifunctional RNA-binding protein pivotal to virus replication. J. Gen. Virol. 2002, 83, 723–734. [Google Scholar] [CrossRef]
- Noda, T.; Kawaoka, Y. Structure of influenza virus ribonucleoprotein complexes and their packaging into virions. Rev. Med. Virol. 2010, 20, 380–391. [Google Scholar] [CrossRef]
- Gallagher, J.R.; Torian, U.; McCraw, D.M.; Harris, A.K. Structural studies of influenza virus RNPs by electron microscopy indicate molecular contortions within NP supra-structures. J. Struct. Biol. 2017, 197, 294–307. [Google Scholar] [CrossRef]
- Reich, S.; Guilligay, D.; Pflug, A.; Malet, H.; Berger, I.; Crépin, T.; Hart, D.; Lunardi, T.; Nanao, M.; Ruigrok, R.W.H.; et al. Structural insight into cap-snatching and RNA synthesis by influenza polymerase. Nature 2014, 516, 361–366. [Google Scholar] [CrossRef]
- Yamanaka, K.; Ishihama, A.; Nagata, K. Reconstitution of influenza virus RNA-nucleoprotein complexes structurally resembling native viral ribonucleoprotein cores. J. Biol. Chem. 1990, 265, 11151–11155. [Google Scholar] [CrossRef]
- Honda, A.; Mukaigawa, J.; Yokoiyama, A.; Kato, A.; Ueda, S.; Nagata, K.; Krystal, M.; Nayak, D.P.; Ishihama, A. Purification and Molecular Structure of RNA Polymerase from Influenza Virus A/PRS1. J. Biochem. 1990, 107, 624–628. [Google Scholar] [CrossRef]
- Kingsbury, D.W.; Jones, I.M.; Murti, K. Assembly of influenza ribonucleoprotein in vitro using recombinant nucleoprotein. Virology 1987, 156, 396–403. [Google Scholar] [CrossRef]
- Parvin, J.D.; Palese, P.; Honda, A.; Ishihama, A.; Krystal, M. Promoter analysis of influenza virus RNA polymerase. J. Virol. 1989, 63, 5142–5152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.F.; Chen, Y.H.; Chu, S.Y.; Lin, M.I.; Hsu, H.T.; Wu, P.Y.; Wu, C.J.; Liu, H.W.; Lin, F.Y.; Lin, G.; et al. E339...R416 salt bridge of nucleoprotein as a feasible target for influenza virus inhibitors. Proc. Natl. Acad. Sci. USA 2011, 108, 16515–16520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nero, T.L.; Parker, M.W.; Morton, C.J. Protein structure and computational drug discovery. Biochem. Soc. Trans. 2018, 46, 1367–1379. [Google Scholar] [CrossRef] [PubMed]
- Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Huang, C.C.; Ferrin, T.E. Tools for integrated sequence-structure analysis with UCSF Chimera. BMC Bioinform. 2006, 7, 339. [Google Scholar]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Wang, J.; Cieplak, P.; Kollman, P.A. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000, 21, 1049–1074. [Google Scholar] [CrossRef]
- Chen, A.A.; Garcia, A.E. High-resolution reversible folding of hyperstable RNA tetraloops using molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2013, 110, 16820–16825. [Google Scholar] [CrossRef] [Green Version]
- Miner, J.C.; Chen, A.A.; García, A.E. Free-energy landscape of a hyperstable RNA tetraloop. Proc. Natl. Acad. Sci. USA 2016, 113, 6665–6670. [Google Scholar] [CrossRef] [Green Version]
- Seebald, L.M.; DeMott, C.M.; Ranganathan, S.; Asare-Okai, P.N.; Glazunova, A.; Chen, A.; Shekhtman, A.; Royzen, M. Cobalt-based paramagnetic probe to study RNA-protein interactions by NMR. J. Inorg. Biochem. 2017, 170, 202–208. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera: A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coloma, R.; Valpuesta, J.M.; Arranz, R.; Carrascosa, J.L.; Ortín, J.; Martín-Benito, J. The Structure of a Biologically Active Influenza Virus Ribonucleoprotein Complex. PLoS Pathog. 2009, 5, e1000491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, W.K.; Lazniewski, M.; Plewczynski, D. RNA structure interactions and ribonucleoprotein processes of the influenza A virus. Briefings Funct. Genom. 2018, 17, 402–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazniewski, M.; Dawson, W.K.; Szczepińska, T.; Plewczynski, D. The structural variability of the influenza A hemagglutinin receptor-binding site. Briefings Funct. Genom. 2017, 17, 415–427. [Google Scholar]
- Flick, R.; Hobom, G. Interaction of influenza virus polymerase with viral RNA in the ‘corkscrew’ conformation. J. Gen. Virol. 1999, 80, 2565–2572. [Google Scholar] [CrossRef]
- Fodor, E.; Pritlove, D.C.; Brownlee, G.G. The influenza virus panhandle is involved in the initiation of transcription. J. Virol. 1994, 68, 4092–4096. [Google Scholar] [CrossRef] [Green Version]
- Fodor, E.; Pritlove, D.C.; Brownlee, G.G. Characterization of the RNA-fork model of virion RNA in the initiation of transcription in influenza A virus. J. Virol. 1995, 69, 4012–4019. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Sneyd, H.; Dekant, R.; Wang, J. Influenza A Virus Nucleoprotein: A Highly Conserved Multi-Functional Viral Protein as a Hot Antiviral Drug Target. Curr. Top. Med. Chem. 2017, 17. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.K.; Boutz, P.L.; Nayak, D.P. Influenza Virus Nucleoprotein Interacts with Influenza Virus Polymerase Proteins. J. Virol. 1998, 72, 5493–5501. [Google Scholar] [CrossRef] [Green Version]
- Fodor, E.; Smith, M. The PA Subunit Is Required for Efficient Nuclear Accumulation of the PB1 Subunit of the Influenza A Virus RNA Polymerase Complex. J. Virol. 2004, 78, 9144–9153. [Google Scholar] [CrossRef] [Green Version]
- Arai, Y.; Kawashita, N.; Daidoji, T.; Ibrahim, M.S.; El-Gendy, E.M.; Takagi, T.; Takahashi, K.; Suzuki, Y.; Ikuta, K.; Nakaya, T.; et al. Novel Polymerase Gene Mutations for Human Adaptation in Clinical Isolates of Avian H5N1 Influenza Viruses. PLOS Pathog. 2016, 12, e1005583. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, E.C.; Orr, O.E.; Man Liu, S.; Engelhardt, O.G.; Fodor, E. Characterization of the interaction between the influenza A virus polymerase subunit PB1 and the host nuclear import factor Ran-binding protein 5. J. Gen. Virol. 2011, 92, 1859–1869. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RNP Model Target | Data Source(s) | Data Content | Critical Components to Model |
|---|---|---|---|
| vRNA (helical domain) | PDB_ID: 4BBL PDB RNA fragments | Phosphorous atoms in RNP helical region | All-atom vRNA |
| vRNA (sequence) | Influenza Virus Resource [19], GenBank [7], PDB [9] | Primary sequence of vRNA | All-atom vRNA structure with a target Influenza A sequence |
| NP (helical domain) | PDB_ID: 4BBL, EMDB_ID: emd_2205 | NPs in helical conformation | NP folded domain orientation |
| NP (circular domain) | PDB_ID: 2WFS, EMDB_ID: emd_1603 | NPs in hairpin loop conformation | NP folded domain orientation |
| POL + vRNA (promoter) | PDB_ID: 4WSB | POL with docked RNA promoter | POL docked with “Half-corkscrew” promoter conformation |
| NP + RNA + POL | EMDB_ID: emd_2208 | vRNA + NP + POL targets | POL docking to RNP |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miner, J.C.; Lappala, A.; Fenimore, P.W.; Fischer, W.M.; McMahon, B.H.; Hengartner, N.W.; Sanbonmatsu, K.Y.; Tung, C.-S. Modeling the Influenza A NP-vRNA-Polymerase Complex in Atomic Detail. Biomolecules 2021, 11, 124. https://doi.org/10.3390/biom11010124
Miner JC, Lappala A, Fenimore PW, Fischer WM, McMahon BH, Hengartner NW, Sanbonmatsu KY, Tung C-S. Modeling the Influenza A NP-vRNA-Polymerase Complex in Atomic Detail. Biomolecules. 2021; 11(1):124. https://doi.org/10.3390/biom11010124
Chicago/Turabian StyleMiner, Jacob C., Anna Lappala, Paul W. Fenimore, William M. Fischer, Benjamin H. McMahon, Nicolas W. Hengartner, Karissa Y. Sanbonmatsu, and Chang-Shung Tung. 2021. "Modeling the Influenza A NP-vRNA-Polymerase Complex in Atomic Detail" Biomolecules 11, no. 1: 124. https://doi.org/10.3390/biom11010124