
Abstract
Maximizing crop yields relies on the use of agrochemicals to control insect pests. One of the most widely used classes of insecticides are neonicotinoids that interfere with signalling of the neurotransmitter acetylcholine, but these can also disrupt crop-pollination services provided by bees. Here, we analysed whether chronic low dose long-term exposure to the neonicotinoid thiamethoxam alters gene expression and alternative splicing in brains of Africanized honey bees, Apis mellifera, as adaptation to altered neuronal signalling. We find differentially regulated genes that show concentration-dependent responses to thiamethoxam, but no changes in alternative splicing. Most differentially expressed genes have no annotated function but encode short Open Reading Frames, a characteristic feature of anti-microbial peptides. As this suggested that immune responses may be compromised by thiamethoxam exposure, we tested the impact of thiamethoxam on bee immunity by injecting bacteria. We show that intrinsically sub-lethal thiamethoxam exposure makes bees more vulnerable to normally non-pathogenic bacteria. Our findings imply a synergistic mechanism for the observed bee population declines that concern agriculturists, conservation ecologists and the public.
Similar content being viewed by others


Introduction
The western honey bee Apis mellifera is highly beneficial for human societies. Besides honey production, this semi-domesticated insect plays a critical role in sustaining global food security through the provision of managed pollination services that contribute to increased yields of many crops. Globally, crop productivity is also enhanced by the application of pesticides, including insecticides. Agrochemicals are, however, among the key factors implicated in contributing to declining bee health and abundance and there is a need to find a balance between the necessity of insecticide applications and the unintended effects to non-target species1,2,3,4,5,6.
Compared to organophosphate pesticides, neonicotinoids are relatively safe for vertebrates and have been one of the most important classes of insecticides for the last three decades7. Neonicotinoids can be applied to seeds and they then spread systemically within growing plants, killing leaf-eating pests. This reduces non-target effects compared to spraying but still represents a toxic burden for pollinators as neonicotinoids are commonly found in wildflowers adjacent to treated fields8,9.
Neonicotinoids act as agonists, competing with the neurotransmitter acetylcholine in binding to nicotinic acetylcholine receptors (nAChR)10,11,12. The increased toxicity for insects is thought to be caused by the characteristically high nAChR density within insect nervous systems13. Detailed knowledge of cellular and molecular effects of insecticide exposure is required to mitigate negative effects or refine target specificity. Changes in gene expression and processing of RNAs, including alternative splicing, are among the mechanisms available to an organism to adapt to environmental perturbations14,15. Sub-lethal exposure to xenobiotics, such as pesticides, can alter alternative splicing16,17,18. Since many ion channel genes and other important neuronal genes undergo extensive alternative splicing, they are prime targets for changes induced by xenobiotics17,19. Moreover, sub-lethal uptake of some neonicotinoids affects the honey bee brain, impairing foraging behaviour, flight, navigation, communication, learning and memory20,21,22,23,24,25.
Neonicotinoid exposure has also been linked to a decline in bee health, including a reduction of immune competence26,27,28. Insects do not have antibodies and rely on the innate immune system to fight microbial infections; both cellular and humoral responses have been identified29, 30. The cellular response leading to phagocytosis or encapsulation of pathogens is mediated by three types of haematopoietic cell lineages29,31,32. The humoral immune response is activated by Toll and Imd33 pathways, leading to the expression of short (10–100 amino acids) antimicrobial peptides (AMPs) which are highly diverse among insect species34. The Toll-pathway is triggered by Gram-positive bacteria and fungi, ultimately leading to expression of AMPs via the NFkappa-B transcription factor Dif, that are then secreted from the fat body into the haemolymph29,35,36. The Immune deficiency (Imd) pathway leads to expression of a different set of AMPs via the NFkappa-B transcription factor relish in response to peptidoglycans from primarily Gram-negative bacterial through the activation of pattern recognition receptors and a complex intracellular signalling cascade29,30,35. In addition to the fatbody, AMP expression activated by the Imd pathway has also been detected in glial cells in the brain37,38,39,40.
We have previously shown that worker-bee larvae in colonies contaminated with the neonicotinoid imidacloprid, have altered expression of genes belonging to lipid-carbohydrate-mitochondrial metabolic networks41. We have further demonstrated that sub-lethal exposure to thiamethoxam, another neonicotinoid, can cause impairment in the midgut and brain of the Africanized Apis mellifera, as well as contribute to a reduction in honey bee lifespan9,42,43,44.
In this study, we analysed the effects of chronic, sub-lethal, thiamethoxam exposure on genome-wide gene expression and alternative splicing in the brains of honey bee workers. We found 52 differentially regulated genes showing a concentration dependent response to thiamethoxam exposure. Most of these genes have no annotated function but the vast majority are characterized by encoding short Open Reading Frames (sORFs), half of which are predicted to encode antimicrobial peptides. This result suggested that immune responses may be compromised by thiamethoxam exposure. Indeed, we found that thiamethoxam-exposed bees that were also infected with bacteria had greatly decreased viability compared to infected but chemically unexposed bees. Overall, our results suggest that thiamethoxam makes bees vulnerable to bacterial infection by compromising immune responses.
Materials and methods
We assessed whether the neonicotinoid thiamethoxam alters gene expression in the brain of Africanized honey bees, Apis mellifera, after long-term exposure to field-relevant concentrations.
Thiamethoxam exposures
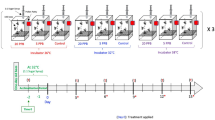
Genetically unrelated bees from three unrelated hives from different locations in the apiary of the Biosciences Institute, UNESP, Rio Claro-SP (Brazil). Brood frames were removed from three different colonies and kept for 24 h in biochemical oxygen demand (BOD) at 34 °C and relative humidity of 80 ± 10%. The newly emerged bees (aged 0–24 h) were marked with special non-toxic pen (Posca pen) and returned to their respective colonies until they were collected after 15 days. All experiments followed the recommendations of the guidelines for xenobiotic assessments on bees45,46.
Thereafter, bees were kept in groups of 20 individuals in small cages (12 cages/treatment) within an incubator at 32 °C. Bees were fed ad libitum with sugar solution (1:1 water and inverted sugar) from pierced 2 ml Eppendorf tubes for the control group (dataset A, Supplemental Data 1) and, for toxin exposure treatments, thiamethoxam (Sigma) was added at 2 ng/ml (low dose, LD, dataset B, Supplemental Data 1) or at 50 ng/ml (high dose, HD, dataset C, Supplemental Data 1). Sugar-feeders were re-filled before they were empty to guarantee continuous consumption. The tested concentrations were chosen based on the range of realistic field concentrations found in pollen and nectar9,47,48,49,50. Assuming that caged worker bees consume about 22 µl of 50% sugar per day51 the bees would have ingested 0.44 ng and 11 ng after ten days for the low and high dose exposure, respectively. After 10 days, bees were cold-anaesthetised and their brains were dissected for RNA extraction.
RNA extraction, illumina sequencing, analysis of differential gene expression and splicing
Total RNA was extracted from ten dissected brains per sample and three replicates were done for the control, the low and the high dose treatment (dataset A, Supplemental Data 1). To extract total RNA, brains were cracked in liquid nitrogen with a pestle and then homogenized in 50 µl of Tri-reagent (SIGMA). Then the volume was increased to 500 µl and proceeded following the manufacturer’s instructions. Total RNA was then stored in 70% ethanol and transported at ambient temperature from Brazil to the UK.
Total RNA was treated with DNase I (Ambion) and stranded libraries for Illumina sequencing were prepared after poly(A) selection from total RNA (1 μg) with the TruSeq stranded mRNA kit (Illumina) using random primers for reverse transcription according to the manufacturer’s instructions. Pooled indexed libraries were sequenced on an Illumina HiSeq2500 to yield 28–41 million paired-end 125 bp reads for three control, three low dose and three high dose samples. After demultiplexing, sequence reads were aligned to the Apis mellifera genome (Amel-4.5-scaffolds) using Tophat2.0.652. Differential gene expression was determined by Cufflinks-Cuffdiff and the FDR-correction for multiple tests to raw p values, with p < 0.05 considered significant53. Illumina sequencing and differential gene expression analysis was carried out by Fasteris (Switzerland). Sequencing data are deposited in GEO under GSE132858.
Alternative splicing was analysed by rMATS54 and manually inspected by uploading bam files into the Integrated Genome Viewer55 and comparing read frequency. Comparison of gene lists was made with Venny 2.1 (https://bioinfogp.cnb.csic.es/tools/venny/). Protein sequences from differentially expressed genes of bees were obtained from ensemble (http://metazoa.ensembl.org/Apis_mellifera/Info/Index) and blasted against Drosophila annotated proteins using flybase (http://flybase.org) to assign gene functions. Proteins with no assigned functions were scanned for motifs using the Interpro server (https://www.ebi.ac.uk/interpro/)56. Short ORFs were analysed for antimicrobial peptide prediction using the following server: https://www.dveltri.com/ascan/v2/ascan.html57.
Reverse transcription quantitative polymerase chain reaction (RT-qPCR)
Reverse transcription was carried out with Superscript II (Invitrogen) as previously described58. PCR was performed as described and PCR products were analysed on ethidium bromide stained 3% agarose gels19. Neuronal genes used as reference for gene expression comparison were bee erect wing (ewg, GB44653) and Amyloid Precursor Protein-like (Appl, GB48454), based on expression analysis in Drosophila59,60. To validate cDNAs obtained from RT, the following primers were used to amplify bee ewg with AM ewgG F (CCTGATGGTACCGTATCAATTATTCAAGTTG) and AM ewgH R (CCGTGTCCATCTTCTCCTGTGAGAATGATTTG), bee Appl with AM Appl F (GCGCGATTCCAGGAAACTGTTGCTGCTC) and AM Appl R2 (CTGCTGTCCAGCAGATGTTTGTAATGAG). Additional primers were GB45995 (GTTGCATTTTTACGCGTACAGTTACACGACAG) and GB45995 R (GGGAAATCCCCGGGAAGAGAGCAACTGGAG), GB45995 F (GTTGCATTTTTACGCGTACAGTTACACGACAG) and GB45995 R (GGGAAATCCCCGGGAAGAGAGCAACTGGAG), and GB47479 F (GGGCTATTTGCTATCTAAGTGATCCTCC) and GB47479 R (GGGTTTAGGAGTTTTCGTTTTAGCTGCTG) and PCR amplifications were done by 30 s initial denaturation at 94 °C, and then 40 cycles in total with 30 s at 94 °C, annealing at 48 °C with 30 s extension at 72 °C for 2 cycles, then at 50 °C with 30 s extension at 72 °C for 2 cycles and then at 52 °C with and 60 s extension at 72 °C for 36 cycles with a final extension of 2 min at 72 °C.
RNA quality and quantity were assessed using an Agilent 6000Nano Kit. 250 ng of DNAse-treated RNA (the same samples used for deep seq) was reversed transcribed using a SuperScript III Invitrogen kit at 55 °C, following manufacturer’s instructions. Primers for quantitative real-time PCR (RT-qPCR) were designed by Primer Express software (exon/exon junctions included except for GB41813). Amplicon sizes were assessed in 15% acrylamide gel, PCR products were then sequenced and efficiency of every pair of primers was included in the analysis.
Duplicate samples of cDNA were amplified in Real Time PCR with SybrGreen chemistry under following conditions: denaturation 93 °C for 30 s, annealing 60 °C for 30 s, elongation 72 °C for 30 s. GB41923 was amplified with primers GB41923 F1 (CGCGTTGATCGTCATGATATTG) and GB41923 R1 (CTATAAGGAAATTTTGAGCCTTCGA), GB40669 with primers GB40669 F1 (GGCCGGATATCGCTTCAAA) and GB40669 R1 (GTCTCTTTTATCTTTTCCTCGGAATTC), and GB48969 with primers GB48969 F1 (TTGCAGCCGTAGCAAAAGGTA), GB48969 R1 (ACCGATTTGAGCACCTTGGT) and bubblegum (bgm, GB51680) with primers bgm F1 (CATGCACAAAGAGTACAAAAATTTCA) and bgm R1 (TGGTCCCAATTCTCCAGTAACA). Analysis of CT values was performed in Light Cycler480 software and normalization and differential expression was determined with the 2−∆∆Ct method61 by normalizing to the expression of ewg (GB44653), Appl (GB48454) and actin (GB44311) genes. Actin and ewg were amplified with primers ewg Fw2 (CCGCGTCTCCTACAGCTCTT) and ewg Rv2 (TGTAAAACTGCCGTAGGATATTGG) and actF1 (TTCCCATCTATCGTCGGAAGA) and actR1 (TTTGTCCCATGCCAACCAT), and Appl with primers AM appl F and AM appl R2, following41,62.
Bacterial infection assays
Thiamethoxam-induced alteration of anti-microbial peptide gene expression could disrupt bees’ immune response. To evaluate the effect of thiamethoxam on immunity, we adopted an assay procedure, initially developed for Drosophila, which assesses how efficiently injected non-pathogenic bacteria are cleared by anti-microbial peptides63. To assay clearance in bees we used Bacillus badius, a normally non-pathogenic bacterium commonly found in the environment and Ochrobactrum anthropi, which are Gram-positive and Gram-negative members of the honey bee microbiome64,65. O. anthropi were isolated from worker bee gut cultures by plating the gut content on LB agar plates incubated at 30 °C.
Bacterial species were identified by colony PCR and ribosomal 16S sequencing: a colony was picked from an LB agar plate with a yellow tip and placed into 10 µl TE in a PCR tube and heated to 94 °C for 5 min. The PCR mix was added adjusting the MgCl concentration to 1.5 mM. PCR was done for 20–40 cycles with 54 °C annealing for 40 s and 1 min extension at 72 °C. A 490 bp fragment of the ribosomal 16S gene was amplified with primers 16S F (ACTGAGACACGGYCCAGACTCCTACGTC) and 16S R (GCGTGGACTACCAGGGTATCTAATCC) and sequenced with primer 16S Fseq (CTCCTACGGGAGGCAGCAGTRGGGTC). If sequences did not yield a single species, primers 16S F2 (GTGGACTACCAGGGTATCTAATCCTG) and 16S R2 (CCTACGGTTACCTTGTTACGACTTCAC) were used for amplification of a 733 bp fragment, which was sequenced by 16S R2seq (CCATGGTGTGACGGGCGGTGTGTAC).
Forager honey bees for infection assays were collected from colonies of the Winterbourne Garden of the University of Birmingham (UK). They were kept and injected with bacteria as we described previously19. Bacteria for injections were freshly plated and grown overnight on LB plates. Then a single colony was used to inoculate a 5 ml LB in a 50 ml Falcon tube and grown overnight to saturation: 2 µl of this culture was then injected. Bacterial titres of cultures were determined at the time of injections by plating 100 µl of 105, 106 and 107 dilutions in LB and counting the colonies the next day: this showed that 2–8 × 106 B. badius or O. anthropi were typically injected. In some treatments the injected bacteria were diluted tenfold (Fig. 3). Groups of 8 to 12 bees were exposed to combinations of bacterial and thiamethoxam doses and survival was assessed at 24 h and 48 h.
Data on survival were analysed using backwards stepwise logistic ANOVAs adopting a logit-link function and assuming quasi-binomial distributed errors. These analyses were carried out using GenStat v.19 (VSN International, Hemel Hempstead). Statistical analysis of viability were done with GraphPad prism using ANOVA followed by Tukey–Kramer post-hoc tests.
Results
Chronic thiamethoxam exposure effects gene expression
After low (LD) and high dose (HD) exposure to thiamethoxam, there were 222 up- and 181 down-regulated genes for LD (Fig. 1) and 233 up- and 114 down-regulated genes for HD with a 1.5 fold difference in expression compared to the control treatment (Fig. 1; Supplemental Data 1). From these differentially regulated genes, 37 were up-regulated and 15 were down-regulated in a dose-sensitive manner (Fig. 1; Supplemental Data 1).

Thiamethoxam induces differential expression in a subset of genes. Venn diagrams indicating the number of differentially expressed genes between control bees and bees exposed to a low dose (left) and high dose (right) of thiamethoxam that were up- (A) or down- (B) regulated. 37 genes were up-regulated and 15 genes were down-regulated in a dose-sensitive manner.
To validate these results from the Illumina sequencing, we performed RT-qPCR for three of the dose-responsive genes: GB41923, a putative sodium-chloride co-transporter, and GB48969, GB40669, two genes with unknown function. We detected an expression difference for all three genes upon thiamethoxam exposure (Supplemental Fig. 1). We also validated and confirmed differential expression of bubblegum, encoding a very long-chain acyl-CoA synthetase, which has been found to be down-regulated in honey bee larvae exposed to the neonicotinoid imidacloprid41 (Supplemental Fig. 1).
Alternative splicing has been suggested as a mechanism to adapt gene expression to environmental changes17,66. We analysed the RNA-seq data for changes in alternative splicing but found no conclusive patterns (Supplemental Information; Supplemental data 2).
Dose-responsive expression occurs mostly in genes encoding uncharacterized ORFs
Next, we categorized the genes dose-responsive to thiamethoxam according to their functions, taking into account known functions of orthologues in Drosophila and functions deduced from annotated protein domains retrieved by BLAST analysis. Amongst the dose-sensitive genes that were up-regulated (Fig. 1A), 14% (5/37) were assigned roles in cellular signalling (with potential links to altered neuronal function, such as olfactory and taste perception) and as structural components (cytoskeleton), and 8% (3/37) were assigned functions in transcriptional regulation of gene expression (Fig. 2A). However, 59% (22/37) of dose-sensitive up-regulated genes and 73% (11/15) of dose-sensitive down-regulated genes had neither clear orthologues in Drosophila nor any recognizable protein domains that would indicate a biological function (Fig. 2A), which is in contrast to about 20% of genes with unknown function in gene expression studies in Drosophila67,68.

Classification of thiamethoxam induced differentially expressed genes according to function and size. (A) Numbers of genes are plotted according to functions for thiamethoxam induced differentially expressed genes with annotated functions for up- (top) and down-regulated (bottom) genes. (B) Pie charts indicating the fraction of thiamethoxam induced differentially expressed genes according the ORF length for up- (left) and down- (right) regulated genes.
Most thiamethoxam dose-responsive genes encode short proteins
Many of the dose-responsive, differentially expressed genes with unknown function coded for short ORFs. For thiamethoxam-induced differentially up- and down-regulated genes, respectively 73% (27/37) and 87% (13/15) encode for genes with ORFs of 250 amino acids or shorter (Fig. 2). Using a machine learning algorithm57, we predicted from the 40 genes coding for peptides of ≤ 250 amino acids, that 17 (43%), have antimicrobial function (11/17 genes were up-regulated and 6/17 were down-regulated, Fig. 2).
Thiamethoxam makes worker bees more vulnerable to bacterial infection
When saturated liquid cultures (2–8 Mio bacteria in 2 µl) of B. badius and O. anthropi were injected into bees that were not exposed to thiamethoxam, viability was little affected, indicating that the bee immune system usually clears the infection efficiently (Fig. 3, Table 1). In contrast, co-injection of normally sub-lethal doses of thiamethoxam together with either B. badius or O. anthropi resulted in bee death (Fig. 3, Table 1). In particular, at a dose of 1 µM thiamethoxam (arrow in Fig. 3) bees generally survived for 48 h. In contrast, presence of either B. badius or O. anthropi together with thiamethoxam killed almost all bees within 48 h.

Thiamethoxam exposure makes bees vulnerable to infection by Bacillus badius and Ochrobactrum anthropi. Viability of groups of 8–12 bees 24 h (grey bars) and 48 h (black bars) after injection with B. badius (A) or O. anthropi (B) (2–8 × 106 and diluted 1:10) alone or together with thiamethoxam (at a range of doses, µM) shown as means and standard errors of three replicates. Arrows indicate injection of a 1 µM thiamethoxam solution with (A right and B) or without bacteria (A left). Statistical significance is indicated by **(p < 0.01) and ***(p < 0.001).
Using data on each bacterial species separately, along with replicates without bacterial exposure, showed that bee viability was negatively affected by increasing doses of thiamethoxam and of bacteria (Table 1: 2-way logistic ANOVAs). At 48 h, a synergistic interaction between these main effects was detected when the bacterial species was B. badius: bee viability declined more rapidly in response to bacterial dose when bees were exposed to higher doses of thiamethoxam (Table 1, Fig. 3).
To test for differences between the effects of the two bacterial species, replicates without bacterial exposure were excluded and the effects of species, non-zero bacterial dose, thiamethoxam and their interactions were explored (Table 1: 3-way logistic ANOVAs). At 24 h, bee viability declined with both thiamethoxam dose and bacterial dose and was lower when the bacterium applied was O. anthropi, without interactions (Table 1). At 48 h, the same dosage effects were found but the species effect was not significant (Table 1). There was, however, a marginally non-significant interaction between bacterial species and bacterial dose: as probability estimates from logistic analyses are inexact, we explored the consequences of retaining this interaction in the model. This generated a further significant interaction between thiamethoxam dose and the species of bacteria applied (F1,21 = 5.85, p = 0.025): O. anthropi affected bees more negatively than did B. badius as pesticide dose increased.
We conclude that thiamethoxam suppresses the abilities of bees to cope with natural challenges of the immune system, which would normally not be fatal.
Discussion
Our study addresses the effect of thiamethoxam on gene expression at field relevant doses of thiamethoxam found in pollen and nectar. In particular we chose for these experiments a low and a high dose, which is about 0.04 ng and 1.1 ng per bee per day for a 10 day chronic long-term exposure. A number of studies have used thiamethoxam doses in the same range for analysing changes in gene expression or observing an impact on behaviour following short term exposure18,20,21,22,23,24,69,70. In fact, it was found that chronic low-dose long-term thiamethoxam exposure altered bee activity, motor function and movement to light24, but to our knowledge no previous analysis of gene expression under such conditions has been done.
A key finding of our analysis of honey bee transcriptomes is the highly enriched fraction of dose-responsive, uncharacterised genes encoding short open reading frames (sORFs). Such sORFs have only recently been recognized to encode functional peptides71,72,73, some of which play important roles during development or have a function in the immune system74,75. Intriguingly, the majority of the dose-responsive sORFs we have identified are predicted to encode peptides with antimicrobial function (anti-microbial peptides, AMPs). The sORFs we identified have not been reported in prior whole-transcriptome evaluations of neonicotinoid exposure in bee brains despite using similar concentrations of neonicotinoids18,69,70. A possible explanation for the different gene set we obtained after thiamethoxan exposure is that we analysed changes after long-term, low dose exposure, while prior studies used shorter exposures.
The finding that mostly sORFs, and among them many predicted AMPs are differentially expressed points to the immune system for being dysregulated by neonicotinoids. The best-characterized AMPs are expressed in the fat body upon pathogen exposure, but it is now recognized that glial cells in the brain also exert immune functions and express AMPs37,39,40. Accordingly, the sORFs that we found to be differentially expressed upon chronic low dose thiamethoxam exposure might form a basal immune defence, similar to the antimicrobial environment present in saliva in vertebrates which contains many AMPs.
Glia are support cells of neurons and are protective in a number of ways. Dysregulated AMP’s in the brain have been linked to neurodegenerative disease37,38,40. Hence, the dysregulation of sORFs in response to thiamethoxam exposure could indicate disturbed communication between these two cell types beyond immune functions. Likewise, antimicrobial peptides have also been identified in having a role in learning and memory in Drosophila, but the molecular mechanism how AMPs enhance learning and memory remains unknown76.
We noted that other studies found an overrepresentation of down-regulated genes with known function in immune and defence processes upon exposure to different types of neonicotinoids18,26,69,70,77. In contrast we did not find differentially regulated immune genes including the known AMPs in the brain. AMPs are highly expressed in the fat body. For the analysis of gene expression in bee brains we dissected them from cold-anaesthetized bees and ensured that all fat body surrounding the brain was removed. It is possible, that freezing of brains prior to dissection69 or residual fat body adhering to the brain contributed considerably to a different profile of immune gene expression in other studies.
Immunosuppression has also observed been upon exposure to neonicotinoids co-infecting bees with viruses of Nosema, a unicellular parasite of bees. In particular, this affected the cellular response, but also expression of AMPs18,26,69,70,77. Since we did not detect differential expression of known immune genes, the bees in our study were not infected with these known pathogens, but we cannot exclude an impact of other microbiota that are non-pathogenic in healthy bees. Moreover, our analysis focused on gene expression in the brain, where glial cells govern immune functions, which is a different humoral response than mediated by fatbody cells secreting AMPs into the hemolymph.
Bees, including Apis mellifera, are characterised by their limited set of canonical immune genes, compared to non-social insects, such as the fruit fly D. melanogaster78,79,80. Currently, only six antimicrobial-peptide genes comprising four gene families have been described in honey bees80. In contrast, Drosophila has 20 antimicrobial-peptide genes comprising eight gene families29,80. From these genes, only defensin is conserved between honey bees and Drosophila, consistent with the idea that antimicrobial peptides evolve fast to adapt to species-specific environmental conditions34. Given the low number of known antimicrobial peptides in bees, it is conceivable that new (currently uncharacterised) genes encoding antimicrobial peptides are evolving. Consistent with this idea, some of the sORFs differentially regulated by thiamethoxam are bee or hymenopteran specific.
Various agrochemicals, have been shown to alter the gut microbiome81,82. Our results are consistent with previous findings where neonicotinoid exposure adversely affects insect immunity82,83. Specifically, we have shown that immune challenges from what are normally non-pathogenic bacteria become fatal to bees when combined with thiamethoxam exposure. How foreign bacteria enter the body of bees appears to be crucial. For example, Dickel and colleagues found that oral ingestion of Enterococcus faecalis bacteria, increases the survival neonicotinoid-exposed, caged worker bees84. In our study, the bacteria were injected between segments, which mimics punctures sustained by attacks of the Varroa mite. We note that pathogens can enter bee haemolymph through punctures inflicted by Varroa destructor mites65 and thus there may be considerable mortality within hives infested with Varroa and also exposed to thiamethoxam.
In summary, the most prominent changes in gene expression upon long-term low-dose thiamethoxam exposure identified mostly genes that encode short ORFs, around half of which are predicted to code for antimicrobial peptides. Furthermore, thiamethoxam exposure reduced the capacity of bees to withstand microbial infection. Taken together, these findings imply that low doses of neonicotinoids may be intrinsically sub-lethal to bees but can be ultimately fatal via a weakened immune response to extrinsic pathogens. The roles of the identified genes in the immune response of bees will need to be identified to establish how bee immunity might be strengthened to resist bacterial infections.
References
Klein, S., Cabirol, A., Devaud, J.-M., Barron, A. B. & Lihoreau, M. Why bees are so vulnerable to environmental stressors. Trends Ecol. Evol. 32, 268–278 (2017).
Steinhauer, N. et al. Drivers of colony losses. Curr. Opin. Insect Sci. 26, 142–148 (2018).
Sánchez-Bayo, F. & Wyckhuys, K. A. G. Worldwide decline of the entomofauna: a review of its drivers. Biol. Conserv. 232, 8–27 (2019).
Rundlöf, M. et al. Seed coating with a neonicotinoid insecticide negatively affects wild bees. Nature 521, 77–80 (2015).
Shah, F. M. et al. Field evaluation of synthetic and neem-derived alternative insecticides in developing action thresholds against cauliflower pests. Sci. Rep. 9, 7684 (2019).
Decourtye, A. & Devillers, J. Ecotoxicity of neonicotinoid insecticides to bees. Adv. Exp. Med. Biol. 683, 85–95 (2010).
Tomizawa, M. & Casida, J. E. Neonicotinoid insecticide toxicology: mechanisms of selective action. Annu. Rev. Pharmacol. Toxicol. 45, 247–268 (2005).
Wood, T. J. & Goulson, D. The environmental risks of neonicotinoid pesticides: a review of the evidence post 2013. Environ. Sci. Pollut. Res. Int. 24, 17285–17325 (2017).
Tavares, D. A., Roat, T. C., Silva-Zacarin, E. C. M., Nocelli, R. C. F. & Malaspina, O. Exposure to thiamethoxam during the larval phase affects synapsin levels in the brain of the honey bee. Ecotoxicol. Environ. Saf. 169, 523–528 (2019).
Matsuda, K. et al. Neonicotinoids: insecticides acting on insect nicotinic acetylcholine receptors. Trends Pharmacol. Sci. 22, 573–580 (2001).
Jeschke, P. & Nauen, R. Neonicotinoids-from zero to hero in insecticide chemistry. Pest Manag. Sci. 64, 1084–1098 (2008).
Crossthwaite, A. J. et al. The invertebrate pharmacology of insecticides acting at nicotinic acetylcholine receptors. J. Pestic. Sci. 42, 67–83 (2017).
Gauthier, M. State of the art on insect nicotinic acetylcholine receptor function in learning and memory. In Insect Nicotinic Acetylcholine Receptors. Advances in Experimental Medicine and Biology (ed. Thany, S. H.) 97–115 (Springer, Berlin, 2010).
Nilsen, T. W. & Graveley, B. R. Expansion of the eukaryotic proteome by alternative splicing. Nature 463, 457–463 (2010).
Pai, A. A. & Luca, F. Environmental influences on RNA processing: biochemical, molecular and genetic regulators of cellular response. Wiley Interdiscip. Rev. RNA 10, e1503 (2019).
Sumanasekera, C., Watt, D. S. & Stamm, S. Substances that can change alternative splice-site selection. Biochem. Soc. Trans. 36, 483–490 (2008).
Zaharieva, E., Chipman, J. K. & Soller, M. Alternative splicing interference by xenobiotics. Toxicology 296, 1–12 (2012).
Colgan, T. J. et al. Caste- and pesticide-specific effects of neonicotinoid pesticide exposure on gene expression in bumblebees. Mol. Ecol. 28, 1964–1974 (2019).
Decio, P. et al. Acute thiamethoxam toxicity in honeybees is not enhanced by common fungicide and herbicide and lacks stress-induced changes in mRNA splicing. Sci. Rep. 9, 19196 (2019).
Cabirol, A. & Haase, A. The neurophysiological bases of the impact of neonicotinoid pesticides on the behaviour of honeybees. Insects 10, 344 (2019).
Fischer, J. et al. Neonicotinoids interfere with specific components of navigation in honeybees. PLoS ONE 9, e91364 (2014).
Tison, L. et al. Effects of sublethal doses of thiacloprid and its formulation Calypso on the learning and memory performance of honey bees. J. Exp. Biol. 220, 3695–3705 (2017).
Williamson, S. M. & Wright, G. A. Exposure to multiple cholinergic pesticides impairs olfactory learning and memory in honeybees. J. Exp. Biol. 216, 1799–1807 (2013).
Tosi, S., Burgio, G. & Nieh, J. C. A common neonicotinoid pesticide, thiamethoxam, impairs honey bee flight ability. Sci. Rep. 7, 1–8 (2017).
Aliouane, Y. et al. Subchronic exposure of honeybees to sublethal doses of pesticides: effects on behavior. Environ. Toxicol. Chem. 28, 113–122 (2009).
Brandt, A., Gorenflo, A., Siede, R., Meixner, M. & Büchler, R. The neonicotinoids thiacloprid, imidacloprid, and clothianidin affect the immunocompetence of honey bees (Apis mellifera L.). J. Insect Physiol. 86, 40–47 (2016).
Perveen, N. & Ahmad, M. Toxicity of some insecticides to the haemocytes of giant honeybee, Apis dorsata F. under laboratory conditions. Saudi J. Biol. Sci. 24, 1016–1022 (2017).
Tesovnik, T. et al. Immune related gene expression in worker honey bee (Apis mellifera carnica) pupae exposed to neonicotinoid thiamethoxam and Varroa mites (Varroa destructor). PLoS ONE 12, e0187079 (2017).
Lemaitre, B. & Hoffmann, J. The host defense of Drosophila melanogaster. Annu. Rev. Immunol. 25, 697–743 (2007).
Buchon, N., Silverman, N. & Cherry, S. Immunity in Drosophila melanogaster: from microbial recognition to whole-organism physiology. Nat. Rev. Immunol. 14, 796–810 (2014).
Govind, S. Innate immunity in Drosophila: pathogens and pathways. Insect Sci. 15, 29–43 (2008).
Parsons, B. & Foley, E. Cellular immune defenses of Drosophila melanogaster. Dev. Comp. Immunol. 58, 95–101 (2016).
Sheehan, G., Garvey, A., Croke, M. & Kavanagh, K. Innate humoral immune defences in mammals and insects: the same, with differences?. Virulence 9, 1625–1639 (2018).
Vilcinskas, A. Evolutionary plasticity of insect immunity. J. Insect Physiol. 59, 123–129 (2013).
Ferrandon, D., Imler, J.-L., Hetru, C. & Hoffmann, J. A. The Drosophila systemic immune response: sensing and signalling during bacterial and fungal infections. Nat. Rev. Immunol. 7, 862–874 (2007).
Kleino, A. & Silverman, N. The Drosophila IMD pathway in the activation of the humoral immune response. Dev. Comp. Immunol. 42, 25–35 (2014).
Kounatidis, I. & Chtarbanova, S. Role of glial immunity in lifespan determination: a Drosophila perspective. Front. Immunol. 9, 1362 (2018).
Kounatidis, I. et al. NF-κB immunity in the brain determines fly lifespan in healthy aging and age-related neurodegeneration. Cell Rep. 19, 836–848 (2017).
Petersen, A. J., Rimkus, S. A. & Wassarman, D. A. ATM kinase inhibition in glial cells activates the innate immune response and causes neurodegeneration in Drosophila. Proc. Natl. Acad. Sci. USA 109, E656–E664 (2012).
Lye, S. H. & Chtarbanova, S. Drosophila as a model to study brain innate immunity in health and disease. Int. J. Mol. Sci. 19, 12 (2018).
Derecka, K. et al. Transient exposure to low levels of insecticide affects metabolic networks of honeybee larvae. PLoS ONE 8, e68191 (2013).
Oliveira, R. A., Roat, T. C., Carvalho, S. M. & Malaspina, O. Side-effects of thiamethoxam on the brain andmidgut of the africanized honeybee Apis mellifera (Hymenopptera: Apidae). Environ. Toxicol. 29, 1122–1133 (2014).
Tavares, D. A., Roat, T. C., Carvalho, S. M., Silva-Zacarin, E. C. M. & Malaspina, O. In vitro effects of thiamethoxam on larvae of Africanized honey bee Apis mellifera (Hymenoptera: Apidae). Chemosphere 135, 370–378 (2015).
Friol, P. S., Catae, A. F., Tavares, D. A., Malaspina, O. & Roat, T. C. Can the exposure of Apis mellifera (Hymenoptera, Apiadae) larvae to a field concentration of thiamethoxam affect newly emerged bees?. Chemosphere 185, 56–66 (2017).
OCDE (1998), Test No. 213: Honeybees, Acute Oral Toxicity Test, OECD Guidelines for the Testing of Chemicals, Section 2, Éditions OCDE, Paris, https://doi.org/10.1787/9789264070165-en
OCDE (2017), Test No. 245: Honey Bee (Apis Mellifera L.), Chronic Oral Toxicity Test (10-Day Feeding), OECD Guidelines for the Testing of Chemicals, Section 2, Éditions OCDE, Paris, https://doi.org/10.1787/9789264284081-en
Krupke, C. H., Hunt, G. J., Eitzer, B. D., Andino, G. & Given, K. Multiple routes of pesticide exposure for honey bees living near agricultural fields. PLoS ONE 7, e29268 (2012).
Mullin, C. A. et al. High levels of miticides and agrochemicals in North American apiaries: implications for honey bee health. PLoS ONE 5, e9754 (2010).
Pilling, E., Campbell, P., Coulson, M., Ruddle, N. & Tornier, I. A four-year field program investigating long-term effects of repeated exposure of honey bee colonies to flowering crops treated with thiamethoxam. PLoS ONE 8, e77193 (2013).
Stoner, K. A. & Eitzer, B. D. Movement of soil-applied imidacloprid and thiamethoxam into nectar and pollen of squash (Cucurbita pepo). PLoS ONE 7, e39114 (2012).
Free, J. B. & Spencer-Booth, Y. Observations on the temperature regulation and food consumption of honeybees (Apis mellifera). J. Exp. Biol. 35, 930 (1958).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 (2013).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012).
Shen, S. et al. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. 111, E5593–E5601 (2014).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Veltri, D., Kamath, U. & Shehu, A. Deep learning improves antimicrobial peptide recognition. Bioinformatics 34, 2740–2747 (2018).
Koushika, S. P., Soller, M., DeSimone, S. M., Daub, D. M. & White, K. Differential and inefficient splicing of a broadly expressed Drosophila erect wing transcript results in tissue-specific enrichment of the vital EWG protein isoform. Mol. Cell. Biol. 19, 3998–4007 (1999).
Koushika, S. P., Soller, M. & White, K. The neuron-enriched splicing pattern of Drosophila erect wing is dependent on the presence of ELAV protein. Mol. Cell. Biol. 20, 1836–1845 (2000).
Haussmann, I. U., Li, M. & Soller, M. ELAV-mediated 3′-end processing of ewg transcripts is evolutionarily conserved despite sequence degeneration of the ELAV-binding site. Genetics 189, 97–107 (2011).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 (2001).
Scharlaken, B. et al. Reference gene selection for insect expression studies using quantitative real-time PCR: the head of the honeybee, Apis mellifera, after a bacterial challenge. J. Insect Sci. 8, 1–10 (2008).
Nehme, N. T. et al. Relative roles of the cellular and humoral responses in the Drosophila host defense against three gram-positive bacterial infections. PLoS ONE 6, e14743 (2011).
Engel, P., Martinson, V. G. & Moran, N. A. Functional diversity within the simple gut microbiota of the honey bee. Proc. Natl. Acad. Sci. USA 109, 11002–11007 (2012).
Raymann, K. & Moran, N. A. The role of the gut microbiome in health and disease of adult honey bee workers. Curr. Opin. Insect Sci. 26, 97–104 (2018).
Soller, M. Pre-messenger RNA processing and its regulation: a genomic perspective. Cell. Mol. Life Sci. 63, 796–819 (2006).
Haussmann, I. U., White, K. & Soller, M. Erect wing regulates synaptic growth in Drosophila by integration of multiple signaling pathways. Genome Biol. 9, R73 (2008).
Haussmann, I. U. & Soller, M. Differential activity of EWG transcription factor isoforms identifies a subset of differentially regulated genes important for synaptic growth regulation. Dev. Biol. 348, 224–230 (2010).
Christen, V., Schirrmann, M., Frey, J. E. & Fent, K. Global transcriptomic effects of environmentally relevant concentrations of the neonicotinoids clothianidin, imidacloprid, and thiamethoxam in the brain of honey bees (Apis mellifera ). Environ. Sci. Technol. 52, 7534–7544 (2018).
Li, Z. et al. Brain transcriptome of honey bees (Apis mellifera) exhibiting impaired olfactory learning induced by a sublethal dose of imidacloprid. Pestic. Biochem. Physiol. 156, 36–43 (2019).
Couso, J.-P. & Patraquim, P. Classification and function of small open reading frames. Nat. Rev. Mol. Cell Biol. 18, 575–589 (2017).
Pueyo, J. I., Magny, E. G. & Couso, J. P. New peptides under the s(ORF)ace of the genome. Trends Biochem. Sci. 41, 665–678 (2016).
Andrews, S. J. & Rothnagel, J. A. Emerging evidence for functional peptides encoded by short open reading frames. Nat. Rev. Genet. 15, 193–204 (2014).
Pueyo, J. I. et al. Hemotin, a regulator of phagocytosis encoded by a small ORF and conserved across metazoans. PLoS Biol. 14, e1002395 (2016).
Galindo, I., Pueyo, I., Fouix, S., Bishop, S. A. & Couso, J. P. Peptides encoded by short ORFs control development and define a new eukaryotic gene family. PLoS Biol. 5, e106 (2007).
Barajas-Azpeleta, R. et al. Antimicrobial peptides modulate long-term memory. PLoS Genet. 14, e1007440 (2018).
De Smet, L. et al. Stress indicator gene expression profiles, colony dynamics and tissue development of honey bees exposed to sub-lethal doses of imidacloprid in laboratory and field experiments. PLoS ONE 12, e0171529 (2017).
Barribeau, S. M. et al. A depauperate immune repertoire precedes evolution of sociality in bees. Genome Biol. 16, 83 (2015).
López-Uribe, M. M., Sconiers, W. B., Frank, S. D., Dunn, R. R. & Tarpy, D. R. Reduced cellular immune response in social insect lineages. Biol. Lett. 12, 20150984 (2016).
Evans, J. D. et al. Immune pathways and defence mechanisms in honey bees Apis mellifera. Insect Mol. Biol. 15, 645–656 (2006).
Motta, E. V. S., Raymann, K. & Moran, N. A. Glyphosate perturbs the gut microbiota of honey bees. Proc. Natl. Acad. Sci. 115, 10305–10310 (2018).
Chmiel, J. A., Daisley, B. A., Burton, J. P. & Reid, G. Deleterious effects of neonicotinoid pesticides on Drosophila melanogaster immune pathways. MBio 10, e01395-e1419 (2019).
Di Prisco, G. et al. Neonicotinoid clothianidin adversely affects insect immunity and promotes replication of a viral pathogen in honey bees. Proc. Natl. Acad. Sci. 110, 18466–18471 (2013).
Dickel, F., Münch, D., Amdam, G. V., Mappes, J. & Freitak, D. Increased survival of honeybees in the laboratory after simultaneous exposure to low doses of pesticides and bacteria. PLoS ONE 13, e0191256 (2018).
Acknowledgements
We thank N. Parker and the Winterbourne garden for bees, N. Parker for bee suits, D. Scocchia, V. Soller-Haussmann and K. Nallasivan for help with bee collections. G. Salmond for bacterial strains, D. Scocchia for help with bacterial culturing and genotyping, and L. Orsini, E. Davies and I. Haussmann for comments on the manuscript. For this work we acknowledge funding from the Foundation for Research Support of the State of São Paulo, FAPESP (2012/13370-8; 2013/07251-9; 2014/23197-7; 2015/22368-5), the Biotechnology and Biological Sciences Research Council (BBSRC), the Nottingham-Birmingham Fund, and the Sukran Sinan Memory Fund.
Author information
Authors and Affiliations
Contributions
P.D., P.U. and K.D. performed the experiments, T.C.R, I.C.W.H., O.M., N.M. analysed data, R.S. and M.S. supervised experiments and analysed data, R.S. and M.S. wrote the manuscript with help from P.D., P.U. and I.C.W.H.
Corresponding authors
Ethics declarations
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Decio, P., Ustaoglu, P., Derecka, K. et al. Thiamethoxam exposure deregulates short ORF gene expression in the honey bee and compromises immune response to bacteria. Sci Rep 11, 1489 (2021). https://doi.org/10.1038/s41598-020-80620-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-80620-7
This article is cited by
-
Gene expression in bumble bee larvae differs qualitatively between high and low concentration imidacloprid exposure levels
Scientific Reports (2023)
-
A systematic review of research conducted by pioneer groups in ecotoxicological studies with bees in Brazil: advances and perspectives
Environmental Science and Pollution Research (2022)
-
Dynamically expressed single ELAV/Hu orthologue elavl2 of bees is required for learning and memory
Communications Biology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
