Metagenomics Approaches for the Detection and Surveillance of Emerging and Recurrent Plant Pathogens

 ,
,  ,
,  and
and
Abstract
:1. Plant Disease Diagnosis
1.1. A Growing Global Problem
1.2. Current Challenges
1.3. Current Diagnostic Techniques
2. High-Throughput Sequencing (HTS)
2.1. Applications
2.2. Sample Preparation
2.3. Metabarcoding
2.4. Shotgun Metagenomics
2.5. Sequencing Methods
2.6. Databases
3. Application of HTS in Plant Disease Diagnosis
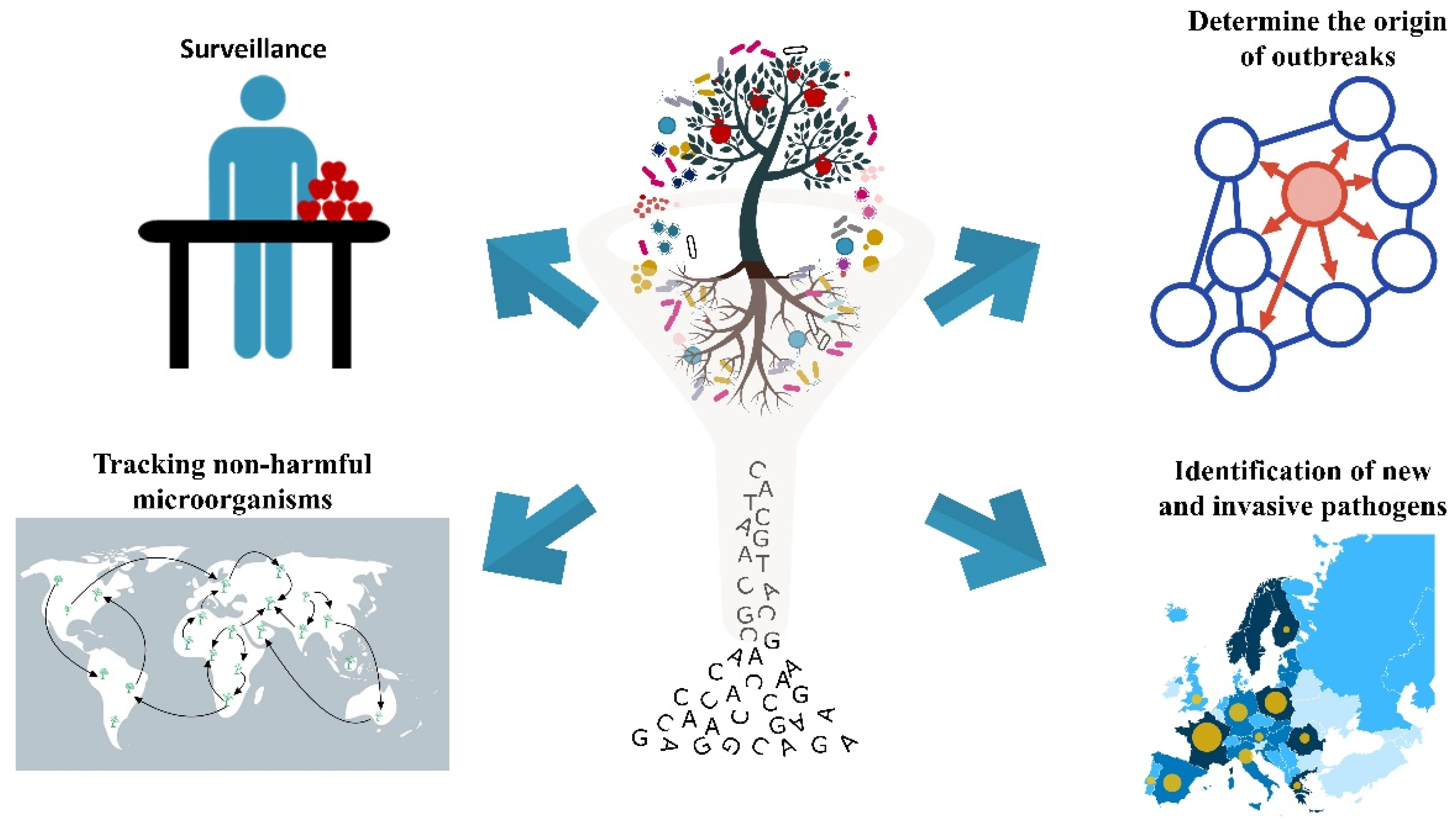
3.1. Surveillance
3.2. Identification of Emerging Pathogens
3.3. Determine the Origin of Outbreaks
3.4. Tracking Non-Harmful Microorganisms
3.5. Limitations
4. Conclusions and Future Studies
Author Contributions
Funding
Conflicts of Interest
References
- Lichtenberg, E.; Olson, L.J. The fruit and vegetable import pathway for potential invasive pest arrivals. PLoS ONE 2018, 13, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- EUROPHYT EUROPHYT-Interceptions–European Union Notification System for Plant Health Interceptions–Annual Reports. Available online: https://ec.europa.eu/food/plant/plant_health_biosecurity/europhyt/annual_reports_en (accessed on 15 January 2021).
- Oerke, E.-C.; Dehne, H.-W.; Schönbeck, F.; Weber, A. Crop Production and Crop Protection: Estimated Losses in Major Food and Cash Crops; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Savary, S.; Ficke, A.; Aubertot, J.N.; Hollier, C. Crop losses due to diseases and their implications for global food production losses and food security. Food Secur. 2012, 4, 519–537. [Google Scholar] [CrossRef]
- Godfray, H.C.J.; Beddington, J.R.; Crute, I.R.; Haddad, L.; Lawrence, D.; Muir, J.F.; Pretty, J.; Robinson, S.; Thomas, S.M.; Toulmin, C. Food security: The challenge of feeding 9 billion people. Science 2010, 327, 812–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godfray, H.C.J.; Crute, I.R.; Haddad, L.; Muir, J.F.; Nisbett, N.; Lawrence, D.; Pretty, J.; Robinson, S.; Toulmin, C.; Whiteley, R. The future of the global food system. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 2769–2777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilman, D.; Balzer, C.; Hill, J.; Befort, B.L. Global food demand and the sustainable intensification of agriculture. Proc. Natl. Acad. Sci. USA 2011, 108, 20260–20264. [Google Scholar] [CrossRef] [Green Version]
- United Nations. Population Prospects 2019: Ten Key Findings. 2019. Available online: https://population.un.org/wpp/Publications/Files/WPP2019_10KeyFindings.pdf (accessed on 15 January 2021).
- Ghadge, A.; Wurtmann, H.; Seuring, S. Managing climate change risks in global supply chains: A review and research agenda. Int. J. Prod. Res. 2020, 58, 44–64. [Google Scholar] [CrossRef]
- Crane-Droesch, A.; Marshall, E.; Rosch, S.; Riddle, A.; Cooper, J.; Wallander, S. Climate Change and Agricultural Risk Management into the 21st Century; United States Department of Agriculture—Economic Research Service: Washington, DC, USA, 2019. [Google Scholar]
- European Union REGULATION (EU) 2016/2031. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32016R2031 (accessed on 15 January 2021).
- Cruz, C.D.; Valent, B. Wheat blast disease: Danger on the move. Trop. Plant Pathol. 2017, 42, 210–222. [Google Scholar] [CrossRef] [Green Version]
- Gitaitis, R.; Walcott, R. The epidemiology and management of seedborne bacterial diseases. Annu. Rev. Phytopathol. 2008, 45, 371–397. [Google Scholar] [CrossRef]
- Gupta, A.K.; Solanki, I.S.; Bashyal, B.M.; Singh, Y.; Srivastava, K. Bakanae of rice—An emerging disease in Asia. J. Anim. Plant Sci. 2015, 25, 1499–1514. [Google Scholar]
- Brown, J.K.M.; Hovmøller, M.S. Aerial dispersal of fungi on the global and continental scales and its consequences for plant disease. Science 2002, 297, 537–541. [Google Scholar] [CrossRef] [Green Version]
- Keller, M.D.; Bergstrom, G.C.; Shields, E.J. The aerobiology of Fusarium graminearum. Aerobiologia 2014, 30, 123–136. [Google Scholar] [CrossRef]
- Womiloju, T.O.; Miller, J.D.; Mayer, P.M.; Brook, J.R. Methods to determine the biological composition of particulate matter collected from outdoor air. Atmos. Environ. 2003, 37, 1352–2310. [Google Scholar] [CrossRef]
- Franić, I.; Prospero, S.; Hartmann, M.; Allan, E.; Auger-Rozenberg, M.A.; Grünwald, N.J.; Kenis, M.; Roques, A.; Schneider, S.; Sniezko, R.; et al. Are traded forest tree seeds a potential source of nonnative pests? Ecol. Appl. 2019, 29, e01971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saponari, M.; Giampetruzzi, A.; Loconsole, G.; Boscia, D.; Saldarelli, P. Xylella fastidiosa in olive in apulia: Where we stand. Phytopathology 2019, 109, 175–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brasier, C.M.; Vettraino, A.M.; Chang, T.T.; Vannini, A. Phytophthora lateralis discovered in an old growth Chamaecyparis forest in Taiwan. Plant Pathol. 2010, 59, 595–603. [Google Scholar] [CrossRef]
- Hopkins, D.L. Natural Hosts of Xylella fastidiosa in Florida. Plant Dis. 1988, 72, 429. [Google Scholar] [CrossRef]
- McElrone, A.J.; Sherald, J.L.; Pooler, M.R. Identification of alternative hosts of Xylella fastidiosa in the Washington, D.C., area using nested polymerase chain reaction (PCR). J. Arboric. 1999, 25, 258. [Google Scholar]
- Lins, S.R.O.; de Abreu, M.S.; Alves, E.; Barbosa, J.F.; de Souza, R.M. Report of Xylella fastidiosa in petioles and hypocotyls of coffee plants with symptoms of buttery spot. Cienc. Agrotecnol. 2008, 32, 42–47. [Google Scholar] [CrossRef] [Green Version]
- Widmer, T.L.; McMahon, M.B.; Luster, D.G. Plant pathogenic fungi are harbored as endophytes in Rhododendron spp. native to the Eastern U.S.A. Fungal Ecol. 2020, 47, 100949. [Google Scholar] [CrossRef]
- Cooke, D.E.L.; Schena, L.; Cacciola, S.O. Tools to detect, identify and monitor Phytophthora species in natural ecosystems. J. Plant Pathol. 2007, 89, 13–28. [Google Scholar]
- Tedersoo, L.; Drenkhan, R.; Anslan, S.; Morales-Rodriguez, C.; Cleary, M. High-throughput identification and diagnostics of pathogens and pests: Overview and practical recommendations. Mol. Ecol. Resour. 2019, 19, 47–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelfattah, A.; Sanzani, S.M.; Wisniewski, M.; Berg, G.; Cacciola, S.O.; Schena, L. Revealing Cues for Fungal Interplay in the Plant–Air Interface in Vineyards. Front. Plant Sci. 2019, 10, 922. [Google Scholar] [CrossRef] [PubMed]
- Erwin, D.C.; Ribeiro, O.K. Phytophthora Diseases Worldwide; American Phytopathological Society (APS Press): St. Paul, MA, USA, 1996. [Google Scholar]
- Spadaro, D.; Agustí, N.; Ortega, S.F.; Hurtado Ruiz, M.A. Diagnostics and Identification of Diseases, Insects and Mites. In Integrated Pest and Disease Management in Greenhouse Crops; Springer International Publishing: Cham, Switzerland, 2020. [Google Scholar]
- Sanzani, S.M.; Li Destri Nicosia, M.G.; Faedda, R.; Cacciola, S.O.; Schena, L. Use of quantitative PCR detection methods to study biocontrol agents and phytopathogenic fungi and oomycetes in environmental samples. J. Phytopathol. 2014, 162, 1–13. [Google Scholar] [CrossRef]
- Schena, L.; Li Destri Nicosia, M.G.; Sanzani, S.M.; Faedda, R.; Ippolito, A.; Cacciola, S.O. Development of quantitative PCR detection methods for phytopathogenic fungi and oomycetes. J. Plant Pathol. 2013, 95, 7–24. [Google Scholar]
- Prencipe, S.; Sillo, F.; Garibaldi, A.; Gullino, M.L.; Spadaro, D. Development of a Sensitive TaqMan qPCR Assay for Detection and Quantification of Venturia inaequalis in Apple Leaves and Fruit and in Air Samples. Plant Dis. 2020, 104, 2851–2859. [Google Scholar] [CrossRef]
- Carneiro, G.A.; Matíc, S.; Ortu, G.; Garibaldi, A.; Spadaro, D.; Gullino, M.L. Development and validation of a TaqMan real-time PCR assay for the specific detection and quantification of Fusarium fujikuroi in Rice plants and seeds. Phytopathology 2017, 107, 885–892. [Google Scholar] [CrossRef] [Green Version]
- Notomi, T.; Okayama, H.; Masubuchi, H.; Yonekawa, T.; Watanabe, K.; Amino, N.; Hase, T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000, 28, e63. [Google Scholar] [CrossRef] [Green Version]
- Chomczynski, P.; Rymaszewski, M. Alkaline polyethylene glycol-based method for direct PCR from bacteria, eukaryotic tissue samples, and whole blood. Biotechniques 2006, 40, 454–458. [Google Scholar] [CrossRef]
- Franco Ortega, S.; Del Pilar Bustos López, M.; Nari, L.; Boonham, N.; Gullino, M.L.; Spadaro, D. Rapid Detection of Monilinia fructicola and Monilinia laxa on Peach and Nectarine using Loop-Mediated Isothermal Amplification. Plant Dis. 2019, 9, 2305–2314. [Google Scholar] [CrossRef]
- Franco Ortega, S.; Tomlinson, J.; Hodgetts, J.; Spadaro, D.; Gullino, M.L.; Boonham, N. Development of loop-mediated isothermal amplification assays for the detection of seedborne fungal pathogens Fusarium fujikuroi and Magnaporthe oryzae in rice seed. Plant Dis. 2018, 102, 1549–1558. [Google Scholar] [CrossRef] [Green Version]
- Franco Ortega, S.; Tomlinson, J.; Gilardi, G.; Spadaro, D.; Gullino, M.L.; Garibaldi, A.; Boonham, N. Rapid detection of Fusarium oxysporum f. sp. lactucae on soil, lettuce seeds and plants using loop-mediated isothermal amplification. Plant Pathol. 2018, 67, 1462–1473. [Google Scholar] [CrossRef] [Green Version]
- Franco Ortega, S.; Prencipe, S.; Gullino, M.L.; Spadaro, D. New molecular tool for a quick and easy detection of apple scab in the field. Agronomy 2020, 10, 581. [Google Scholar] [CrossRef] [Green Version]
- Lau, H.Y.; Wu, H.; Wee, E.J.H.; Trau, M.; Wang, Y.; Botella, J.R. Specific and sensitive isothermal electrochemical biosensor for plant pathogen DNA detection with colloidal gold nanoparticles as probes. Sci. Rep. 2017, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sanzari, I.; Leone, A.; Ambrosone, A. Nanotechnology in Plant Science: To Make a Long Story Short. Front. Bioeng. Biotechnol. 2019, 7, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, K.S.; Li, S.J.; Tzeng, K.C.; Cheng, T.C.; Chang, C.Y.; Chiu, C.Y.; Liao, C.Y.; Hsu, J.J.; Lin, Z.P. Fluorescence silica nanoprobe as a biomarker for rapid detection of plant pathogens. Adv. Mater. Res. 2009, 79, 513–516. [Google Scholar] [CrossRef]
- Chen, L.; Ren, Y.; Zhang, Y.; Xu, J.; Zhang, Z.; Wang, Y. Genome-wide profiling of novel and conserved Populus microRNAs involved in pathogen stress response by deep sequencing. Planta 2012, 235, 873–883. [Google Scholar] [CrossRef]
- Chaudhary, V.; Jangra, S.; Yadav, N.R. Nanotechnology based approaches for detection and delivery of microRNA in healthcare and crop protection. J. Nanobiotechnol. 2018, 16, 40. [Google Scholar] [CrossRef] [Green Version]
- Schena, L.; Nigro, F.; Ippolito, A. Real-time PCR detection and quantification of soilborne fungal pathogens: The case of Rosellinia necatrix, Phytophthora nicotianae, P. citrophthora, and Verticillium dahliae. Phytopathol. Mediterr. 2004, 43, 273–280. [Google Scholar] [CrossRef]
- Anderson, P.K.; Cunningham, A.A.; Patel, N.G.; Morales, F.J.; Epstein, P.R.; Daszak, P. Emerging infectious diseases of plants: Pathogen pollution, climate change and agrotechnology drivers. Trends Ecol. Evol. 2004, 19, 535–544. [Google Scholar] [CrossRef]
- Creer, S.; Deiner, K.; Frey, S.; Porazinska, D.; Taberlet, P.; Thomas, W.K.; Potter, C.; Bik, H.M. The ecologist’s field guide to sequence-based identification of biodiversity. Methods Ecol. Evol. 2016, 7, 1008–1018. [Google Scholar] [CrossRef]
- Esposito, A.; Colantuono, C.; Ruggieri, V.; Chiusano, M.L. Bioinformatics for agriculture in the next-generation sequencing era. Chem. Biol. Technol. Agric. 2016, 3, 9. [Google Scholar] [CrossRef] [Green Version]
- Lindahl, B.D.; Nilsson, R.H.; Tedersoo, L.; Abarenkov, K.; Carlsen, T.; Kjøller, R.; Kõljalg, U.; Pennanen, T.; Rosendahl, S.; Stenlid, J.; et al. Fungal community analysis by high-throughput sequencing of amplified —A user’s guide. New Phytol. 2013, 199, 288–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowrousian, M. Next-generation sequencing techniques for eukaryotic microorganisms: Sequencing-based solutions to biological problems. Eukaryot. Cell 2010, 9, 1300–1310. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, R.H.; Anslan, S.; Bahram, M.; Wurzbacher, C.; Baldrian, P.; Tedersoo, L. Mycobiome diversity: High-throughput sequencing and identification of fungi. Nat. Rev. Microbiol. 2019, 17, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Wesolowska-Andersen, A.; Bahl, M.I.; Carvalho, V.; Kristiansen, K.; Sicheritz-Pontén, T.; Gupta, R.; Licht, T.R. Choice of bacterial DNA extraction method from fecal material influences community structure as evaluated by metagenomic analysis. Microbiome 2014, 2, 19. [Google Scholar] [CrossRef] [Green Version]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 87. [Google Scholar] [CrossRef] [Green Version]
- Bakker, M.G. A fungal mock community control for amplicon sequencing experiments. Mol. Ecol. Resour. 2018, 18, 541–556. [Google Scholar] [CrossRef]
- Porter, T.M.; Hajibabaei, M. Scaling up: A guide to high-throughput genomic approaches for biodiversity analysis. Mol. Ecol. 2018, 27, 313–338. [Google Scholar] [CrossRef] [Green Version]
- Tkacz, A.; Hortala, M.; Poole, P.S. Absolute quantitation of microbiota abundance in environmental samples. Microbiome 2018, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Lane, D.J.; Pace, B.; Olsen, G.J.; Stahl, D.A.; Sogin, M.L.; Pace, N.R. Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc. Natl. Acad. Sci. USA 1985, 82, 6955–6959. [Google Scholar] [CrossRef] [Green Version]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Bolchacova, E.; Voigt, K.; Crous, P.W.; et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelfattah, A.; Malacrinò, A.; Wisniewski, M.; Cacciola, S.O.; Schena, L. Metabarcoding: A powerful tool to investigate microbial communities and shape future plant protection strategies. Biol. Control 2018, 120, 1–10. [Google Scholar] [CrossRef]
- Stielow, J.B.; Lévesque, C.A.; Seifert, K.A.; Meyer, W.; Irinyi, L.; Smits, D.; Renfurm, R.; Verkley, G.J.M.; Groenewald, M.; Chaduli, D.; et al. One fungus, which genes? Development and assessment of universal primers for potential secondary fungal DNA barcodes. Pers. Mol. Phylogeny Evol. Fungi 2015, 35, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samson, R.A.; Visagie, C.M.; Houbraken, J.; Hong, S.B.; Hubka, V.; Klaassen, C.H.W.; Perrone, G.; Seifert, K.A.; Susca, A.; Tanney, J.B.; et al. Phylogeny, identification and nomenclature of the genus Aspergillus. Stud. Mycol. 2014, 78, 141–173. [Google Scholar] [CrossRef] [Green Version]
- Lalucat, J.; Mulet, M.; Gomila, M.; García-Valdés, E. Genomics in bacterial taxonomy: Impact on the genus pseudomonas. Genes 2020, 11, 139. [Google Scholar] [CrossRef] [Green Version]
- Scibetta, S.; Schena, L.; Abdelfattah, A.; Pangallo, S.; Cacciola, S.O. Selection and experimental evaluation of universal primers to study the fungal microbiome of higher plants. Phytobiomes J. 2018, 2, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Porter, T.M.; Shokralla, S.; Baird, D.; Brian Golding, G.; Hajibabaei, M. Ribosomal DNA and plastid markers used to sample fungal and plant communities from wetland soils reveals complementary biotas. PLoS ONE 2016, 11, e0142759. [Google Scholar] [CrossRef] [Green Version]
- Amatulli, M.T.; Spadaro, D.; Gullino, M.L.; Garibaldi, A. Molecular identification of Fusarium spp. associated with bakanae disease of rice in Italy and assessment of their pathogenicity. Plant Pathol. 2010, 59, 839–844. [Google Scholar] [CrossRef]
- Hibbett, D.; Abarenkov, K.; Kõljalg, U.; Öpik, M.; Chai, B.; Cole, J.; Wang, Q.; Crous, P.; Robert, V.; Helgason, T.; et al. Sequence-based classification and identification of Fungi. Mycologia 2016, 108, 1049–1068. [Google Scholar] [CrossRef]
- Ramdial, H.; Latchoo, R.K.; Hosein, F.N.; Rampersad, S.N. Phylogeny and haplotype analysis of fungi within the Fusarium incarnatum-equiseti species complex. Phytopathology 2017, 107, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Boutigny, A.L.; Gautier, A.; Basler, R.; Dauthieux, F.; Leite, S.; Valade, R.; Aguayo, J.; Ioos, R.; Laval, V. Metabarcoding targeting the EF1 alpha region to assess Fusarium diversity on cereals. PLoS ONE 2019, 14, e0207988. [Google Scholar] [CrossRef] [PubMed]
- Houbraken, J.; Samson, R.A. Phylogeny of Penicillium and the segregation of Trichocomaceae into three families. Stud. Mycol. 2011, 70, 1–51. [Google Scholar] [CrossRef] [PubMed]
- Prencipe, S.; Siciliano, I.; Gatti, C.; Garibaldi, A.; Gullino, M.L.; Botta, R.; Spadaro, D. Several species of Penicillium isolated from chestnut flour processing are pathogenic on fresh chestnuts and produce mycotoxins. Food Microbiol. 2018, 76, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Prencipe, S.; Siciliano, I.; Contessa, C.; Botta, R.; Garibaldi, A.; Gullino, M.L.; Spadaro, D. Characterization of Aspergillus section Flavi isolated from fresh chestnuts and along the chestnut flour process. Food Microbiol. 2018, 69, 159–169. [Google Scholar] [CrossRef]
- Schena, L.; Cooke, D.E.L. Assessing the potential of regions of the nuclear and mitochondrial genome to develop a “molecular tool box” for the detection and characterization of Phytophthora species. J. Microbiol. Methods 2006, 67, 70–85. [Google Scholar] [CrossRef]
- Legeay, J.; Husson, C.; Cordier, T.; Vacher, C.; Marcais, B.; Buée, M. Comparison and validation of Oomycetes metabarcoding primers for Phytophthora high throughput sequencing. J. Plant Pathol. 2019, 101, 743–748. [Google Scholar] [CrossRef]
- James, T.Y.; Kauff, F.; Schoch, C.L.; Matheny, P.B.; Hofstetter, V.; Cox, C.J.; Celio, G.; Gueidan, C.; Fraker, E.; Miadlikowska, J.; et al. Reconstructing the early evolution of Fungi using a six-gene phylogeny. Nature 2006, 443, 818–822. [Google Scholar] [CrossRef]
- Schoch, C.L.; Shoemaker, R.A.; Seifert, K.A.; Hambleton, S.; Spatafora, J.W.; Crous, P.W. A multigene phylogeny of the Dothideomycetes using four nuclear loci. Mycologia 2006, 98, 1041–1052. [Google Scholar] [CrossRef]
- Anelli, P.; Peterson, S.W.; Haidukowski, M.; Logrieco, A.F.; Moretti, A.; Epifani, F.; Susca, A. Penicillium gravinicasei, a new species isolated from cave cheese in Apulia, Italy. Int. J. Food Microbiol. 2018, 282, 66–70. [Google Scholar] [CrossRef]
- Karlsson, I.; Edel-Hermann, V.; Gautheron, N.; Durling, M.B.; Kolseth, A.K.; Steinberg, C.; Persson, P.; Friberg, H. Genus-specific primers for study of Fusarium communities in field samples. Appl. Environ. Microbiol. 2016, 82, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Tedersoo, L.; Anslan, S.; Bahram, M.; Põlme, S.; Riit, T.; Liiv, I.; Kõljalg, U.; Kisand, V.; Nilsson, R.H.; Hildebrand, F.; et al. Shotgun metagenomes and multiple primer pair-barcode combinations of amplicons reveal biases in metabarcoding analyses of fungi. MycoKeys 2015, 10, 1–43. [Google Scholar] [CrossRef]
- Nafian, F.; Gharavi, S.; Soudi, M.R. Degenerate primers as biomarker for gene-targeted metagenomics of the catechol 1, 2-dioxygenase-encoding gene in microbial populations of petroleum-contaminated environments. Ann. Microbiol. 2016, 66, 1127–1136. [Google Scholar] [CrossRef]
- Sauvage, T.; Schmidt, W.E.; Suda, S.; Fredericq, S. A metabarcoding framework for facilitated survey of endolithic phototrophs with tufA. BMC Ecol. 2016, 16, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbrecht, V.; Hebert, P.D.N.; Steinke, D. Slippage of degenerate primers can cause variation in amplicon length. Sci. Rep. 2018, 8, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Bodenhausen, N.; Horton, M.W.; Bergelson, J. Bacterial Communities Associated with the Leaves and the Roots of Arabidopsis thaliana. PLoS ONE 2013, 8, e56329. [Google Scholar] [CrossRef]
- Chelius, M.K.; Triplett, E.W. The diversity of archaea and bacteria in association with the roots of Zea mays L. Microb. Ecol. 2001, 41, 252–263. [Google Scholar] [CrossRef]
- Hanshew, A.S.; Mason, C.J.; Raffa, K.F.; Currie, C.R. Minimization of chloroplast contamination in 16S rRNA gene pyrosequencing of insect herbivore bacterial communities. J. Microbiol. Methods 2013, 95, 149–155. [Google Scholar] [CrossRef] [Green Version]
- Abdelfattah, A.; Whitehead, S.R.; Macarisin, D.; Liu, J.; Burchard, E.; Freilich, S.; Dardick, C.; Droby, S.; Wisniewski, M. Effect of washing, waxing and low-temperature storage on the postharvest microbiome of apple. Microorganisms 2020, 8, 944. [Google Scholar] [CrossRef]
- Lundberg, D.S.; Yourstone, S.; Mieczkowski, P.; Jones, C.D.; Dangl, J.L. Practical innovations for high-throughput amplicon sequencing. Nat. Methods 2013, 10, 999–1002. [Google Scholar] [CrossRef]
- Scibetta, S.; Schena, L.; Chimento, A.; Cacciola, S.O.; Cooke, D.E.L. A molecular method to assess Phytophthora diversity in environmental samples. J. Microbiol. Methods 2012, 88, 356–368. [Google Scholar] [CrossRef]
- Prigigallo, M.I.; Mosca, S.; Cacciola, S.O.; Cooke, D.E.L.; Schena, L. Molecular analysis of Phytophthora diversity in nursery-grown ornamental and fruit plants. Plant Pathol. 2015, 64, 1308–1319. [Google Scholar] [CrossRef] [Green Version]
- Prigigallo, M.I.; Abdelfattah, A.; Cacciola, S.O.; Faedda, R.; Sanzani, S.M.; Cooke, D.E.L.; Schena, L. Metabarcoding analysis of phytophthora diversity using genus-specific primers and 454 pyrosequencing. Phytopathology 2016, 106, 305–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mora-Sala, B.; Gramaje, D.; Abad-Campos, P.; Berbegal, M. Diversity of Phytophthora species associated with Quercus ilex L. in three Spanish regions evaluated by NGS. Forests 2019, 10, 979. [Google Scholar] [CrossRef] [Green Version]
- Mosca, S.; Li Destri Nicosia, M.G.; Cacciola, S.O.; Schena, L. Molecular analysis of Colletotrichum species in the carposphere and phyllosphere of olive. PLoS ONE 2014, 9, e114031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedl, M.A.; Druzhinina, I.S. Taxon-specific metagenomics of Trichoderma reveals a narrow community of opportunistic species that regulate each other’s development. Microbiology 2012, 158 Pt 1, 69. [Google Scholar] [CrossRef] [Green Version]
- Knight, R.; Vrbanac, A.; Taylor, B.C.; Aksenov, A.; Callewaert, C.; Debelius, J.; Gonzalez, A.; Kosciolek, T.; McCall, L.I.; McDonald, D.; et al. Best practices for analysing microbiomes. Nat. Rev. Microbiol. 2018, 16, 410–422. [Google Scholar] [CrossRef] [Green Version]
- Amir, A.; McDonald, D.; Navas-Molina, J.A.; Kopylova, E.; Morton, J.T.; Zech Xu, Z.; Kightley, E.P.; Thompson, L.R.; Hyde, E.R.; Gonzalez, A.; et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R. UNOISE2: Improved error-correction for Illumina 16S and ITS amplicon sequencing. bioRxiv 2016. [Google Scholar] [CrossRef] [Green Version]
- Pauvert, C.; Buée, M.; Laval, V.; Edel-Hermann, V.; Fauchery, L.; Gautier, A.; Lesur, I.; Vallance, J.; Vacher, C. Bioinformatics matters: The accuracy of plant and soil fungal community data is highly dependent on the metabarcoding pipeline. Fungal Ecol. 2019, 41, 23–33. [Google Scholar] [CrossRef]
- Eren, A.M.; Maignien, L.; Sul, W.J.; Murphy, L.G.; Grim, S.L.; Morrison, H.G.; Sogin, M.L. Oligotyping: Differentiating between closely related microbial taxa using 16S rRNA gene data. Methods Ecol. Evol. 2013, 4, 1111–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joishy, T.K.; Dehingia, M.; Khan, M.R. Bacterial diversity and metabolite profiles of curd prepared by natural fermentation of raw milk and back sloping of boiled milk. World J. Microbiol. Biotechnol. 2019, 35, 102. [Google Scholar] [CrossRef] [PubMed]
- Berry, M.A.; White, J.D.; Davis, T.W.; Jain, S.; Johengen, T.H.; Dick, G.J.; Sarnelle, O.; Denef, V.J. Are oligotypes meaningful ecological and phylogenetic units? A case study of Microcystis in Freshwater lakes. Front. Microbiol. 2017, 8, 365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okazaki, Y.; Fujinaga, S.; Tanaka, A.; Kohzu, A.; Oyagi, H.; Nakano, S.I. Ubiquity and quantitative significance of bacterioplankton lineages inhabiting the oxygenated hypolimnion of deep freshwater lakes. ISME J. 2017, 11, 2279–2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quijada, N.M.; De Filippis, F.; Sanz, J.J.; García-Fernández, M. del C.; Rodríguez-Lázaro, D.; Ercolini, D.; Hernández, M. Different Lactobacillus populations dominate in “Chorizo de León” manufacturing performed in different production plants. Food Microbiol. 2018, 70, 94–102. [Google Scholar] [CrossRef]
- Franco Ortega, S.; Ferrocino, I.; Adams, I.; Silvestri, S.; Spadaro, D.; Gullino, M.L.; Boonham, N. Monitoring and Surveillance of Aerial Mycobiota of Rice Paddy through DNA Metabarcoding and qPCR. J. Fungi 2020, 6, 372. [Google Scholar] [CrossRef]
- Abdelfattah, A.; Li Destri Nicosia, M.G.; Cacciola, S.O.; Droby, S.; Schena, L. Metabarcoding analysis of fungal diversity in the phyllosphere and carposphere of olive (Olea europaea). PLoS ONE 2015, 10, e0131069. [Google Scholar] [CrossRef] [Green Version]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef] [Green Version]
- White, R.A., III; Brown, J.; Colby, S.; Overall, C.; Lee, J.-Y.; Zucker, J.; Glaesemann, K.; Jansson, C.; Jansson, J. ATLAS (Automatic Tool for Local Assembly Structures)—A comprehensive infrastructure for assembly, annotation, and genomic binning of metagenomic and metatranscriptomic data. PeerJ Prepr. 2017, 5, e2843v1. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.; Zhou, L.; Hall, D.G.; Li, W.; Doddapaneni, H.; Lin, H.; Liu, L.; Vahling, C.M.; Gabriel, D.W.; Williams, K.P.; et al. Complete genome sequence of citrus huanglongbing bacterium, “Candidatus Liberibacter asiaticus” obtained through metagenomics. Mol. Plant.-Microbe Interact. 2009, 22, 1011–1020. [Google Scholar] [CrossRef] [Green Version]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrington, C.T.; Lin, E.I.; Olson, M.T.; Eshleman, J.R. Fundamentals of pyrosequencing. Arch. Pathol. Lab. Med. 2013, 137, 1296–1303. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, É.D.; Duceppe, M.O.; Bérubé, J.A.; Kimoto, T.; Lemieux, C.; Bilodeau, G.J. Screening for exotic forest pathogens to increase survey capacity using metagenomics. Phytopathology 2018, 108, 1509–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, J.A.; Pan, Y.; Tooming-Klunderud, A.; Eijsink, V.G.H.; McHardy, A.C.; Nederbragt, A.J.; Pope, P.B. Improved metagenome assemblies and taxonomic binning using long-read circular consensus sequence data. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]
- James, T.Y.; Marino, J.A.; Perfecto, I.; Vandermeer, J. Identification of putative coffee rust mycoparasites via single-molecule DNA sequencing of infected pustules. Appl. Environ. Microbiol. 2016, 82, 631–639. [Google Scholar] [CrossRef] [Green Version]
- Schlaeppi, K.; Bender, S.F.; Mascher, F.; Russo, G.; Patrignani, A.; Camenzind, T.; Hempel, S.; Rillig, M.C.; van der Heijden, M.G.A. High-resolution community profiling of arbuscular mycorrhizal fungi. New Phytol. 2016, 212, 780–791. [Google Scholar] [CrossRef]
- Leggett, R.M.; Clark, M.D. A world of opportunities with nanopore sequencing. J. Exp. Bot. 2017, 68, 5419–5429. [Google Scholar] [CrossRef]
- Sanderson, N.D.; Street, T.L.; Foster, D.; Swann, J.; Atkins, B.L.; Brent, A.J.; McNally, M.A.; Oakley, S.; Taylor, A.; Peto, T.E.A.; et al. Real-time analysis of nanopore-based metagenomic sequencing from infected orthopaedic devices. BMC Genom. 2018, 19, 714. [Google Scholar] [CrossRef] [Green Version]
- Menegon, M.; Cantaloni, C.; Rodriguez-Prieto, A.; Centomo, C.; Abdelfattah, A.; Rossato, M.; Bernardi, M.; Xumerle, L.; Loader, S.; Delledonne, M. On site DNA barcoding by nanopore sequencing. PLoS ONE 2017, 12, e0184741. [Google Scholar] [CrossRef] [Green Version]
- Abarenkov, K.; Nilsson, R.H.; Larsson, K.H.; Alexander, I.J.; Eberhardt, U.; Erland, S.; Høiland, K.; Kjøller, R.; Larsson, E.; Pennanen, T.; et al. The UNITE database for molecular identification of fungi—Recent updates and future perspectives. New Phytol. 2010, 186, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Berney, C.; Ciuprina, A.; Bender, S.; Brodie, J.; Edgcomb, V.; Kim, E.; Rajan, J.; Parfrey, L.W.; Adl, S.; Audic, S.; et al. UniEuk: Time to Speak a Common Language in Protistology! J. Eukaryot. Microbiol. 2017, 64, 407–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; Desantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, R.H.; Ryberg, M.; Kristiansson, E.; Abarenkov, K.; Larsson, K.H.; Köljalg, U. Taxonomic reliability of DNA sequences in public sequences databases: A fungal perspective. PLoS ONE 2006, 1, e59. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [Green Version]
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Apweiler, R.; Alpi, E.; Antunes, R.; Arganiska, J.; Bely, B.; Bingley, M.; et al. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar] [CrossRef]
- Keepers, K.G.; Pogoda, C.S.; White, K.H.; Anderson Stewart, C.R.; Hoffman, J.R.; Ruiz, A.M.; McCain, C.M.; Lendemer, J.C.; Kane, N.C.; Tripp, E.A. Whole Genome Shotgun Sequencing Detects Greater Lichen Fungal Diversity Than Amplicon-Based Methods in Environmental Samples. Front. Ecol. Evol. 2019, 7, 484. [Google Scholar] [CrossRef] [Green Version]
- Català, S.; Pérez-Sierra, A.; Abad-Campos, P. The use of genus-specific amplicon pyrosequencing to assess Phytophthora species diversity using eDNA from soil and water in northern spain. PLoS ONE 2015, 10, e0119311. [Google Scholar] [CrossRef] [Green Version]
- Lewis, J.A.; Papavizas, G.C. Biocontrol of cotton damping-off caused by Rhizoctonia solani in the field with formulations of Trichoderma spp. and Gliocladium virens. Crop. Prot. 1991, 10, 396–402. [Google Scholar] [CrossRef]
- Baysal-Gurel, F.; Kabir, M.N. Evaluation of fungicides and biocontrol products for the control of Phytophthora root rot of hydrangeas. Arch. Phytopathol. Plant. Prot. 2019, 52, 481–496. [Google Scholar] [CrossRef]
- Panth, M.; Hassler, S.C.; Baysal-Gurel, F. Methods for management of soilborne diseases in crop production. Agriculture 2020, 10, 16. [Google Scholar] [CrossRef] [Green Version]
- Mihajlovic, M.; Rekanovic, E.; Hrustic, J.; Grahovac, M.; Tanovic, B. Methods for management of soilborne plant pathogens. Pestic. Fitomed. 2017, 32, 9–24. [Google Scholar] [CrossRef]
- Åström, B.; Gerhardson, B. Differential reactions of wheat and pea genotypes to root inoculation with growth-affecting rhizosphere bacteria. Plant. Soil 1988, 109, 263–269. [Google Scholar] [CrossRef]
- West, J.S.; Kimber, R.B.E. Innovations in air sampling to detect plant pathogens. Ann. Appl. Biol. 2015, 166, 4–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirst, J.M. An automatic volumetric spore trap. Ann. Appl. Biol. 1952, 39, 257–265. [Google Scholar] [CrossRef]
- Chien, C.C.; Tsai, W.H.; Yang, Y.Z.; Liu, C. Epidemiology of rice blast disease in central areas of Taiwan. Chung Hua Nung Yeh Yen Chiu J. Agric. Res. China 1984, 33, 169–180. [Google Scholar]
- Comtois, P.; Alcazar, P.; Néron, D. Pollen counts statistics and its relevance to precision. Aerobiologia 1999, 15, 19–28. [Google Scholar] [CrossRef]
- Garbelotto, M.; Gonthier, P. Biology, epidemiology, and control of Heterobasidion species worldwide. Annu. Rev. Phytopathol. 2013, 51, 39–59. [Google Scholar] [CrossRef] [Green Version]
- Tiedeman, S.V. Heterobasidion annosum: Biology, ecology, impact, and control. J. Phytopathol. 2000, 148, 127–128. [Google Scholar] [CrossRef]
- Worrall, J.J.; Harrington, T.C.; Blodgett, J.T.; Conklin, D.A.; Fairweather, M.L. Heterobasidion annosum and H. parviporum in the Southern Rocky Mountains and Adjoining States. Plant. Dis. 2010, 94, 115–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguayo, J.; Fourrier-Jeandel, C.; Husson, C.; Ioos, R. Assessment of passive traps combined with high-throughput sequencing to study airborne fungal communities. Appl. Environ. Microbiol. 2018, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, I.P.; Skelton, A.; Macarthur, R.; Hodges, T.; Hinds, H.; Flint, L.; Nath, P.D.; Boonham, N.; Fox, A. Carrot yellow leaf virus is associated with carrot internal necrosis. PLoS ONE 2014, 9, e109125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbard, A.; Lewis, C.M.; Yoshida, K.; Ramirez-Gonzalez, R.H.; de Vallavieille-Pope, C.; Thomas, J.; Kamoun, S.; Bayles, R.; Uauy, C.; Saunders, D.G.O. Field pathogenomics reveals the emergence of a diverse wheat yellow rust population. Genome Biol. 2015, 16, 23. [Google Scholar] [CrossRef] [Green Version]
- Adams, I.P.; Fox, A.; Boonham, N.; Massart, S.; De Jonghe, K. The impact of high throughput sequencing on plant health diagnostics. Eur. J. Plant. Pathol. 2018, 152, 909–919. [Google Scholar] [CrossRef]
- Fredricks, D.N.; Relman, D.A. Sequence-based identification of microbial pathogens: A reconsideration of Koch’s postulates. Clin. Microbiol. Rev. 1996, 9, 18–33. [Google Scholar] [CrossRef]
- Gerner-Smidt, P.; Besser, J.; Concepción-Acevedo, J.; Folster, J.P.; Huffman, J.; Joseph, L.A.; Kucerova, Z.; Nichols, M.C.; Schwensohn, C.A.; Tolar, B. Whole genome sequencing: Bridging one-health surveillance of foodborne diseases. Front. Public Health 2019, 7, 172. [Google Scholar] [CrossRef]
- World Health Organization. Whole Genome Sequencing for Foodborne Disease Surveillance: Landscape Paper; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- Antwerpen, M.H.; Georgi, E.; Nikolic, A.; Zoeller, G.; Wohlsein, P.; Baumgärtner, W.; Peyrefitte, C.; Charrel, R.; Meyer, H. Use of next generation sequencing to study two cowpox virus outbreaks. PeerJ 2019, 7, e6561. [Google Scholar] [CrossRef] [Green Version]
- Børsting, C.; Morling, N. Next generation sequencing and its applications in forensic genetics. Forensic Sci. Int. Genet. 2015, 18, 78–89. [Google Scholar] [CrossRef]
- FDA. Whole Genome Sequencing (WGS) Program. Available online: https://www.fda.gov/food/science-research-food/whole-genome-sequencing-wgs-program (accessed on 15 January 2021).
- Gaudin, M.; Desnues, C. Hybrid capture-based next generation sequencing and its application to human infectious diseases. Front. Microbiol. 2018, 9, 2924. [Google Scholar] [CrossRef] [Green Version]
- Rai, M.; Agarkar, G. Plant-fungal interactions: What triggers the fungi to switch among lifestyles? Crit. Rev. Microbiol. 2016, 42, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Tun, H.M.; Jahan, M.; Zhang, Z.; Kumar, A.; Fernando, D.; Farenhorst, A.; Khafipour, E. Comparison of DNA-, PMA-, and RNA-based 16S rRNA Illumina sequencing for detection of live bacteria in water. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afshinnekoo, E.; Meydan, C.; Chowdhury, S.; Jaroudi, D.; Boyer, C.; Bernstein, N.; Maritz, J.M.; Reeves, D.; Gandara, J.; Chhangawala, S.; et al. Geospatial Resolution of Human and Bacterial Diversity with City-Scale Metagenomics. Cell Syst. 2015, 1, 72–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olmos, A.; Boonham, N.; Candresse, T.; Gentit, P.; Giovani, B.; Kutnjak, D.; Liefting, L.; Maree, H.J.; Minafra, A.; Moreira, A.; et al. High-throughput sequencing technologies for plant pest diagnosis: Challenges and opportunities. EPPO Bull. 2018, 48, 219–224. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pẽa, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Blankenberg, D.; Gordon, A.; Von Kuster, G.; Coraor, N.; Taylor, J.; Nekrutenko, A.; Team, G. Manipulation of FASTQ data with galaxy. Bioinformatics 2010, 26, 1783–1785. [Google Scholar] [CrossRef]
- FAO. International Standard for Phytosanitary Measures 11. Pest Risk Analysis for Quarantine Pests. Available online: http://www.fao.org/3/a-j1302e.pdf (accessed on 15 January 2021).


{kind=link}
| Field of Application | Application Scope | Techniques Used | Main Detected Pathogens | Ref. |
|---|---|---|---|---|
| Traded seed lots of gymnosperm and angiosperm tree species | General surveillance | Illumina sequencing, metabarcoding, ASV (amplicon sequence variants) assembly | Several seedborne pathogens | [18] |
| Aerial samples in rice field. | General surveillance | Illumina sequencing, metabarcoding, olygotyping, OTU (operational taxonomic unit) assembly. | Magnaporthe oryzae, Magnaporthe grisea | [103] |
| Leaves, flowers and fruits of olive | General surveillance | 454 Pyrosequencing, Metabarcoding, OTU assembly, classical phylogenetic analysis of sequences. | Collethotricum spp., Pseudocercospora cladosporioides | [104] |
| Aerial and insect trap samples, soil samples | General surveillance | Metabarcoding, IonTorrent sequencing, OTU assembly. | Heterobasidium annosum sensu stricto, Heterobasidium abietinum/parviporum, Phytophthora spp. | [111] |
| Field samples of barley, durum and soft wheat | Specific surveillance | Illumina sequencing, metabarcoding, Genus-specific primers, OTU assembly. | Fusarium spp. | [68] |
| Soil and root samples of ornamental potted plants | Specific surveillance | 454 Pyrosequencing, metabarcoding, genus-specific primers, classical phylogenetic analysis of sequences. | Phytophthora spp. | [89] |
| Soil and water samples from forests and plantations | Specific surveillance | 454 Pyrosequencing, metabarcoding, genus-specific primers, classical phylogenetic analysis of sequences. | Phytophthora spp. | [126] |
| Infected citrus infected psyllid | Pathogen identification | 454 Pyrosequencing, shotgun metagenomics, complete pathogen genome sequencing. | Candidatus Liberibacter asiaticus | [107] |
| Carrot samples | Identification of emerging pathogens | Illumina sequencing, shotgun metagenomics. | Carrot Yellow Leaf Virus | [140] |
| Infected wheat leaves | Determining outbreak origin | Illumina sequencing, shotgun metatranscriptomics. | Puccinia striiformis f. sp. Tritici | [141] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piombo, E.; Abdelfattah, A.; Droby, S.; Wisniewski, M.; Spadaro, D.; Schena, L. Metagenomics Approaches for the Detection and Surveillance of Emerging and Recurrent Plant Pathogens. Microorganisms 2021, 9, 188. https://doi.org/10.3390/microorganisms9010188
Piombo E, Abdelfattah A, Droby S, Wisniewski M, Spadaro D, Schena L. Metagenomics Approaches for the Detection and Surveillance of Emerging and Recurrent Plant Pathogens. Microorganisms. 2021; 9(1):188. https://doi.org/10.3390/microorganisms9010188
Chicago/Turabian StylePiombo, Edoardo, Ahmed Abdelfattah, Samir Droby, Michael Wisniewski, Davide Spadaro, and Leonardo Schena. 2021. "Metagenomics Approaches for the Detection and Surveillance of Emerging and Recurrent Plant Pathogens" Microorganisms 9, no. 1: 188. https://doi.org/10.3390/microorganisms9010188