New Nanoparticle Formulation for Cyclosporin A: In Vitro Assessment

 , , ,
, , ,
Abstract
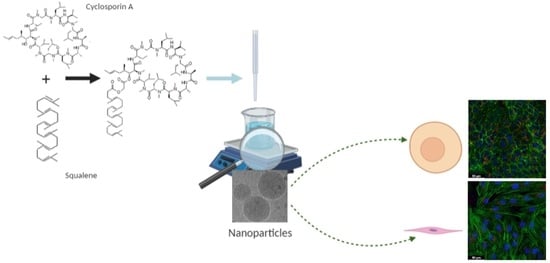
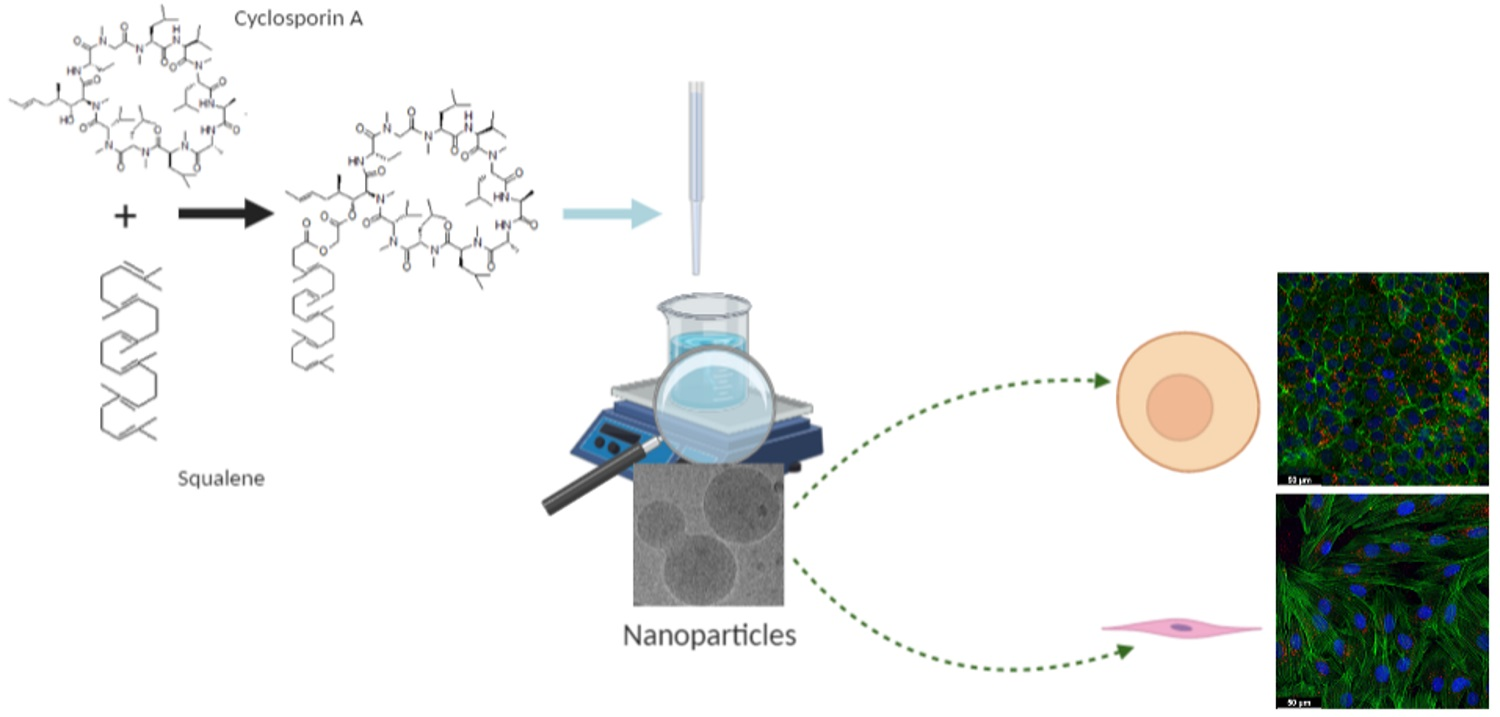
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
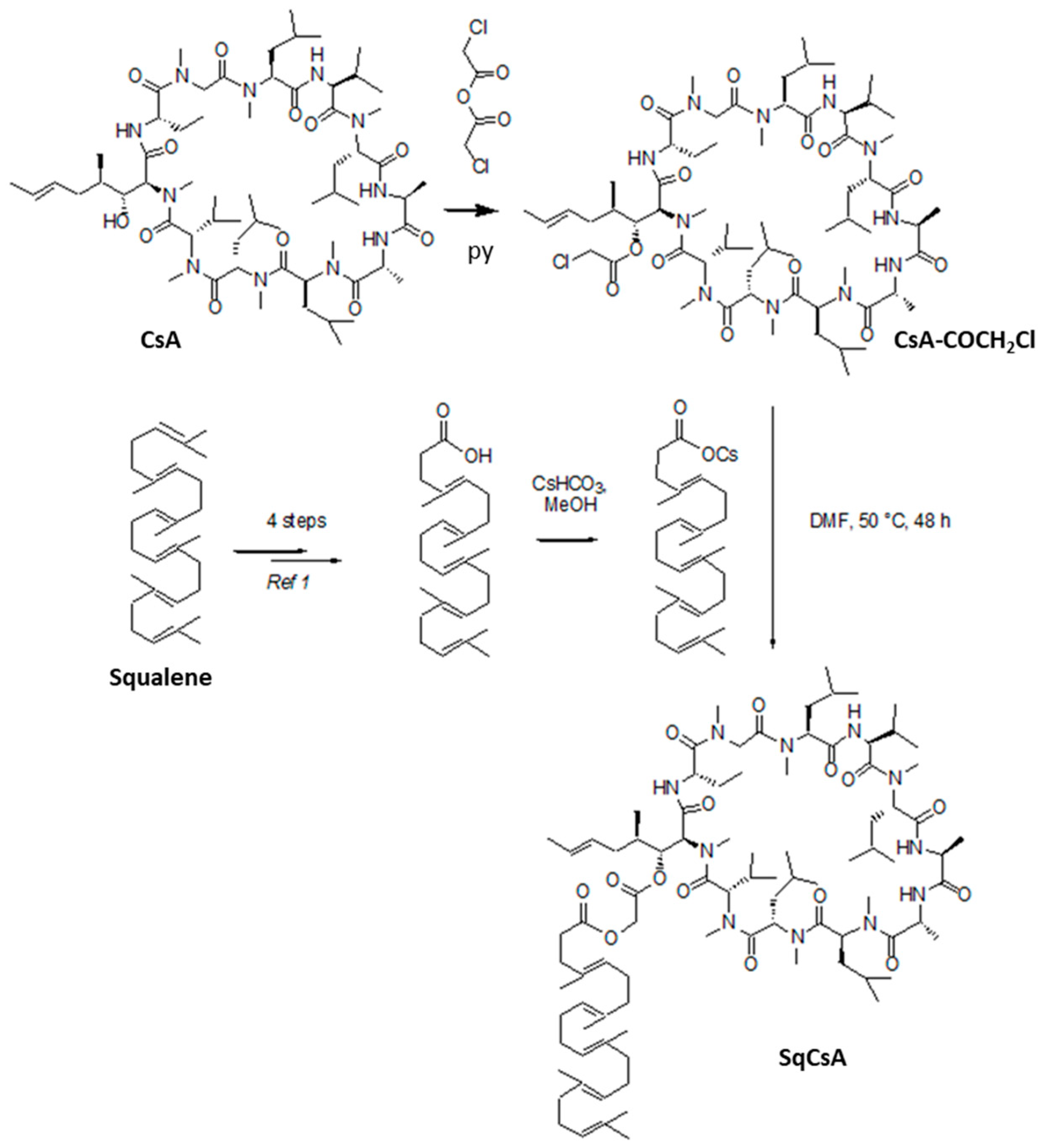
2.2. Synthesis of the SqCsA Conjugate
2.2.1. General
2.2.2. Synthesis of Cyclosporin A Chloroacetic Ester (CsA-COCH2Cl)
2.2.3. Synthesis of 1,1′,2-Trisnorsqualenic Acid Cesium Salt
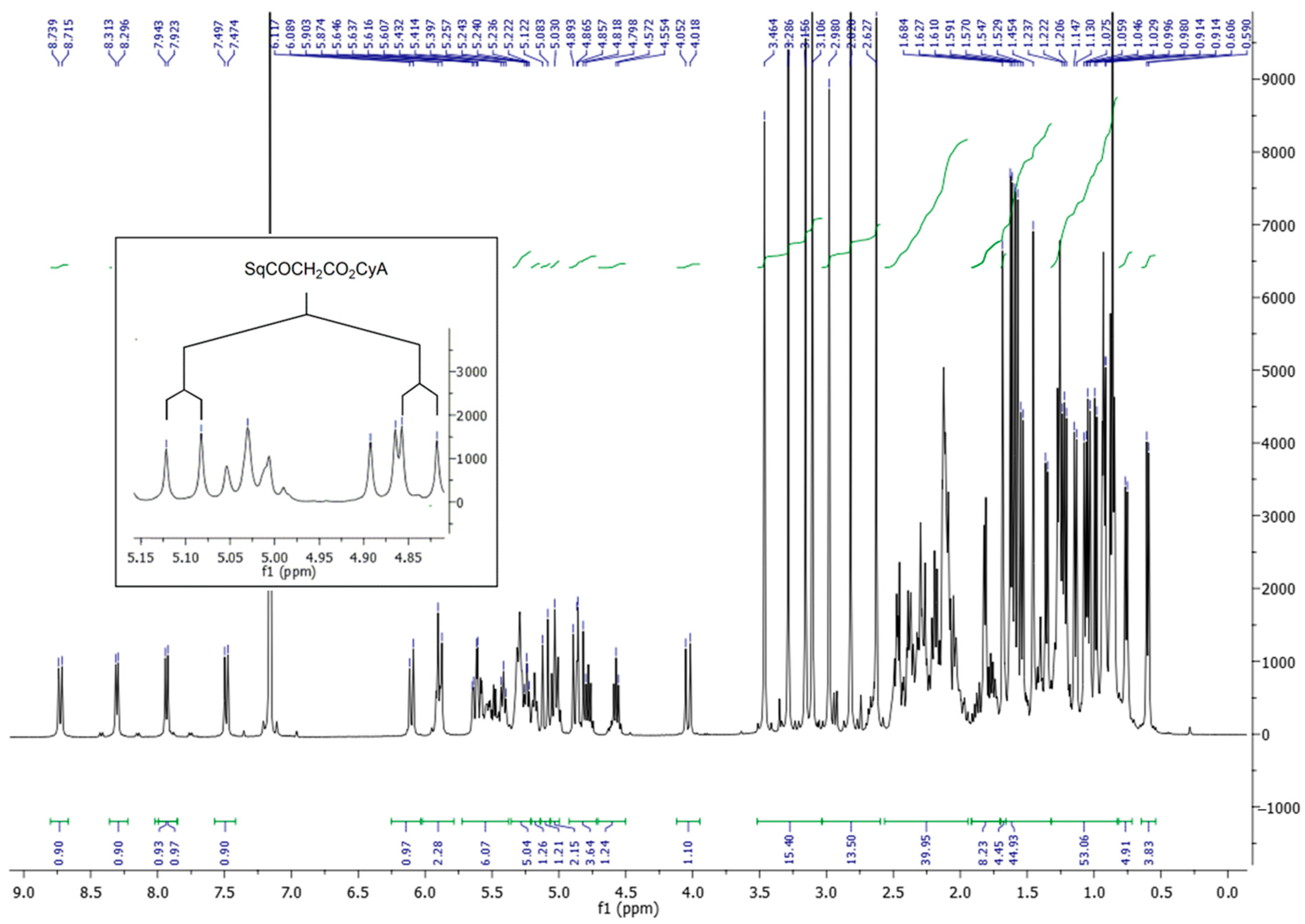
2.2.4. Synthesis of SqCsA Bioconjugate (SqCsA)
2.3. Preparation and Characterization of SqCsA Nanoparticles
2.4. Morphology by CryoTEM
2.5. Stability of NPs
2.6. HPLC Analysis
2.6.1. Sample Preparation
2.6.2. Chromatographic System
2.7. Cell Culture
2.8. Cytotoxicity of SqCsA NPs
2.9. Cell Uptake of Fluorescently Labeled SqCsA NPs
2.10. Cardioprotective Effect Assessment
2.11. Statistical Analysis
3. Results and Discussion
3.1. Synthesis of the SqCsA Conjugate
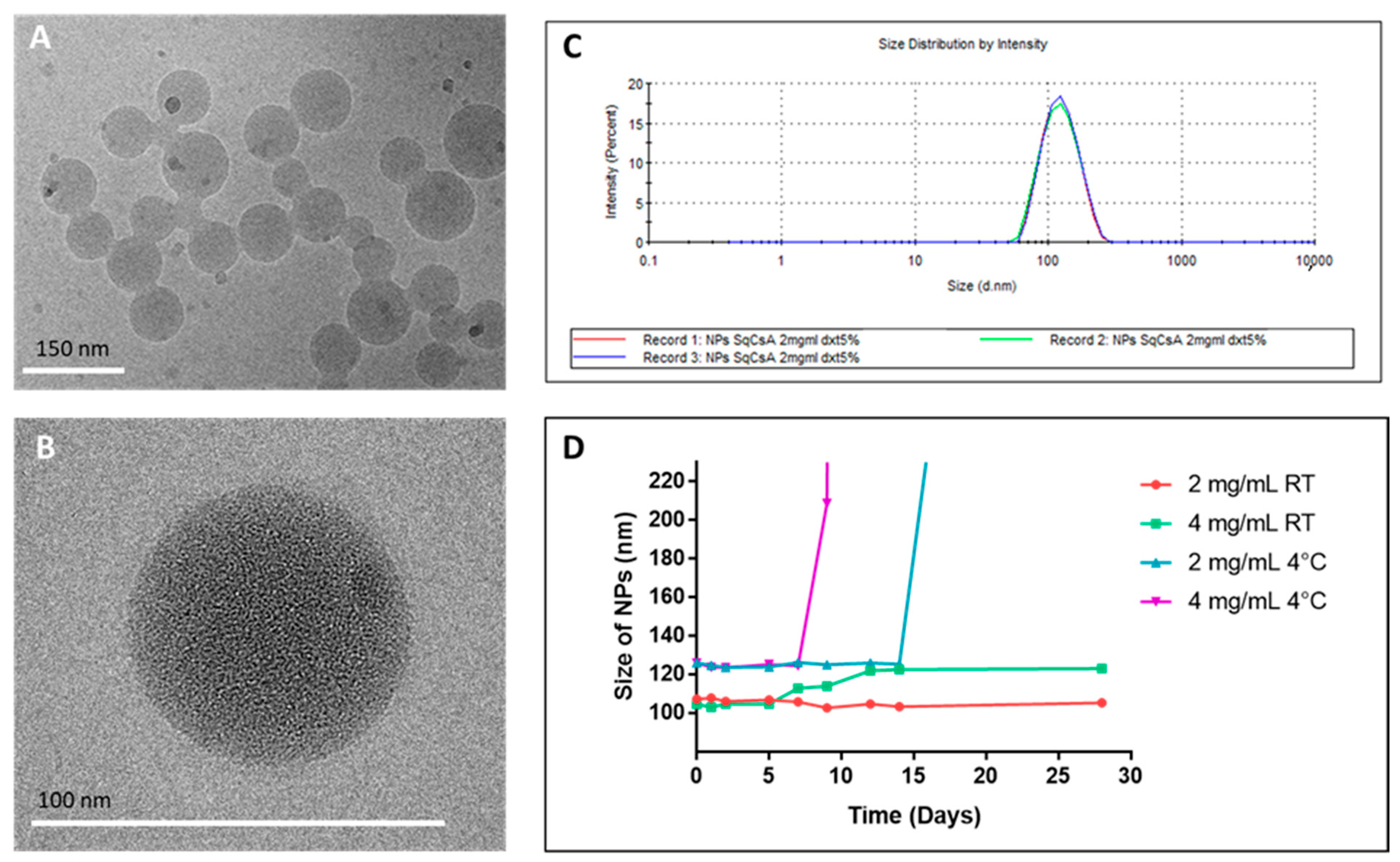
3.2. Formulation and Characterization of SqCsA NPs
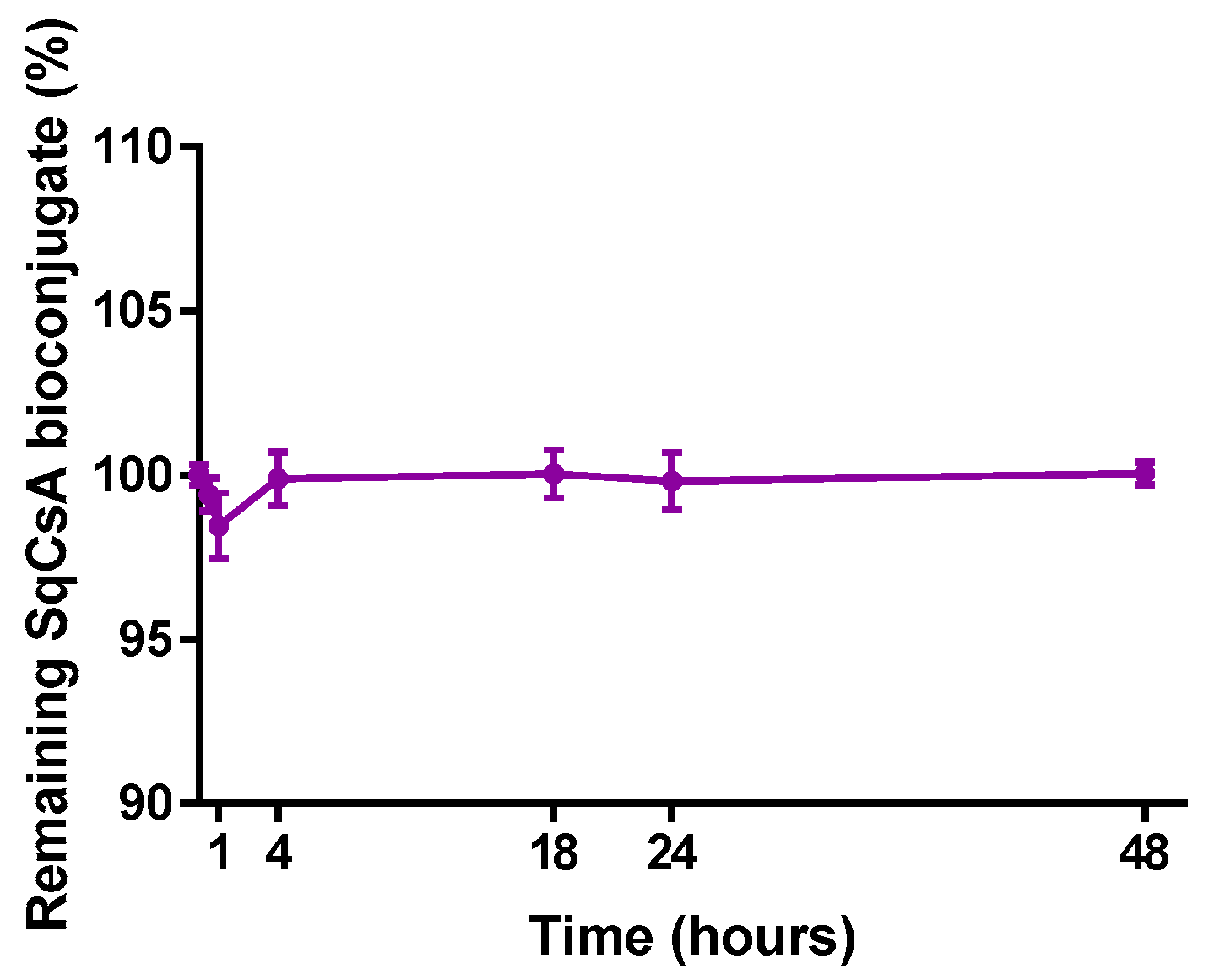
3.3. HPLC Analysis
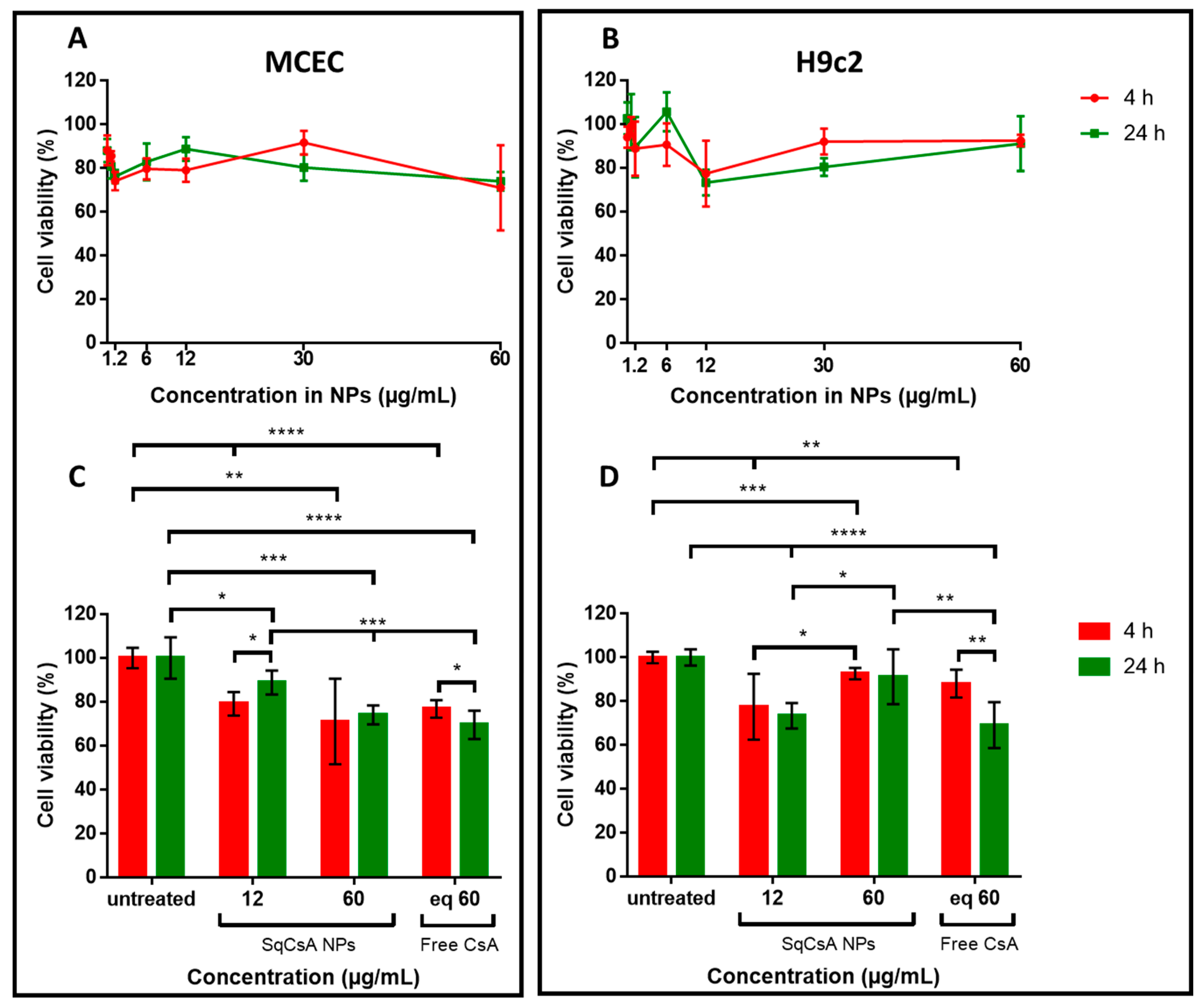
3.4. Cytotoxicity of SqCsA NPs
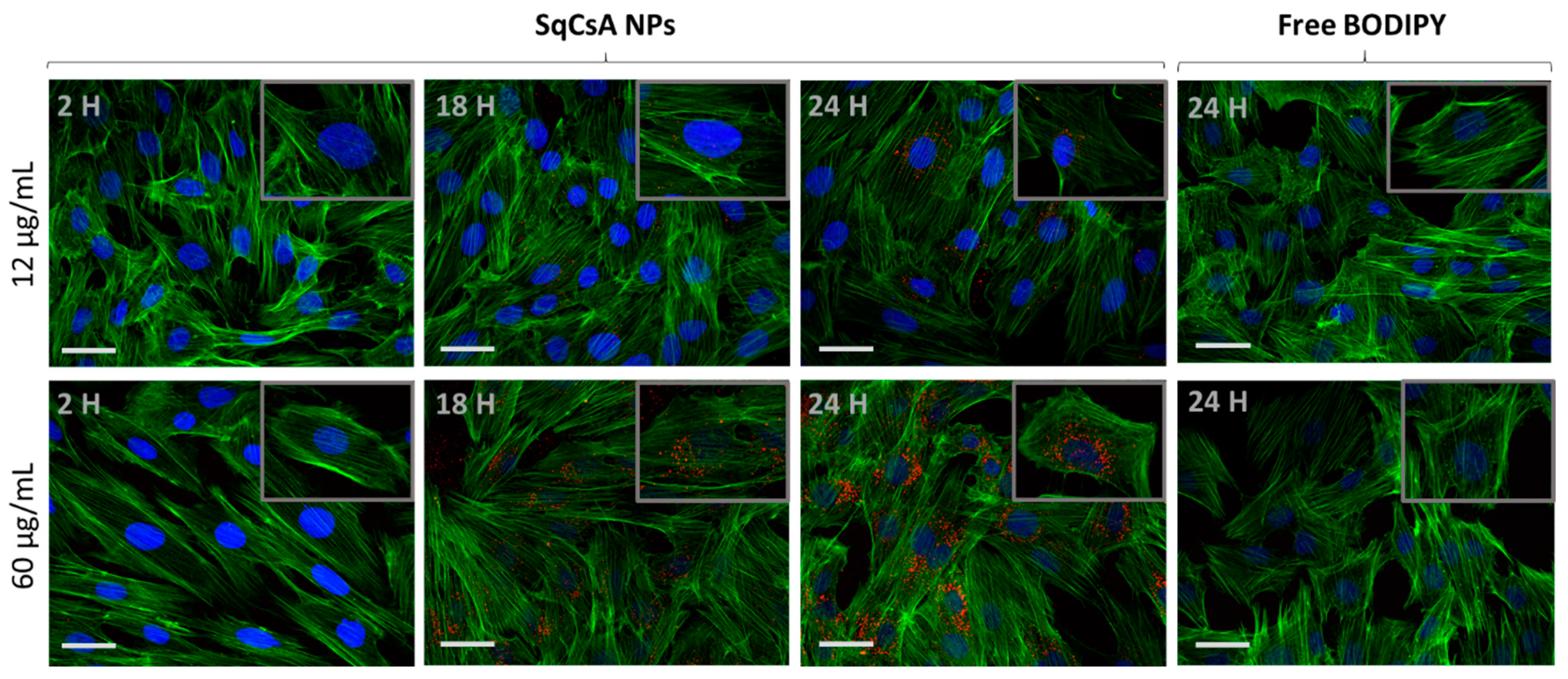
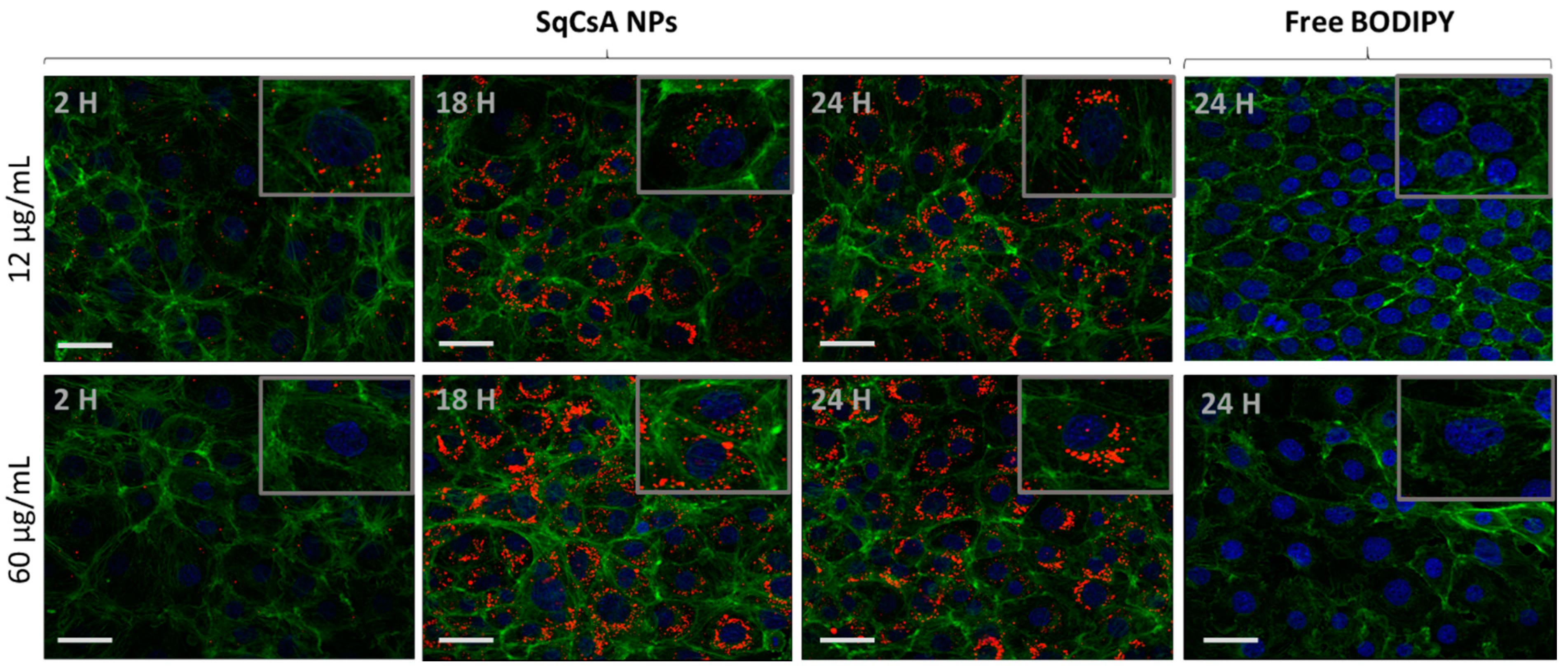
3.5. Cell Uptake of Fluorescently Labeled SqCsA NPs
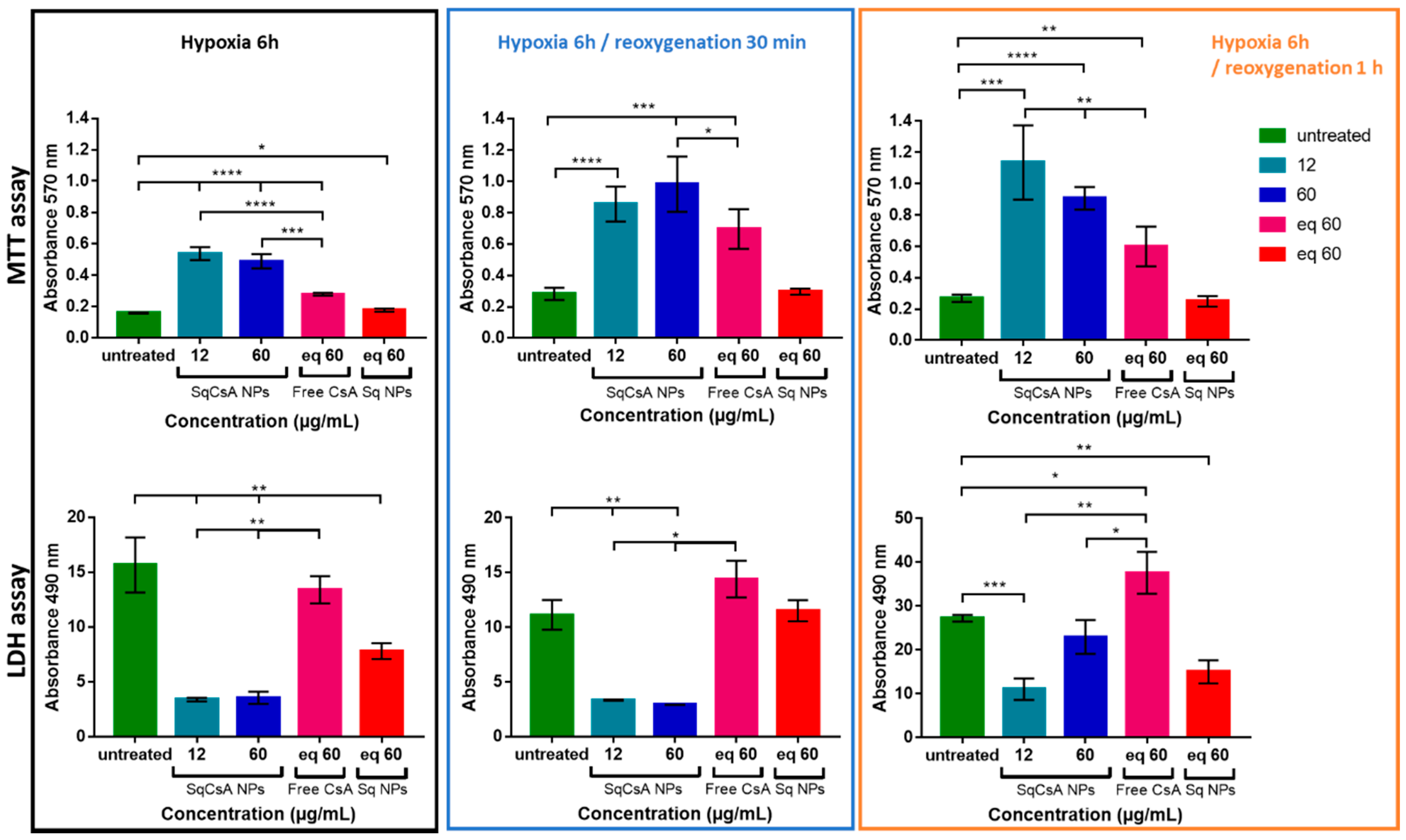
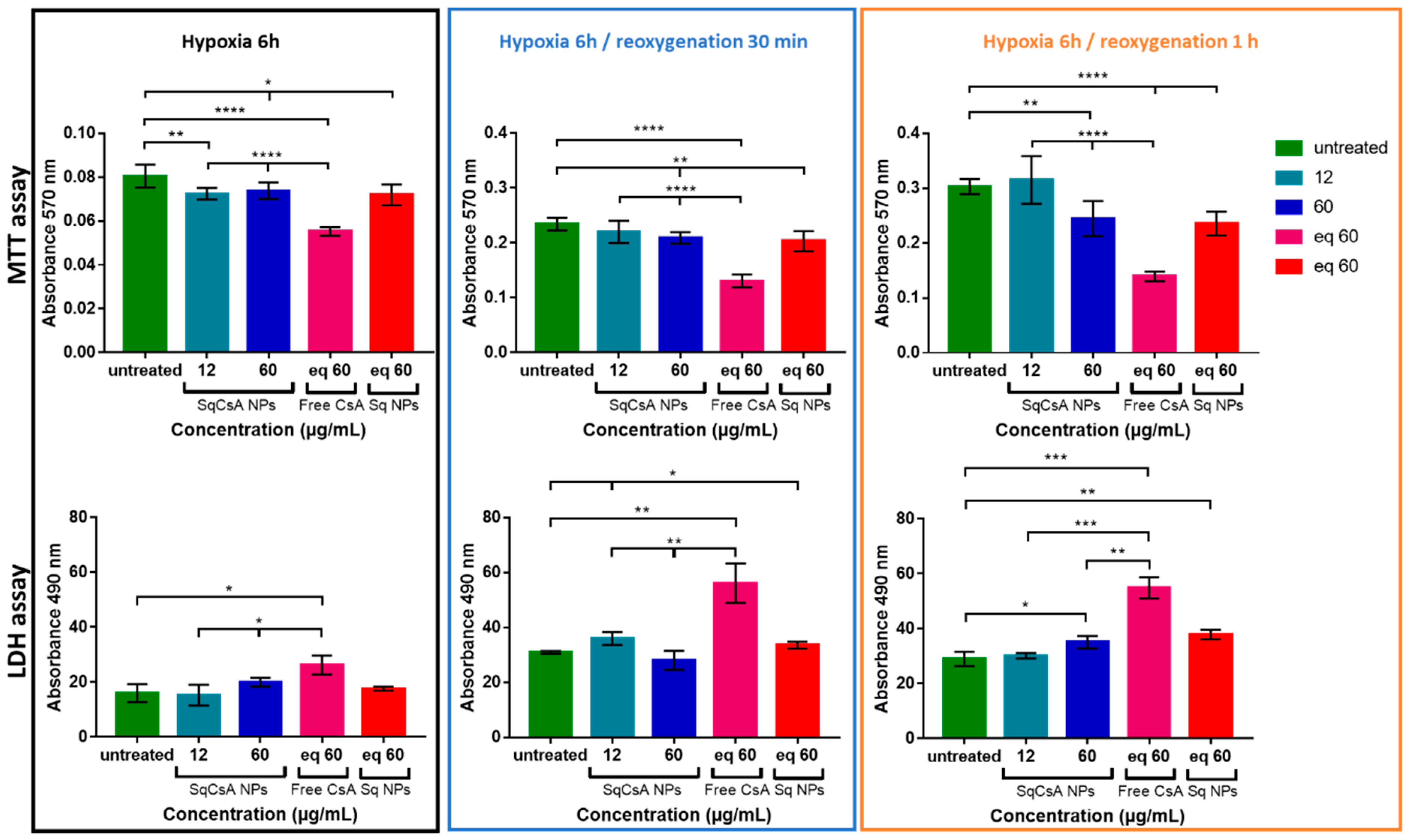
3.6. Cardioprotective Effect Assessment
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fahr, A. Cyclosporin Clinical Pharmacokinetics. Clin. Pharmacokinet. 1993, 24, 472–495. [Google Scholar] [CrossRef] [PubMed]
- Czogalla, A. Oral cyclosporine A the current picture of its liposomal and other delivery systems. Cell. Mol. Biol. Lett. 2009, 14, 139–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faulds, D.; Goa, K.L.; Benfield, P. Cyclosporin: A Review of its Pharmacodynamic and Pharmacokinetic Properties, and Therapeutic Use in Immunoregulatory Disorders. Drugs 1993, 45, 953–1040. [Google Scholar] [CrossRef] [PubMed]
- Italia, J.L.; Bhardwaj, V.; Ravi Kumar, M.N.V. Disease, destination, dose and delivery aspects of ciclosporin: The state of the art. Drug Discov. Today 2006, 11, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Yu, Y.; Chaurasiya, B.; Li, X.; Xu, Y.; Webster, T.; Tu, J.; Sun, R. Stability, safety, and transcorneal mechanistic studies of ophthalmic lyophilized cyclosporine-loaded polymeric micelles. Int. J. Nanomed. 2018, 13, 8281–8296. [Google Scholar] [CrossRef] [Green Version]
- Gäckler, A.; Dolff, S.; Rohn, H.; Korth, J.; Wilde, B.; Eisenberger, U.; Mitchell, A.; Kribben, A.; Witzke, O. Randomized, open-label, comparative phase IV study on the bioavailability of Ciclosporin Pro (Teva) versus Sandimmun® Optoral (Novartis) under fasting versus fed conditions in patients with stable renal transplants. BMC Nephrol. 2019, 20, 167. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Choi, J.-O.; Oh, J.; Cho, H.J.; Jung, S.-H.; Lee, H.-Y.; Kang, S.-M.; Kim, J.-J.; Jeon, E.-S. The Korean Organ Transplant Registry (KOTRY): Second Official Adult Heart Transplant Report. Korean Circ. J. 2019, 49, 724–737. [Google Scholar] [CrossRef]
- Møller-Bisgaard, S.; Ejbjerg, B.; Eshed, I.; Hørslev-Petersen, K.; Hetland, M.; Jurik, A.; Thomsen, H.; Torfing, T.; Stengaard-Pedersen, K.; Junker, P.; et al. Effect of a treat-to-target strategy based on methotrexate and intra-articular betamethasone with or without additional cyclosporin on MRI-assessed synovitis, osteitis, tenosynovitis, bone erosion, and joint space narrowing in early rheumatoid arthritis: Results from a 2-year randomized double-blind placebo-controlled trial (CIMESTRA). Scand. J. Rheumatol. 2017, 46, 335–345. [Google Scholar] [CrossRef] [Green Version]
- Essaghraoui, A.; Belfkira, A.; Hamdaoui, B.; Nunes, C.; Lima, S.A.C.; Reis, S. Improved Dermal Delivery of Cyclosporine A Loaded in Solid Lipid Nanoparticles. Nanomaterials 2019, 9, 1204. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, D.F.; Malhotra, R.P.; Schechter, B.A.; Justice, A.; Weiss, S.L.; Sheppard, J.D. A Phase 3, Randomized, Double-Masked Study of OTX-101 Ophthalmic Solution 0.09% in the Treatment of Dry Eye Disease. Ophthalmology 2019, 126, 1230–1237. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.-J.; Kim, Y.-H.; Chou, M.; Hwang, J.; Cheon, E.-J.; Lee, H.-J.; Chung, S.-H. Evaluation of the Efficacy and Safety of A Novel 0.05% Cyclosporin A Topical Nanoemulsion in Primary Sjögren’s Syndrome Dry Eye. Ocul. Immunol. Inflamm. 2020, 28, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Tauber, J.; Schechter, B.A.; Bacharach, J.; Toyos, M.M.; Smyth-Medina, R.; Weiss, S.L.; Luchs, J.I. A Phase II/III, randomized, double-masked, vehicle-controlled, dose-ranging study of the safety and efficacy of OTX-101 in the treatment of dry eye disease. Clin. Ophthalmol. 2018, 12, 1921–1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guada, M.; Beloqui, A.; Kumar, M.N.V.R.; Préat, V.; del Carmen Dios-Viéitez, M.; Blanco-Prieto, M.J. Reformulating cyclosporine A (CsA): More than just a life cycle management strategy. J. Control. Release 2016, 225, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Fukuzawa, M.; Shearer, G.M. Effect of cyclosporin A on T cell immunity I. Dose-dependent suppression of different murine T helper cell pathways. Eur. J. Immunol. 1989, 19, 49–56. [Google Scholar] [CrossRef]
- Hausenloy, D.; Boston-Griffiths, E.; Yellon, D. Cyclosporin A and cardioprotection: From investigative tool to therapeutic agent: CsA and cardioprotection. Br. J. Pharmacol. 2012, 165, 1235–1245. [Google Scholar] [CrossRef] [Green Version]
- Leshnower, B.G.; Kanemoto, S.; Matsubara, M.; Sakamoto, H.; Hinmon, R.; Gorman, J.H.; Gorman, R.C. Cyclosporine Preserves Mitochondrial Morphology After Myocardial Ischemia/Reperfusion Independent of Calcineurin Inhibition. Ann. Thorac. Surg. 2008, 86, 1286–1292. [Google Scholar] [CrossRef] [Green Version]
- Pagel, P.S.; Krolikowski, J.G. Transient Metabolic Alkalosis During Early Reperfusion Abolishes Helium Preconditioning Against Myocardial Infarction: Restoration of Cardioprotection by Cyclosporin A in Rabbits. Anesth. Analg. 2009, 108, 1076–1082. [Google Scholar] [CrossRef]
- Xie, J.-R.; Yu, L.-N. Cardioprotective effects of cyclosporine A in an in vivo model of myocardial ischemia and reperfusion. Acta Anaesthesiol. Scand. 2007, 51, 909–913. [Google Scholar] [CrossRef]
- Piot, C.; Croisille, P.; Staat, P.; Thibault, H.; Rioufol, G.; Mewton, N.; Elbelghiti, R.; Cung, T.T.; Bonnefoy, E.; Angoulvant, D.; et al. Effect of Cyclosporine on Reperfusion Injury in Acute Myocardial Infarction. N. Engl. J. Med. 2008, 359, 473–481. [Google Scholar] [CrossRef] [Green Version]
- Cung, T.-T.; Morel, O.; Cayla, G.; Rioufol, G.; Garcia-Dorado, D.; Angoulvant, D.; Bonnefoy-Cudraz, E.; Guérin, P.; Elbaz, M.; Delarche, N.; et al. Cyclosporine before PCI in Patients with Acute Myocardial Infarction. N. Engl. J. Med. 2015, 373, 1021–1031. [Google Scholar] [CrossRef]
- Ottani, F.; Latini, R.; Staszewsky, L.; La Vecchia, L.; Locuratolo, N.; Sicuro, M.; Masson, S.; Barlera, S.; Milani, V.; Lombardi, M.; et al. Cyclosporine A in Reperfused Myocardial Infarction. J. Am. Coll. Cardiol. 2016, 67, 365–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guada, M.; Lasa-Saracíbar, B.; Lana, H.; del Carmen Dios-Viéitez, M.; Blanco-Prieto, M.J. Lipid nanoparticles enhance the absorption of cyclosporine A through the gastrointestinal barrier: In vitro and in vivo studies. Int. J. Pharm. 2016, 500, 154–161. [Google Scholar] [CrossRef]
- Partoazar, A.; Nasoohi, S.; Rezayat, S.M.; Gilani, K.; Mehr, S.E.; Amani, A.; Rahimi, N.; Dehpour, A.R. Nanoliposome containing cyclosporine A reduced neuroinflammation responses and improved neurological activities in cerebral ischemia/reperfusion in rat. Fundam. Clin. Pharmacol. 2017, 31, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Lahiani-Skiba, M.; Hallouard, F.; Bounoure, F.; Milon, N.; Karrout, Y.; Skiba, M. Enhanced Dissolution and Oral Bioavailability of Cyclosporine A: Microspheres Based on αβ-Cyclodextrins Polymers. Pharmaceutics 2018, 10, 285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, T.-T.; Wen, B.-F.; Liu, K.; Qin, M.; Gao, Y.-Y.; Ding, D.-J.; Li, W.-T.; Zhang, Y.-X.; Zhang, W.-F. Cyclosporine A/porous quaternized chitosan microspheres as a novel pulmonary drug delivery system. Artif. Cells Nanomed. Biotechnol. 2018, 46, 552–564. [Google Scholar] [CrossRef] [Green Version]
- Goyal, R.; Macri, L.; Kohn, J. Formulation Strategy for the Delivery of Cyclosporine A: Comparison of Two Polymeric Nanospheres. Sci. Rep. 2015, 5, 13065. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, G.; Matoba, T.; Nakano, Y.; Nagaoka, K.; Ishikita, A.; Nakano, K.; Funamoto, D.; Sunagawa, K.; Egashira, K. Nanoparticle-Mediated Targeting of Cyclosporine A Enhances Cardioprotection Against Ischemia-Reperfusion Injury Through Inhibition of Mitochondrial Permeability Transition Pore Opening. Sci. Rep. 2016, 6, 20467. [Google Scholar] [CrossRef] [Green Version]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.Y.; Sood, A.K.; Hua, S. Advances and Challenges of Liposome Assisted Drug Delivery. Front. Pharmacol. 2015, 6, 286. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Jain, A.; Gulbake, A.; Shilpi, S.; Hurkat, P.; Jain, S.K. Peptide and Protein Delivery Using New Drug Delivery Systems. Crit. Rev. Ther. Drug Carrier Syst. 2013, 30, 293–329. [Google Scholar] [CrossRef]
- Kumari, A.; Yadav, S.K.; Yadav, S.C. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf. B Biointerfaces 2010, 75, 1–18. [Google Scholar] [CrossRef]
- Couvreur, P.; Stella, B.; Reddy, L.H.; Hillaireau, H.; Dubernet, C.; Desmaële, D.; Lepêtre-Mouelhi, S.; Rocco, F.; Dereuddre-Bosquet, N.; Clayette, P.; et al. Squalenoyl Nanomedicines as Potential Therapeutics. Nano Lett. 2006, 6, 2544–2548. [Google Scholar] [CrossRef] [PubMed]
- Gaudin, A.; Yemisci, M.; Eroglu, H.; Lepetre-Mouelhi, S.; Turkoglu, O.F.; Dönmez-Demir, B.; Caban, S.; Sargon, M.F.; Garcia-Argote, S.; Pieters, G.; et al. Squalenoyl adenosine nanoparticles provide neuroprotection after stroke and spinal cord injury. Nat. Nanotechnol. 2014, 9, 1054–1062. [Google Scholar] [CrossRef]
- Maksimenko, A.; Caron, J.; Mougin, J.; Desmaële, D.; Couvreur, P. Gemcitabine-based therapy for pancreatic cancer using the squalenoyl nucleoside monophosphate nanoassemblies. Int. J. Pharm. 2015, 482, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Lepetre-Mouelhi, S.; Gautier, A.; Mura, S.; Cailleau, C.; Coudore, F.; Hamon, M.; Couvreur, P. A new painkiller nanomedicine to bypass the blood-brain barrier and the use of morphine. Sci. Adv. 2019, 5, eaau5148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobot, D.; Mura, S.; Rouquette, M.; Vukosavljevic, B.; Cayre, F.; Buchy, E.; Pieters, G.; Garcia-Argote, S.; Windbergs, M.; Desmaële, D.; et al. Circulating Lipoproteins: A Trojan Horse Guiding Squalenoylated Drugs to LDL-Accumulating Cancer Cells. Mol. Ther. 2017, 25, 1596–1605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotelevets, L.; Chastre, E.; Caron, J.; Mougin, J.; Bastian, G.; Pineau, A.; Walker, F.; Lehy, T.; Desmaële, D.; Couvreur, P. A Squalene-Based Nanomedicine for Oral Treatment of Colon Cancer. Cancer Res. 2017, 77, 2964–2975. [Google Scholar] [CrossRef] [Green Version]
- Dormont, F.; Brusini, R.; Cailleau, C.; Reynaud, F.; Peramo, A.; Gendron, A.; Mougin, J.; Gaudin, F.; Varna, M.; Couvreur, P. Squalene-based multidrug nanoparticles for improved mitigation of uncontrolled inflammation. Sci. Adv. 2020, 6, eaaz5466. [Google Scholar] [CrossRef]
- Dormont, F.; Rouquette, M.; Mahatsekake, C.; Gobeaux, F.; Peramo, A.; Brusini, R.; Calet, S.; Testard, F.; Lepetre-Mouelhi, S.; Desmaële, D.; et al. Translation of nanomedicines from lab to industrial scale synthesis: The case of squalene-adenosine nanoparticles. J. Control. Release 2019, 307, 302–314. [Google Scholar] [CrossRef]
- Van Tamelen, E.E.; Curphey, T.J. The selective in vitro oxidation of the terminal double bonds in squalene. Tetrahedron Lett. 1962, 3, 121–124. [Google Scholar] [CrossRef]
- Ceruti, M.; Balliano, G.; Viola, F.; Cattel, L.; Gerst, N.; Schuber, F. Synthesis and biological activity of azasqualenes, bis-azasqualenes and derivatives. Eur. J. Med. Chem. 1987, 22, 199–208. [Google Scholar] [CrossRef]
- Paprica, P.A.; Margaritis, A.; Petersen, N.O. Preparation of novel cyclosporin A derivatives. Bioconjug. Chem. 1992, 3, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Eberle, M.K.; Nuninger, F. Synthesis of the main metabolite (OL-17) of cyclosporin A. J. Org. Chem. 1992, 57, 2689–2691. [Google Scholar] [CrossRef]
- Grote, J.; Fishpaugh, J.; Rege, S. A Practical Method for the Synthesis of a Cyclosporine−Fluorescein Conjugate. Org. Process. Res. Dev. 2005, 9, 822–824. [Google Scholar] [CrossRef]
- Hamel, A.R.; Hubler, F.; Carrupt, A.; Wenger, R.M.; Mutter, M. Cyclosporin A prodrugs: Design, synthesis and biophysical properties: Cyclosporin A prodrugs. J. Pept. Res. 2004, 63, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Rothbard, J.B.; Garlington, S.; Lin, Q.; Kirschberg, T.; Kreider, E.; McGrane, P.L.; Wender, P.A.; Khavari, P.A. Conjugation of arginine oligomers to cyclosporin A facilitates topical delivery and inhibition of inflammation. Nat. Med. 2000, 6, 1253–1257. [Google Scholar] [CrossRef]
- Chandran, V.R. Novel Compounds with High Therapeutic Index. US 2006/0241017 A1, 26 October 2006. [Google Scholar]
- Kessler, H.; Loosli, H.-R.; Oschkinat, H. Peptide conformations. Part 30. Assignment of the 1H-,13C-, and 15N-NMR spectra of cyclosporin A in CDCl3 and C6D6 by a combination of homo- and heteronuclear two-dimensional techniques. Helv. Chim. Acta 1985, 68, 661–681. [Google Scholar] [CrossRef]
- Rouquette, M.; Lepetre-Mouelhi, S.; Dufrançais, O.; Yang, X.; Mougin, J.; Pieters, G.; Garcia-Argote, S.; IJzerman, A.P.; Couvreur, P. Squalene-Adenosine Nanoparticles: Ligands of Adenosine Receptors or Adenosine Prodrug? J. Pharmacol. Exp. Ther. 2019, 369, 144–151. [Google Scholar] [CrossRef]
- Maksimenko, A.; Dosio, F.; Mougin, J.; Ferrero, A.; Wack, S.; Reddy, L.H.; Weyn, A.-A.; Lepeltier, E.; Bourgaux, C.; Stella, B.; et al. A unique squalenoylated and nonpegylated doxorubicin nanomedicine with systemic long-circulating properties and anticancer activity. Proc. Natl. Acad. Sci. USA 2014, 111, E217–E226. [Google Scholar] [CrossRef] [Green Version]
- Arora, R.; Katiyar, S.S.; Kushwah, V.; Jain, S. Solid lipid nanoparticles and nanostructured lipid carrier-based nanotherapeutics in treatment of psoriasis: A comparative study. Expert Opin. Drug Deliv. 2017, 14, 165–177. [Google Scholar] [CrossRef]
- Potta, S.G.; Minemi, S.; Nukala, R.K.; Peinado, C.; Lamprou, D.A.; Urquhart, A.; Douroumis, D. Development of Solid Lipid Nanoparticles for Enhanced Solubility of Poorly Soluble Drugs. J. Biomed. Nanotechnol. 2010, 6, 634–640. [Google Scholar] [CrossRef]
- Bohley, M.; Haunberger, A.; Goepferich, A.M. Intracellular availability of poorly soluble drugs from lipid nanocapsules. Eur. J. Pharm. Biopharm. 2019, 139, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Badihi, A.; Frušić-Zlotkin, M.; Soroka, Y.; Benhamron, S.; Tzur, T.; Nassar, T.; Benita, S. Topical nano-encapsulated cyclosporine formulation for atopic dermatitis treatment. Nanomed. Nanotechnol. Biol. Med. 2020, 24, 102140. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Shi, L.; Ren, L.; Zhou, L.; Qiao, Y.; Wang, H. A nanomedicine approach enables co-delivery of cyclosporin A and gefitinib to potentiate the therapeutic efficacy in drug-resistant lung cancer. Signal. Transduct. Target. Ther. 2018, 3, 16. [Google Scholar] [CrossRef] [Green Version]
- Naeem, M.; Bae, J.; Oshi, M.A.; Kim, M.-S.; Moon, H.R.; Lee, B.L.; Im, E.; Jung, Y.; Yoo, J.-W. Colon-targeted delivery of cyclosporine A using dual-functional Eudragit® FS30D/PLGA nanoparticles ameliorates murine experimental colitis. Int. J. Nanomed. 2018, 13, 1225–1240. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Cheng, Y.; Liu, D.; Liu, M.; Cui, H.; Zhang, B.; Mei, Q.; Zhou, S. Mitochondria-targeted cyclosporin A delivery system to treat myocardial ischemia reperfusion injury of rats. J. Nanobiotechnol. 2019, 17, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roese, E.; Bunjes, H. Drug release studies from lipid nanoparticles in physiological media by a new DSC method. J. Control. Release 2017, 256, 92–100. [Google Scholar] [CrossRef]
- Abed, N.; Saïd-Hassane, F.; Zouhiri, F.; Mougin, J.; Nicolas, V.; Desmaële, D.; Gref, R.; Couvreur, P. An efficient system for intracellular delivery of beta-lactam antibiotics to overcome bacterial resistance. Sci. Rep. 2015, 5, 13500. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Cui, J.; Guo, Y.; Wang, Y.; Kang, L.; Liu, L. Cyclosporin A Protects H9c2 Cells Against Chemical Hypoxia-Induced Injury via Inhibition of MAPK Signaling Pathway. Int. Heart J. 2016, 57, 483–489. [Google Scholar] [CrossRef]
- Hwang, E.A.; Kim, H.S.; Ha, E.; Mun, K.C. Apoptosis in Endothelial Cells by Cyclosporine. Transplant. Proc. 2012, 44, 982–984. [Google Scholar] [CrossRef]
- Davda, J.; Labhasetwar, V. Characterization of nanoparticle uptake by endothelial cells. Int. J. Pharm. 2002, 233, 51–59. [Google Scholar] [CrossRef]
- Voigt, J.; Christensen, J.; Shastri, V.P. Differential uptake of nanoparticles by endothelial cells through polyelectrolytes with affinity for caveolae. Proc. Natl. Acad. Sci. USA 2014, 111, 2942–2947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aliyandi, A.; Satchell, S.; Unger, R.E.; Bartosch, B.; Parent, R.; Zuhorn, I.S.; Salvati, A. Effect of endothelial cell heterogeneity on nanoparticle uptake. Int. J. Pharm. 2020, 587, 119699. [Google Scholar] [CrossRef] [PubMed]
- Di Lisa, F.; Menabò, R.; Canton, M.; Barile, M.; Bernardi, P. Opening of the Mitochondrial Permeability Transition Pore Causes Depletion of Mitochondrial and Cytosolic NAD + and Is a Causative Event in the Death of Myocytes in Postischemic Reperfusion of the Heart. J. Biol. Chem. 2001, 276, 2571–2575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yellon, D.M.; Hausenloy, D.J. Myocardial Reperfusion Injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, A.V.; Javadov, S.; Sickinger, S.; Frotschnig, S.; Grimm, M. H9c2 and HL-1 cells demonstrate distinct features of energy metabolism, mitochondrial function and sensitivity to hypoxia-reoxygenation. Biochim. Biophys. Acta BBA Mol. Cell Res. 2015, 1853, 276–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dube, H.; Selwood, D.; Malouitre, S.; Capano, M.; Simone, M.I.; Crompton, M. A mitochondrial-targeted cyclosporin A with high binding affinity for cyclophilin D yields improved cytoprotection of cardiomyocytes. Biochem. J. 2012, 441, 901–907. [Google Scholar] [CrossRef] [Green Version]
- Nazareth, W.; Yafei, N.; Crompton, M. Inhibition of anoxia-induced injury in heart myocytes by cyclosporin A. J. Mol. Cell. Cardiol. 1991, 23, 1351–1354. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gendron, A.; Lan Linh Tran, N.; Laloy, J.; Brusini, R.; Rachet, A.; Gobeaux, F.; Nicolas, V.; Chaminade, P.; Abreu, S.; Desmaële, D.; et al. New Nanoparticle Formulation for Cyclosporin A: In Vitro Assessment. Pharmaceutics 2021, 13, 91. https://doi.org/10.3390/pharmaceutics13010091
Gendron A, Lan Linh Tran N, Laloy J, Brusini R, Rachet A, Gobeaux F, Nicolas V, Chaminade P, Abreu S, Desmaële D, et al. New Nanoparticle Formulation for Cyclosporin A: In Vitro Assessment. Pharmaceutics. 2021; 13(1):91. https://doi.org/10.3390/pharmaceutics13010091
Chicago/Turabian StyleGendron, Amandine, Natalie Lan Linh Tran, Julie Laloy, Romain Brusini, Aurélie Rachet, Frédéric Gobeaux, Valérie Nicolas, Pierre Chaminade, Sonia Abreu, Didier Desmaële, and et al. 2021. "New Nanoparticle Formulation for Cyclosporin A: In Vitro Assessment" Pharmaceutics 13, no. 1: 91. https://doi.org/10.3390/pharmaceutics13010091