Multiple-Monitor HPLC Assays for Rapid Process Development, In-Process Monitoring, and Validation of AAV Production and Purification


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples and Sample Preparation
2.2. HPLC Analysis
3. Results
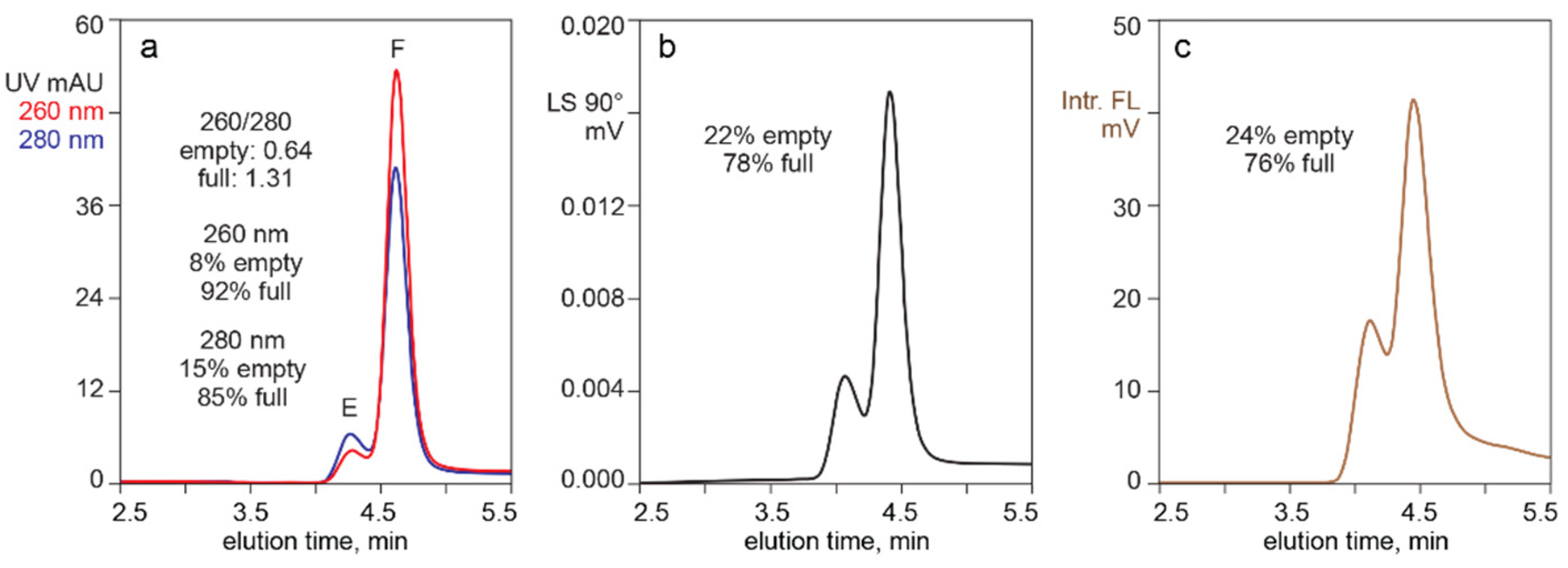
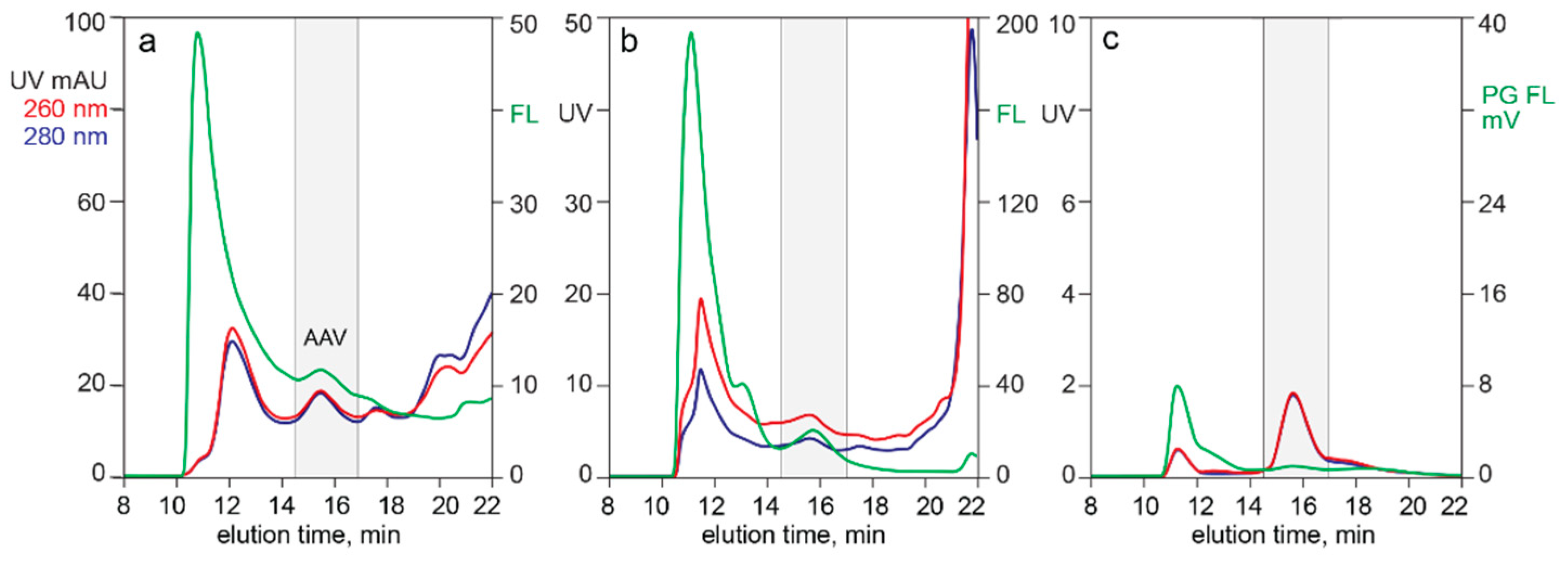
3.1. In-Line HPLC Monitoring of Empty and Full AAV Capsids
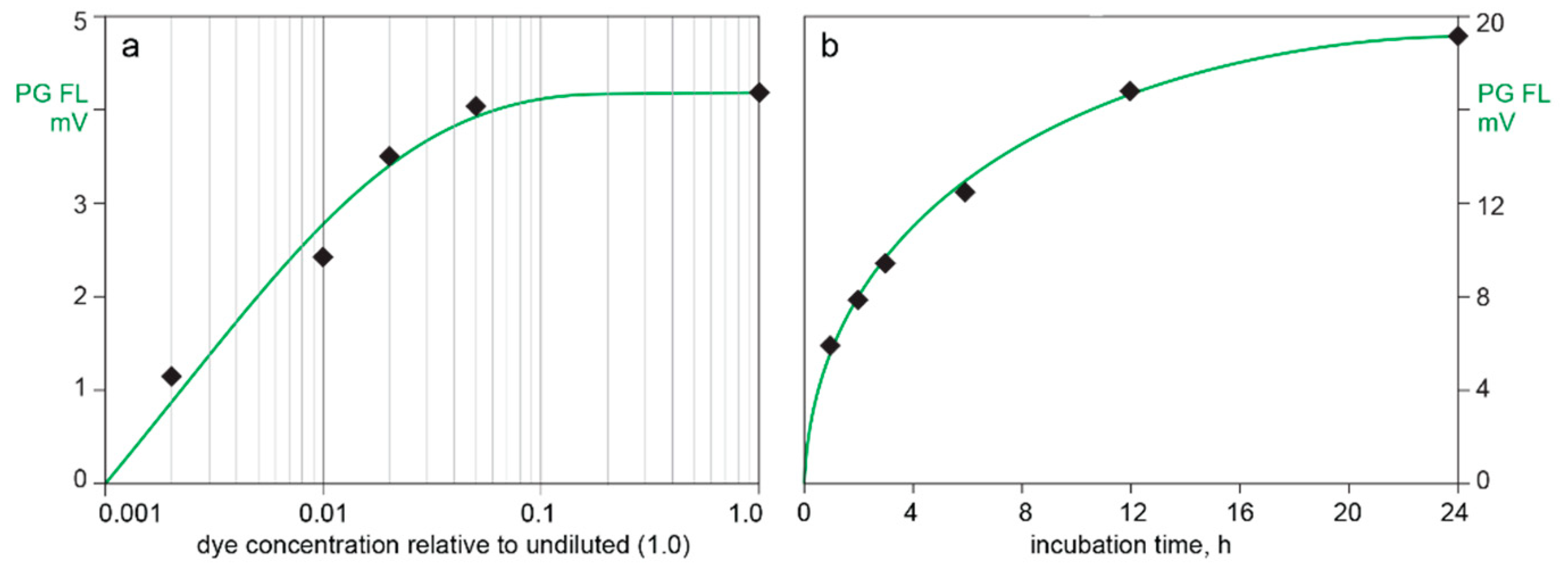
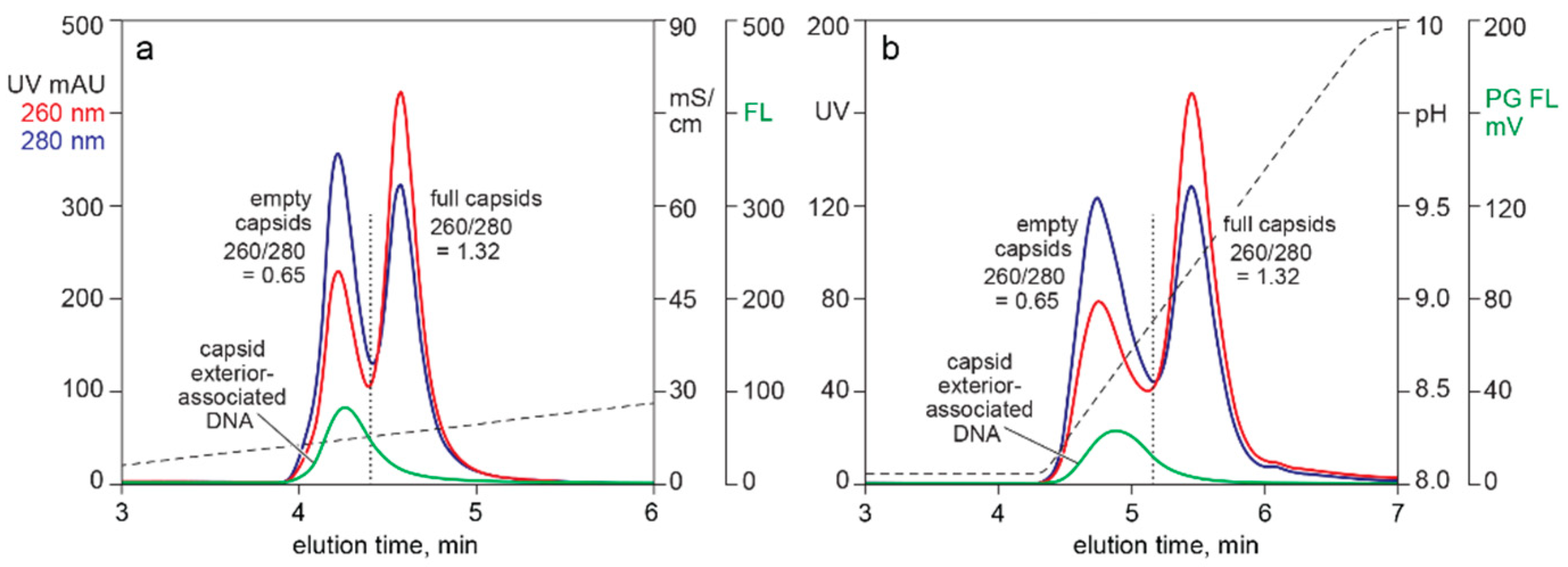
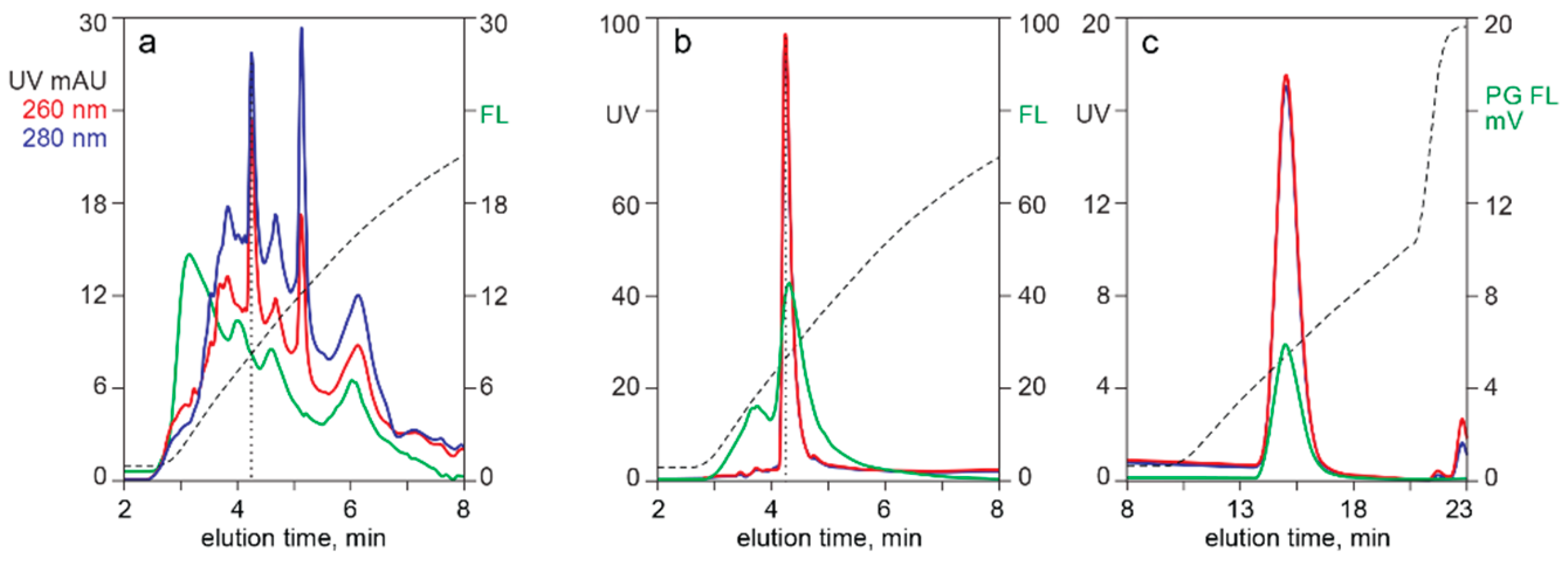
3.2. In-Line HPLC Monitoring of Nucleic Acids
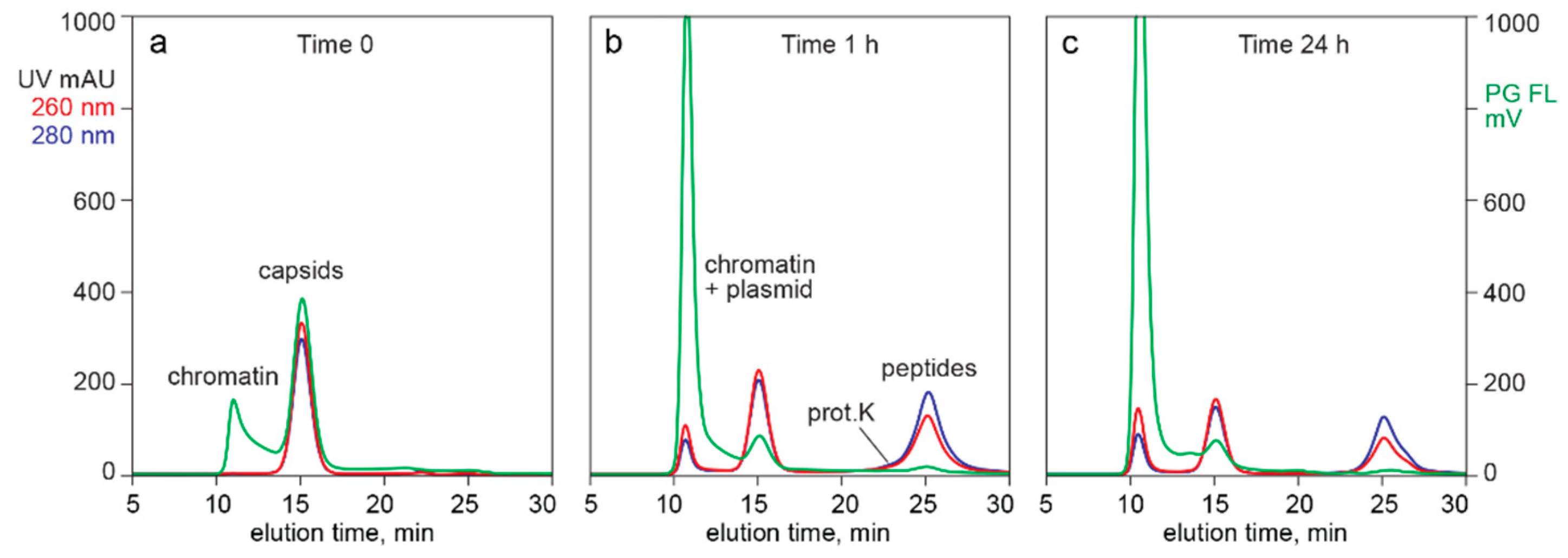
3.3. In-Line HPLC Characterization of Raw and In-Process Materials
4. Discussion and Conclusions
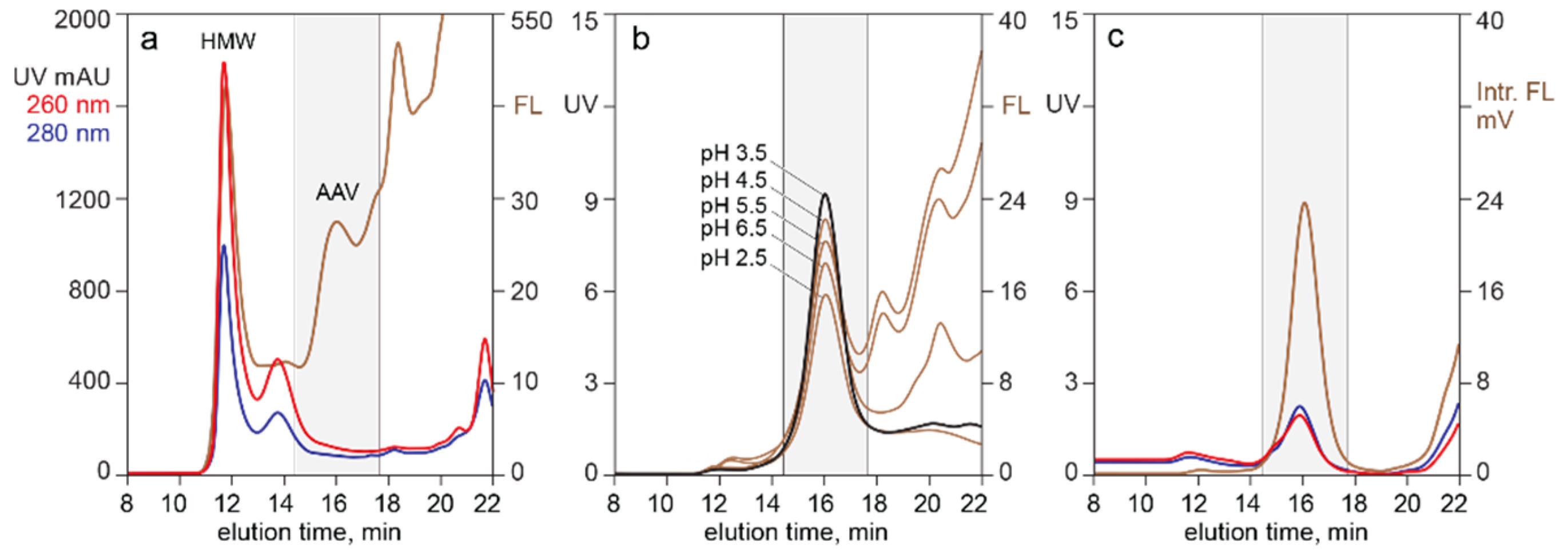
4.1. In-Line HPLC Monitoring by UV Absorbance and Intrinsic Fluorescence
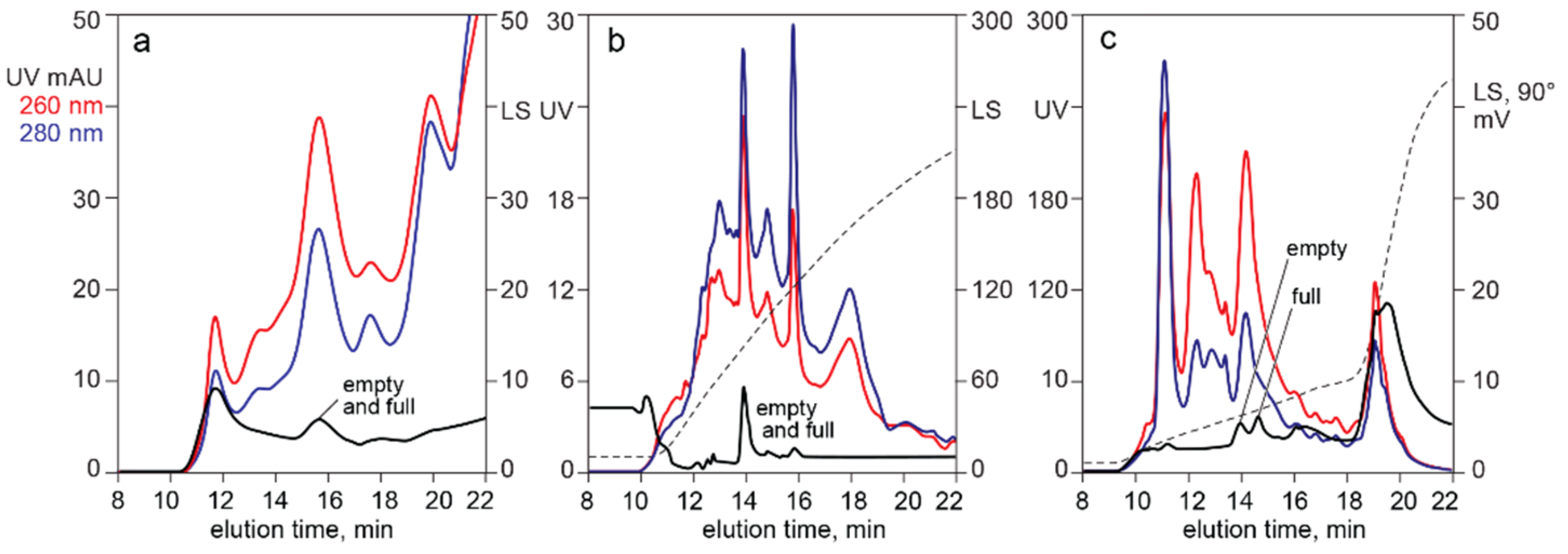
4.2. In-Line HPLC Monitoring by UV Absorbance and Light Scattering
4.3. In-Line HPLC Monitoring by UV Absorbance and Extrinsic Fluorescence
4.4. Applications of Multi-Monitor HPLC Assays
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pierson, E.E.; Keifer, D.Z.; Asokan, A.; Jarrold, M.F. Resolving adeno-associated viral particle diversity with charge detection mass spectrometry. Anal. Chem. 2016, 88, 6718–6725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horowitz, E.D.; Rahman, K.S.; Bower, B.D.; Dismuke, D.J.; Falvo, M.R.; Griffith, J.D.; Harvey, S.C.; Asokan, A. Biophysical and ultrastructural characterization of adeno-associated virus capsid uncoating and genome release. J. Virol. 2013, 87, 2994–3002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wörner, T.P.; Bennet, A.; Habka, S.; Snijder, J.; Friese, O.; Powers, T.; Agbandje-McKenna, M.; Heck, A.J.R. Adeno-associated virus capsid assembly is divergent and stochastic. bioRxiv 2020. [Google Scholar] [CrossRef]
- Bertin, M.; Maurya, S.; Arumugam, S.; Kumar, V.; Jayadharan, G.R. Post-translational modifications in capsid proteins of recombinant Adeno-associated virus (AAV) 1-rh19 serotypes. FEBS J. 2019, 286, 4964–4981. [Google Scholar]
- Giles, A.R.; Sims, J.J.; Turner, K.B.; Govindasamy, L.; Alvira, M.R.; Lock, M.; Wilson, J.M. Deamidation of amino acids on the surface of adeno-associated virus capsids leads to charge heterogeneity and altered vector function. Mol. Ther. 2018, 26, 2848–2862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gagnon, P.; Goricar, B.; Peljhan, S.; Strancar, A. Streamlining industrial purification of adeno-associated virus. BioProcess Int. 2020, 18, S14–S20. [Google Scholar]
- Sommer, J.M.; Smith, P.H.; Parthasarathy, S.; Isaacs, J.; Vijay, S.; Kieran, J.; Powell, S.K.; McClelland, A.; Wright, J.F. Quantification of adeno-associated virus particles and empty capsids by optical density measurement. Mol. Ther. 2003, 7, 122–128. [Google Scholar] [CrossRef]
- Wang, C.; Mulagapati, S.; Chen, Z.; Du, J.; Zhao, X.; Xi, G.; Chen, L.; Linke, T.; Gao, C.; Schmelzer, A.; et al. Developing an anion exchange assay for determining empty and full capsid contents in AAV6.2. Mol. Ther. 2019, 15, 257–263. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.; Chen, W.-C.; Argento, C.; Clarner, P.; Bhatt, V.; Dickerson, R.; Bou-Assaf, G.; Bakhshayeshi, M.; Lu, X.; Bergelson, S.; et al. Analytical strategies of quantitation of adeno-associated virus empty capsids to support process development. Hum. Gene Ther. Met. 2019, 30, 144–152. [Google Scholar] [CrossRef]
- Dickerson, R.; Argento, C.; Pieracci, J.; Bakhshayeshi, M. Separating empty and full recombinant Adeno-associated virus particles using isocratic anion exchange chromatography. Biotechnol. J. 2020. [Google Scholar] [CrossRef]
- Qu, G.; Bahr-Davidson, J.; Proado, J.; Tai, A.; Cataniag, F.; McDonnell, J.; Zhou, J.; Hauck, B.; Luna, J.; Sommer, J.; et al. Separation of adeno-associated virus type 2 empty particles from genome containing vectors by anion exchange chromatography. J. Virol. Met. 2007, 140, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Lock, M.; Alvira, M.; Wilson, J.M. Analysis of particle content of recombinant adeno-associated virus serotype 8 vectors by ion exchange chromatography. Hum. Gene Ther. Met. 2012, 23. [Google Scholar] [CrossRef] [PubMed]
- Lock, M.; Alvira, M. Scalable Purification Method for AAV9. World Patent Application WO2017160360A9, 21 September 2017. Available online: https://patents.google.com/patent/WO2017160360A9/en (accessed on 15 November 2020).
- Urabe, M.; Xin, K.-Q.; Obara, Y.; Nakakura, T.; Mizukami, H.; Kume, A.; Okuda, K.; Ozawa, K. Removal of empty capsids from type 1 adeno-associated virus vector stocks by anion exchange chromatography potentiates transgene expression. Mol. Ther. 2006, 13, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Brument, N.; Morenweiser, R.; Bioquin, V.; Toublanc, E.; Raimbaud, I.; Cherel, Y.; Folliot, S.; Gaden, F.; Boulanger, P.; Kroner-Luz, G.; et al. A versatile and scalable two-step ion exchange chromatography process for the purification of recombinant adeno-associated virus serotypes-2 and -5. Mol. Ther. 2002, 6, 678–686. [Google Scholar] [CrossRef]
- Davidoff, A.M.; Ng, C.Y.C.; Sleep, S.; Gray, J.; Azam, S.; Zhao, Y.; McIntosh, J.H.; Karimipoor, M.; Nathwani, A.C. Purification of recombinant adeno-associated virus type 8 vectors by ion exchange chromatography generates clinical grade vector stock. J. Virol. Met. 2004, 121, 209–215. [Google Scholar] [CrossRef]
- Kaludov, N.; Handelman, B.; Chiorina, J.A. Scalable purification of adeno-associated virus type 2, 4, or 5, using ion exchange chromatography. Hum. Gene Ther. 2004, 13. [Google Scholar] [CrossRef]
- Porterfield, J.Z.; Zlotnick, A. A simple and general method for determining the protein and nucleic acid content of viruses by UV absorbance. Virology 2010, 407, 281–288. [Google Scholar] [CrossRef] [Green Version]
- Tan, L.; Yeo, V.; Yang, Y.-S.; Gagnon, P. Characterization of DNA in cell culture supernatant by fluorescence-detection size exclusion chromatography. Anal. Bioanal. Chem. 2015, 407, 4173–4181. [Google Scholar] [CrossRef]
- Chen, R.F. Fluorescence quantum yield of tryptophan and tyrosine. Analyt. Lett. 1967, 1, 35–42. [Google Scholar] [CrossRef]
- Yang, H.; Xue, X.; Zhao, X.; Wu, Y. Intrinsic fluorescence spectra of tryptophan, tyrosine, and phenylalanine. Advances in Engineering Research. In Proceedings of the 5th International Conference on Advanced Design and Manufacturing Engineering (ICADME 2015), Shenzhen, China, 19–20 September 2015; pp. 224–233. [Google Scholar] [CrossRef] [Green Version]
- Ghisaidoobe, A.B.T.; Chung, S.A. Intrinsic tryptophan fluorescence in the detection and analysis of proteins: A focus on the Förster resonance energy transfer techniques. Intl. J. Mol. Sci. 2014, 15, 22518–22538. [Google Scholar] [CrossRef]
- UniProtKB—P03135 (CAPSD_AAV2S). Available online: https://www.uniprot.org/uniprot/P03135 (accessed on 15 November 2020).
- Pisarevskii, A.N.; Cherenkevich, S.N.; Andrianov, V.T. Fluorescence spectrum and quantum yield of DNA in solution. J. Appl. Spectr. 1966, 5, 452–454. [Google Scholar] [CrossRef]
- Lakowicz, J.R.; Shen, B.; Gryczynski, Z.; D’Auria, S.; Gryczynski, I. Intrinsic fluorescence from DNA can be enhanced by metallic particles. Biochem. Biophys. Res. Comm. 2001, 286, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Vaya, I.; Gustavsson, T.; Miannay, F.-A.; Douk, T.; Markovitsi, D. Fluorescence of natural DNA: From femtosecond to the nanosecond time scales. J. Am. Chem. Soc. 2010, 132, 11834–11835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- UniProtKB—0A140T897 (A0A140T897_BOVIN). Available online: https://www.uniprot.org/uniprot/A0A140T897 (accessed on 15 November 2020).
- UniProtKB—P84243 (H33_HUMAN). Available online: https://www.uniprot.org/uniprot/P84243 (accessed on 15 November 2020).
- UniProtKB—Q02539 (H11_HUMAN). Available online: https://www.uniprot.org/uniprot/Q02539 (accessed on 15 November 2020).
- UniProtKB—P62805 (H4_HUMAN). Available online: https://www.uniprot.org/uniprot/P62805 (accessed on 15 November 2020).
- UniProtKB—P0C0S8 (H2A1_HUMAN). Available online: https://www.uniprot.org/uniprot/P0C0S8 (accessed on 15 November 2020).
- UniProtKB—B4DR52 (B4DR52–HUMAN). Available online: https://www.uniprot.org/uniprot/B4DR52 (accessed on 15 November 2020).
- Libertini, L.J.; Small, E.W. The intrinsic fluorescence of histone H1. Steady state and fluorescence decay studies reveal heterogeneous emission. Biophys. J. 1985, 47, 765–772. [Google Scholar] [CrossRef] [Green Version]
- Vivian, J.T.; Callis, P.R. Mechanisms of tryptophan fluorescence shifts in proteins. Biophys. J. 2001, 80, 2093–2109. [Google Scholar] [CrossRef] [Green Version]
- Zelent, B.; Bialas, C.; Grycznski, I.; Chen, P.; Lewerissa, K.; Corradini, M.G.; Ludescher, R.D.; Vanderkooi, J.M.; Matchinsky, M. Tryptophan fluorescence yields and lifetimes as a probe of conformational changes in human glucokinase. J. Fluoresc. 2017, 27, 1621–1631. [Google Scholar] [CrossRef]
- Singer, V.; Jones, L.; Yue, S.T.; Hauglund, R.P. Characterization of Picogreen reagent and development of a fluorescence-based solution assay for double-stranded DNA quantitation. Anal. Biochem. 1997, 249, 228–238. [Google Scholar] [CrossRef] [Green Version]
- Dragan, A.I.; Casas-Finet, J.R.; Bishop, E.S.; Strouse, R.J.; Schenerman, M.A.; Geddes, C.D. Characterization of Picogreen interaction with dsDNA and the origin of its fluorescence enhancement on binding. Biophys. J. 2010, 99, 3010–3019. [Google Scholar] [CrossRef] [Green Version]
- Dragan, A.I.; Bishop, E.S.; Casas-Finet, J.R.; Strouse, R.J.; Schenerman, M.A.; Gedes, C.D. Metal-enhanced Picogreen fluorescence for double-stranded DNA quantification. Anal. Biochem. 2010, 396, 8–12. [Google Scholar] [CrossRef]
- Amado, A.M.; Pazin, W.M.; Ito, A.S.; Kuzmin, V.A.; Borissevitch, I.E. Acridine orange interaction with DNA: Effect of ionic strength. Biochim. Biophys. Acta 2017, 1861, 900–909. [Google Scholar] [CrossRef]
- Holden, M.J.; Haynes, R.J.; Rabb, S.A.; Satija, N.; Yang, K.; Blasic, J.R., Jr. Factors affecting quantification of total DNA by UV spectroscopy and Picogreen fluorescence. J. Agric. Food Chem. 2009, 57, 7221–7226. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, Y.; Yamaguchi, H.; Einaga, H.; Esumi, M. Pitfalls of DNA quantification using DNA-binding fluorescent dyes and suggested solutions. PLoS ONE 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kremser, L.; Okun, V.M.; Nicodernou, A.; Blaas, D.; Kenndler, E. Binding of fluorescent dye to genomic RNA inside intact human rhinovirus after viral capsid penetration investigated by capillary electrophoresis. Anal. Chem. 2004, 76, 882–887. [Google Scholar] [CrossRef] [PubMed]
- Weiss, V.U.; Bliem, C.B.; Gösler, I.; Fedosyuk, S.; Kratzmeier, M.; Blaas, D.; Allmaier, G. In vitro RNA release from human release rhinovirus monitored by means of a molecular beacon and chip electrophoresis. Anal. Bioanal. Chem. 2016, 408, 4209–4217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- TransFiguracion, J.; Mena, J.A.; Aucoin, M.G.; Kamen, A.A. Development and validation of a HPLC method for quantitation of baculovirus particles. J. Chromatogr. B 2011, 879, 61–68. [Google Scholar] [CrossRef] [Green Version]
- Transfiguracion, J.; Kamen, A. Detection of intact recombinant viruses. World Patent Application WO2006053443A1, 26 May 2006. [Google Scholar]
- Murikami, P.; McCarnan, M.T. Quantitation of adenovirus DNA and virus particles with the Picogreen fluorescent dye. Anal. Biochem. 1999, 274, 283–288. [Google Scholar] [CrossRef]
- Xu, J.; DeVries, S.H.; Zhu, Y. Quantitation of adeno-associated virus with safe nucleic acid dyes. Hum. Gene Ther. 2020, 31, 19–20. [Google Scholar] [CrossRef]
- Steppert, P.; Burgstaller, D.; Klausberger, M.; Tover, A.; Berger, E.; Jungbauer, A. Quantification and characterization of virus-like particles by size-exclusion chromatography and nanoparticle tracking analysis. J. Chromatogr. A 2017, 1487, 89–99. [Google Scholar] [CrossRef]
- Sahin, E.; Roberts, C.J., Jr. Size-exclusion chromatography with multi-angle light scattering for elucidating protein aggregation mechanisms. Met. Molec. Biol. 2012, 899, 403–4023. [Google Scholar]
- Some, D.; Amartely, H.; Tsadok, A.; Lebendiker, M. Characterization of proteins by size-exclusion chromatography coupled to multi-angle light scattering (SEC-MALS). J. Vis. Exp. 2019, 148. [Google Scholar] [CrossRef] [Green Version]
- Aguilar, P.P.; Gonzalez-Dominguez, I.; Schneider, T.A.; Godia, F.; Cervara, L.; Jungbauer, A. At-line multi-angle light scattering detector for faster process development in enveloped virus-like particle purification. J. Sep. Sci. 2019, 42, 2640–2649. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.A.; Gospodarek, A.; Larkin, M.; Kenrick, S.A.; Haverick, M.A.; Tugcu, N.; Brower, M.A.; Richardson, D.D. Multi-angle light scattering as a process analytical technology measuring real-time molecular weight for downstream process control. MABS 2018, 10, 945–950. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Li, D.; Chen, W.; Mu, S.; Chen, Y.; Chai, J. Impact of refractive index increment on the determination of molecular weight of hyaluronic acid by multi-angle laser light scattering technique. Sci. Rep. 2020, 10, 1858. [Google Scholar] [CrossRef] [PubMed]
- Amartely, H.; Avrham, O.; Friedler, A.; Livnah, O.; Lebendiker, M. Coupling multi angle light scattering to ion exchange chromatography (IEX-MALS) for protein characterization. Sci. Rep. 2018, 8, 6907. [Google Scholar] [CrossRef] [PubMed]
- Amartely, H.; Some, D.; Tsadok, A.; Lebendiker, M. Ion exchange chromatography (IEX) coupled to multi-angle light scattering (MALS) for protein separation and characterization. J. Vis. Exp. 2019, 146. [Google Scholar] [CrossRef]
- Staubach, S.; Gagnon, P.; Vrabec, K.; Lojpur, T.; Peljhan, S.; Giebel, B.; Strancar, A. Challenges in industrial process development of exosome-based therapies: Characterizing and managing diversity. Bioprocess Int. 2020, 8. Available online: https://bioprocessintl.com/analytical/downstream-development/classification-of-extracellular-vesicles-using-chromatography-for-exosome-therapy-production/ (accessed on 15 November 2020).
- Gagnon, P.; Nian, R.; Tan, L.; Cheong, J.; Yeo, V.; Yang, Y.-S.; Gan, H.-T. Chromatin-mediated depression of fractionation performance on electronegative multimodal chromatography media, its prevention, and ramifications for purification immunoglobulin G. J. Chromatogr. A 2014, 1374, 145–155. [Google Scholar] [CrossRef]
- Nian, R.; Gagnon, P. Advance chromatin extraction enhances performance and productivity of cation exchange chromatography-based capture of immunoglobulin G monoclonal antibodies. J. Chromatogr. A 2016, 1453, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Gagnon, P.; Nian, R.; Lee, J.; Tan, L.; Abdul-Latiff, S.M.; Lim, C.-L.; Chuah, C.; Bi, X.; Yang, W.S.; Zhang, W.; et al. Nonspecific interactions of chromatin with immunoglobulin G and protein A, and their impact on purification performance. J. Chromatogr. A 2014, 1340, 68–78. [Google Scholar] [CrossRef]
- Nian, R.; Zhang, W.; Tan, L.; Lee, J.; Bi, X.; Yang, Y.-S.; Gan, H.-T.; Gagnon, P. Advance chromatin extraction improves capture performance of protein a affinity chromatography. J. Chromatogr. A 2016, 1431, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Dobnik, D.; Kogovsek, P.; Jakomin, T.; Kosir, N.; Znidaric, M.T.; Leskovec, M.; Kaminsky, S.M.; Mostrom, J.; Lee, H.; Ravnikar, M. Accurate quantification and characterization of adeno-associated viral vectors. Front. Microbiol. 2019, 10, 1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allahverdi, A.; Chen, Q.; Korolev, N.; Nordenskiöld, L. Chromatin compaction under mixed salt conditions: Opposite effects of sodium and potassium ions on nucleosome array folding. Sci. Rep. 2015, 5, 8512. [Google Scholar] [CrossRef] [PubMed]
- De Frutos, M.; Raspaud, E.; Leforestier, A.; Livolant, F. Aggregation of nucleosomes by divalent cations. Biophys. J. 2001, 81, 1127–1132. [Google Scholar] [CrossRef] [Green Version]
- Stacks, P.C.; Schumaker, V.N. Nucleosome dissociation and transfer in concentrated salt solutions. Nucl. Acid. Res. 1979, 7, 2457–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Holde, K.E.; Allen, J.R.; Tatchell, K.; Weischet, W.O.; Lohr, D. DNA histone interactions in nucleosomes. Biophys. J. 1980, 32, 271–282. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gagnon, P.; Goricar, B.; Mencin, N.; Zvanut, T.; Peljhan, S.; Leskovec, M.; Strancar, A. Multiple-Monitor HPLC Assays for Rapid Process Development, In-Process Monitoring, and Validation of AAV Production and Purification. Pharmaceutics 2021, 13, 113. https://doi.org/10.3390/pharmaceutics13010113
Gagnon P, Goricar B, Mencin N, Zvanut T, Peljhan S, Leskovec M, Strancar A. Multiple-Monitor HPLC Assays for Rapid Process Development, In-Process Monitoring, and Validation of AAV Production and Purification. Pharmaceutics. 2021; 13(1):113. https://doi.org/10.3390/pharmaceutics13010113
Chicago/Turabian StyleGagnon, Pete, Blaz Goricar, Nina Mencin, Timotej Zvanut, Sebastijan Peljhan, Maja Leskovec, and Ales Strancar. 2021. "Multiple-Monitor HPLC Assays for Rapid Process Development, In-Process Monitoring, and Validation of AAV Production and Purification" Pharmaceutics 13, no. 1: 113. https://doi.org/10.3390/pharmaceutics13010113