
Abstract
The biological clock is an endogenous biological timing system, which controls metabolic functions in almost all organs. Nutrient metabolism, substrate processing, and detoxification are circadian controlled in livers. However, how the clock genes respond to toxins and influence toxicity keeps unclear. We identified the clock gene Per1 was specifically elevated in mice exposed to toxins such as carbon tetrachloride (CCl4). Mice lacking Per1 slowed down the metabolic rate of toxins including CCl4, capsaicin, and acetaminophen, exhibiting relatively more residues in the plasma. Liver injury and fibrosis induced by acute and chronic CCl4 exposure were markedly alleviated in Per1-deficient mice. These processes involved the binding of PER1 protein and hepatocyte nuclear factor-1alpha (HNF-1α), which enhances the recruitment of HNF-1α to cytochrome P450 2E1 (Cyp2e1) promoter and increases Cyp2e1 expression, thereby promoting metabolism for toxins in the livers. These results indicate that PER1 mediates the metabolism of toxins and appropriate suppression of Per1 response is a potential therapeutic target for toxin-induced hepatotoxicity.
Similar content being viewed by others


Introduction
Most physiological processes in mammals display circadian rhythms that are driven by the endogenous circadian clock. Both central and peripheral clocks are operated by positive- and negative-feedback loops of circadian genes, such as Clock, Bmal1, Period1 (Per1), Period2 (Per2), Cryptochromes1 (Cry1) and Cryptochromes2 (Cry2), Rev-Erb-α, and Rora. In the negative-feedback loops of circadian genes, the PER and CRY form complexes and shut down transcription initiated by CLOCK–BMAL1 heterodimers1,2. Circadian rhythms play an important role in maintaining homeostasis and solid organ function. Although disrupted rhythms can lead to metabolic impairments, the reverse is also true that disrupting metabolism can alter circadian rhythms3. Clock dysfunction accelerates the development of liver diseases such as fatty liver diseases, cirrhosis, hepatitis, and liver cancer, and these disorders also disrupt clock function4. On the other hand, patients with cirrhosis have liver portal hypertension and dysfunctional circadian clock systems5. Hepatic fibrosis induced by carbon tetrachloride (CCl4) in mice leads to alterations in the circadian rhythms of hepatic clock genes6. The overarching evidence that circadian homeostasis is critical to human health, and conversely, that some abnormal metabolism and diseases induce abnormal responses of the circadian clock and negatively affect health, cannot be ignored.
Acute liver injury induced by hepatotoxins has been recognized as one of the most important pharmacovigilance concerns and the leading cause of drug withdrawal on safety grounds7,8. After acute liver injury, parenchymal cells regenerate and replace the necrotic or apoptotic cells. The wound-healing response of the liver to repeated injury leads to liver fibrosis. Advanced liver fibrosis results in cirrhosis, liver failure, and portal hypertension and often requires liver transplantation9. The generation of reactive oxygen species (ROS) and oxidative stress resulted from the metabolism of hepatotoxins is a common mechanism of liver injury. Of the multiple forms of P450s present in the liver endoplasmic reticulum, cytochrome P450 2E1 (CYP2E1) has been implicated as a key metabolizing enzyme for various xenobiotics from food and drugs into toxic metabolites which lead to liver injury. Induction of Cyp2e1 with ethanol, pyrazole, or other chemicals has been shown to promote oxidative stress10,11.
Although endogenous circadian controlled daily metabolism of toxins and hepatic Cyp2e1 expression have been described in mammals12,13, it still was unclear how the clock genes respond toxins, and how these responses influence the capacity to metabolize toxins. In present studies, we identified the clock gene Per1 was specifically elevated in mice exposed to toxins such as CCl4. We demonstrated the PER1 regulated Cyp2e1 expression, thereby promoting metabolism for toxins in the livers. These results suggest that PER1 is a regulator of liver metabolism of toxins. Appropriate suppression of the Per1 response is a potential therapeutic target for toxin-induced hepatotoxicity and dysfunction of the circadian clock.
Materials and methods
Animals and treatment
Male, 8–10-week-old Per1−/− and WT C57BL/6 mice were used in this study. The Per1−/− mice used in this study have been characterized previously14. All mice were maintained under standard laboratory conditions, with 12-h light/12-h dark cycles and free access to food and water at all stages of the experiments. All procedures were approved by the Animal Care and Use Committee at Nanjing University of Science and Technology (ACUC-NUST-20160016).
A single intraperitoneal injection of CCl4 (0.6 mL/kg body weight) was administrated to WT and Per1−/− mice. As controls, animals received the same volume of olive oil intraperitoneally. To determine the statistical significance of any observed differences, we used five male mice per time point following CCl4 administration, which included 24, 48, and 72 h. To detect the circadian mRNA expression of clock genes in response to CCl4, mice were killed at zeitgeber time (ZT)1 (ZT0 corresponds lights on and ZT12 to light off), ZT5, ZT9, ZT13, ZT17, ZT21, ZT24 (n = 5, each time point).To determine the plasma pharmacokinetics of CCl4, the blood samples were obtained from five male mice per time point following CCl4 administration, which included 2, 4, 8, and 12 h. For induction of liver fibrosis, CCl4 was injected twice weekly for 4 weeks. Mice were killed 48 h after the last injection. Serum aspartate transaminase (AST) and alanine transaminase (ALT) activity were measured using an AU2700 automatic biochemical analyzer (Olympus, Tokyo, Japan).
To determine the plasma pharmacokinetics of capsaicin, capsaicin was each separately suspended in 0.5% (w/v) CMC–Na solution to obtain a final concentration of 30 mg/ml. WT and Per1−/− mice were given capsaicin at a dose of 300 mg/kg, via gavage. After oral administration, the blood samples (0.6 mL) were collected at the desired times (30 min, 1, and 3 h) into heparinized centrifuge tubes.
To determine the plasma pharmacokinetics of acetaminophen (APAP), APAP was dissolved in warm saline and injected intraperitoneally at the dose of 500 mg/kg body weight, whereas saline was administered to control animals. After 0.5, 1, 2, or 4 h. Blood samples from six different mice per time point were then quickly centrifuged at 4 °C and a small plasma volume from each mouse was rapidly stored to assess the concentrations of APAP.
Quantitation of CCl4 by the headspace gas chromatographic method
Samples were prepared according to the method described by Jerry et al.15. Stoppered test tubes containing samples for CCl4 analysis were incubated at 60 °C for 15 min. All experiments were carried out by using Bruker GC-450 equipped with an electron capture detector (Bruker, Columbia, MD, USA). Column was 30 m × 0.25 mm i.d. capillary coated with 0.50 mm of 50% phenyl–50% methyl polysiloxane (Rxi-50). The chromatographic conditions were: column temperature, 100 °C: detector temperature, 200°C. Nitrogen was used as carrier gas.
Determination of hepatic oxidative stress and GSH levels
ROS was measured using 2’, 7’-dichlorofluorescein diacetate as a probe. Lipid peroxidation was determined by measuring the formation of the thiobarbituric acid-reactive substances spectrophotometrically and was expressed as malondialdehyde (MDA) concentration. Hepatic glutathione (GSH) levels were estimated by a colorimetric method using Ellman’s reagent and glutathione reductase. Hepatic ROS, MDA, and GSH contents were assayed with commercial kits according to the manufacturers’ instructions (Jiancheng, Nanjing, China).
TUNEL staining
At sacrifice, tissues were rapidly isolated, fixed in 4% paraformaldehyde, cryoprotected with 30% sucrose, and embedded in the optimal cutting temperature compound. The specimens were snap-frozen and sectioned into 15-μm sagittal sections (CM1950; Leica, Germany). Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining was performed using a TUNEL Apoptosis Assay Kit (Beyotime, C1088). Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Fluorescence images of liver slices were observed with fluorescence microscopy (Eclipse 800; Nikon, Tokyo, Japan).
Flow cytometry
After treatment, cells were stained with annexin V and propidium iodide (PI) (Annexin V—early apoptosis detection kit, Beyotime Biotechnology) following the manufacturer’s instructions. In brief, after culture under various conditions, cells were harvested and suspended in the appropriate binding buffer, stained with fluorescein isothiocyanate-conjugated annexin V and propidium iodide at room temperature for 15 minutes, and subsequently analyzed by a FACSCalibur flow cytometer (BD Biosciences, San Diego, CA, USA). Cells positive for annexin V and annexin V/propidium iodide were considered early and late apoptotic cells, respectively. All cells negative for annexin V were considered viable cells.
Histological analysis
Liver tissue was fixed in 10% phosphate-buffered formalin overnight, embedded in paraffin, and cut into 4 μm sections. Sections were stained with hematoxylin and eosin for routine examination or Masson’s trichrome for visualization of hepatic collagen deposition. Immunohistochemical staining was performed according to standard procedures16 using an appropriate specific primary antibody (Anti-CYP2E1, Abcam, Cambridge, UK; Anti-α-SMA, Millipore, Billerica, MA).
Cells and treatment
HepG2 E47 cells expressing Cyp2e1 (Boster Biological Technology Ltd., Wuhan, China) were cultured at 37 °C under 5% CO2 humidified atmosphere, using DMEM, supplemented with 10% FCS, 100 U/ml penicillin, and 100 mg/ml streptomycin.
Primary hepatocytes were isolated from 6 to 8-week-old male C57BL/6 mice using an in situ liver perfusion approach17. In brief, hepatocytes were dissociated from anesthetized adult mice by non-recirculating collagenase perfusion (C5138, Sigma, USA) through the portal vein. The isolated cells are then filtered through a 100 μm pore size mesh nylon filter. Cells were plated in collagen I–coated 6- or 12-well plated (at two or one million cells per well, respectively) in M199 medium plus 10% fetal bovine serum plus penicillin/streptomycin. After 3 hours of attachment, the medium was replaced with the appropriate assay medium.
For Per1 knockdown experiments, the complementary oligonucleotide of small hairpin RNA targeting the 5′‐GGTGCTCCCTAACTATCTATT‐3′ sequence was chemically synthesized, subcloned into the lentiviral vector (GenePharma, Shanghai, China), and transfected into primary hepatocytes or HepG2 E47 cells. Cells were infected with lentivirus at low multiplicity according to the manufacturer’s instructions. Cells were selected in puromycin (1 μg/mL) and polyclonal populations were expanded and analyzed. A hairpin siRNA with no sequence homology to human genes provided by the manufacturer (GenePharma) was used as the negative control. Per1 mRNA knockdown was assessed by real-time PCR. Clones that had sufficient knockdown (>75%) were used for further experiments.
For overexpression of human Per1, cells were transfected with pCMV-Sport2 Per1 or pCMV-Sport2 vector as control using Lipofectamine 2000 Transfection Reagent (Invitrogen, Carlsbad, CA). Construction of pCMV-Sport2 Per1 was characterized previously by Zheng and colleagues18.
The transfected cells were treated with 0.5% (v/v) CCl4 in 0.25% dimethyl sulfoxide prepared in serum-free culture medium for 24 h. All cell lines have been authenticated at the beginning of the study and again within 4 months after completion of the experiments. All cell lines have been tested for mycoplasma contamination frequently. Determination of MDA and GSH levels in cells were performed using commercial kits according to the manufacturers’ instructions (Jiancheng, Nanjing, China).
RNA extraction and quantitative real-time PCR
Total RNA was extracted from samples with Karrol reagent (Karroten Scientific, Nanjing, China) according to the manufacturer’s instructions. Reverse transcript reaction was carried out by reverse transcript kit (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. Real-time PCR was performed with the SYBR Green PCR Kit (Applied Biosystems, Foster City, CA) following the manufacturer’s instructions on an ABI 7300 real-time PCR system (Applied Biosystems) in a 20-μl volume. For an internal standard control, the expression level of glyceraldehyde-3-phosphate dehydrogenase (Gapdh) was simultaneously quantified. All primer sequences used for real-time PCR are shown in Table S1.
RNA-sequencing for detection of differentially expressed genes and pathways
RNA-sequencing (RNA-Seq) analysis and quantification were utilized to investigate changes in liver mRNA profiles among the different treatments performed. Isolated RNA was sent to BGI Co., Ltd. for conducting RNA-seq, which was performed on a BGISEQ-500 (Shenzhen, China). All samples were replicated three times for confirmation purposes. All the generated raw sequencing reads were filtered to remove reads with adapters, reads in which unknown bases are >5%, and low quality reads. Clean reads were then obtained and stored as FASTQ format. HISAT19 was used to map clean reads to the genome of GRCm38.p6. NOISeq20 method was used to screen deferentially expressed genes (DEGs). All DEGs were mapped to terms in Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis.
Western blot analysis
Proteins were extracted following the procedure described previously21. The extraction was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) 8–12% polyacrylamide gel and then electrically transferred to a polyvinylidene difluoride membrane. After blocking with 5% (w/v) BSA in TBST at room temperature for 1 h, the membranes were then incubated with an appropriate specific primary antibody (Anti-CYP2E1, Abcam, ab28146; anti-PER1, Abcam, ab3443; anti-HNF-1α, Cell Signaling Technology, #89670; anti-CBP, Cell Signaling Technology, #7389; anti-β-ACTIN, Bioworld, AP0060) at 4 °C overnight, followed by incubation with HRP-conjugated secondary antibody (Boster Biological Technology Ltd., BA1054) and detected by enhanced chemical luminescence kit (Thermo scientific, Hudson, NH, USA).
Immunoprecipitation assays
Immunoprecipitation assays were performed as described previously22,23 with slight modification. For ChIP assays, cross-linked chromatin was immunoprecipitated with 5 μg of antibody (anti-HNF-1α, Cell Signaling Technology, #89670; anti-PER1, Abcam, ab3443), or negative control rabbit IgG (Beyotime, A7016) at 4 °C overnight. Immunoprecipitated DNA was then used as a template for PCR. All primer sequences used for ChIP-PCRs were listed in Table S1.
For co-immunoprecipitations, liver tissues were homogenized and lysed with a Non-denaturing lysis buffer containing 20 mM Tris-HCl pH 8.0, 137 mM NaCl, 2 mM ethylenediaminetetraacetic acid (EDTA), and 1% NP-40 with protease inhibitor cocktail (Boster Biological Technology). To prepare immunoprecipitates, we incubated lysates with antibody (anti-HNF-1α, Cell Signaling Technology, #89670; anti-PER1, Abcam, ab3443) overnight at 4 °C, and then incubated with Protein A-Sepharose 4B (Invitrogen). Immunoprecipitates were washed five times with wash buffer containing 10 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EGTA, 1 mM EDTA, 1% Triton X-100, 0.2 mM sodium orthovanadate with protease inhibitor cocktail, boiled in SDS–PAGE loading buffer. Proteins were analyzed by western blotting as described above.
Quantitation of capsaicin by HPLC
Samples were prepared according to the method described by Yingying Zhao et al.24. A 200 μL aliquot of plasma was mixed with 50 μL α-naphthol (10 μg/mL; Aladdin, China), 10 μL methanol, 400 μL water, and 500 μL acetonitrile, and vortexing for 1 min. The solution was then extracted with 1.5 mL each of acetic ether and cyclohexane for capsaicin, followed by vortexing for 3 min. After 10 min centrifugation at 3000 rpm, 2.5 ml of the supernatant was transferred to another container and evaporated to dryness via nitrogen at 37 °C. The residue was dissolved in 300 μL of acetonitrile and mixed for 1 min. Following centrifugation at 3000 rpm for 5 min, 50 μL of the supernatant was injected into the High-performance liquid chromatography (HPLC) system for analysis.
Samples were analyzed using an HPLC (Waters 1525 System; Millipore, Bedford, MA, USA) on a reversed-phase C18 column. The mobile phase was acetonitrile–water (43:57) at a flow rate of 1.0 mL/min. The elution of metabolites was monitored at a wavelength of 254 nm.
Quantitation of APAP by HPLC
Samples were prepared according to a method described previously25. A 100 μg/ml methanol solution of theophylline was added to the plasma at a ratio of 1:1. This mixture was allowed to precipitate for at least 10 min and centrifuged twice at 11,000 × g 6–7 min to pellet the precipitated proteins. The mixtures were filtered through 0.45-μm HV Millipore cellulose filters and 20-μ1 samples were injected immediately into the chromatographic system.
Samples were analyzed using an HPLC (Waters 1525 System; Millipore, Bedford, MA, USA) on a reversed-phase C18 column. The gradient elution started with 30% methanol passing through the column at a flow-rate of 1.5 ml/min. After a delay of 0.5 min the methanol concentration was increased linearly to 75% over 7.5 min. The column was returned to 30% methanol after a delay of 1 min. The variable-wavelength detector was set at 254 nm.
Statistics
The single cosinor method was used for analysis of circadian rhythm26, and the cosine function equation was as follows: Y (t) = M + Acos (xt + u). The rhythm characteristics estimated by this method included the mesor (middle value of the fitted cosine representing a rhythm-adjusted mean), the amplitude (half the difference between the minimum and maximum of the fitted cosine function), and the acrophase (time of peak value in the fitted cosine function). Data were presented as means ± S.E.M. Statistical analysis was performed by Student’s t test, one-way ANOVA or two-way ANOVA followed by Tukey’s post hoc test. Significance was defined as P value < 0.05. Sample sizes of all experiments were predetermined by calculations derived from our experience. No sample was excluded from the analyses. Animals were not randomly assigned during collection, but the strain, sex, and age of the mice were the same, and the data analysis was single masked. Investigators were not blinded to the group allocation during the experiment and outcome assessment. The number of replicates was indicated in each figure legend. All qPCR data represented the mean of three technical replicates. The mean of the technical replicates was used per biological replicate. All statistical tests justified as appropriate and the data met the assumptions of the tests. There was an estimate of variation within each group of data.
Results
RNA sequence analysis of gene expression in the liver of mice exposed to CCl4
To search possible response genes following toxins exposure, we performed RNA-Seq to analyze the hepatic gene expression changes between toxin CCl4-treated and oil-controlled mice. A KEGG pathway analysis showed that organismal systems pathways (KEGG level 1) were the major pathways altered in CCl4-treated liver and 2048 DEGs are involved in multiple pathways essential for CCl4-induced liver injury and fibrosis (Fig. 1A), and 70 DEGs in the environmental adaptation pathway on level 2 of the KEGG functional category (Fig. 1C). Circadian rhythm is one of the top 20 affected pathways in the environmental adaptation implicated by DEGs (Fig. 1B). The larger the Rich factor, the greater the enrichment. Circadian rhythm pathway showed the greatest enrichment (Fig. 1B). In KEGG level 3, 294 KEGG pathways including the circadian rhythm pathway between the two groups changed significantly (Supplement Data). A gene that draws our attention was that encoding for the mouse Per1, the core clock gene in negative-feedback loops (Fig. 1C, D). Circadian rhythm pathway contained 10 changed genes (Fig. 1D), eight genes were upregulated and two gene was downregulated (Fig. 1D). However, other core clock genes including Clock, Bmal1, Cry1, Cry2, and Per2, were no obvious changes, and only Per1 was specifically elevated in livers following CCl4 exposure.

Liver tissues were collected 24 h after a single CCl4 treatment. Samples were analyzed by RNA-seq and differentially expressed pathways. A Distribution of the level 2 KEGG pathways in both CK and CCl4. The bar chart showed the numbers of sequences that were assigned within different pathway categories. B Top 20 pathways in the environmental adaptation from A were shown. C Heat map of 70 DEGs in the environmental adaptation pathway from A. D DEGs in the Circadian rhythm pathway from B were shown. CK: control. N = 3 independent biological replicates per group.
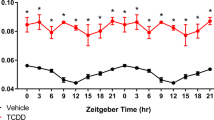
Response changes of clock genes in mice exposed to CCl4
To validate the results obtained from RNA-Seq, we carried out real-time PCR analysis for core clock genes expression in the liver mRNA between CCl4 and control group. As shown in Fig. 2, in both experimental and control groups, the core clock gene expressions were highly significantly changed within 24 hours. The present results confirmed that CCl4 acutely increased the expression of Per1 in livers (Fig. 2C). Table 1 showed the mesor of Per1 was significantly higher in CCl4-treated mice. While Per1 mRNA levels dramatically elevated, the mRNA rhythmicities of other clock genes were maintained and significantly declined in CCl4-treated mice livers (Fig. 2A–F). These findings revealed that CCl4-induced liver injury resulted in changes in the expression of circadian clock genes with a specific elevation of Per1 mRNA. Therefore, these observations raised a fundamental question to the biological relevance for such gene regulations.

A single intraperitoneal injection of CCl4 (0.6 mL/kg body weight) was administrated to WT at ZT1. Mice were killed at ZT1, ZT5, ZT9, ZT13, ZT17, ZT21, and ZT24. Hepatic mRNA levels of Bmal1 A, Clock B, Per1 C, Per2 D, Cry1 E, and Cry2 F were measured by real-time PCR after CCl4 treatment. CK: control. Solid lines indicate experimental response curves; dashed lines, fitted model curves. Red dashed line with the number represents mesor. Data were shown as means ± S.E.M. N = 5 independent biological replicates per group. Data represent cumulative results from three independent experiments.
Per1 deficiency decreased the metabolism rate of toxins
In order to show that the specific expression of Per1 caused by toxins is related to toxin degradation, a biochemical assay was undertaken to measure CCl4 residual levels in the plasma of WT and Per1 knockout (Per1−/−) mice after given a single intraperitoneal injection of CCl4. Plasma extract obtained from CCl4-treated Per1−/− mice displayed a higher level of CCl4 residue compared with that of WT mice, indicating loss of Per1 decreased liver metabolism rate of CCl4 (Fig. 3a). Next, we investigated whether the metabolism of other toxins was slowed down in Per1−/− mice. Certain concentrations capsaicin and APAP were intragastrically or intraperitoneally administrated into two genotypes of mice, respectively. The results showed that the residues of capsaicin in the plasma of Per1−/− mice were significantly higher than those in wild-type mice, either in the morning or at night (Fig. S1A). Moreover, more APAP residues in plasma were observed after the APAP intraperitoneal administration in Per1−/− mouse (Fig. S1B). Together, these results indicated that Per1 deficiency reduced the metabolic rate of toxins.

A The time course of plasma CCI4 concentrations from 0 to 12 h following a single intraperitoneal injection of CCl4 (0.6 mL/kg body weight). B Serum activities of ALT and C AST at 24, 48, and 72 h after single CCl4 treatment were measured. Liver tissues were collected 24, 48, and 72 h after single CCl4 treatment. Hepatic concentrations of D ROS production, E MDA, and F GSH were measured 24 h after single CCl4 treatment. G Representative immunostaining images. Apoptotic cells were visualized by TUNEL staining (green) and counterstained with DAPI (blue). H Representative H&E staining of livers from each treatment group. CK: control. Bar = 100 μm. Representative images from N = 3 biological replicates. Data were shown as means ± S.E.M. N = 5 independent biological replicates per group. * indicates P value < 0.05, ** indicates P value < 0.01, Per1−/− group versus WT group; # indicates P value < 0.05, ## indicates P value < 0.01, CCl4 group versus control group. Data represent cumulative results from three independent experiments.
Per1 deficiency decreased CCl4-induced acute liver injury
Then, we found an alleviated liver injury in Per1−/− mice, reflecting with significantly decreased AST and ALT levels at 24, 48, and 72 h after CCl4 administration (Fig. 3B, C). The levels of hepatic ROS were also significantly lower in Per1−/− mice compared with WT mice (Fig. 3D), but the MDA levels did not significantly change after CCl4 (Fig. 3E). The hepatic GSH level was reduced by CCl4 administration, with a further reduction in WT mice compared with Per1−/− mice (Fig. 3F). Apoptotic cells were visualized by TUNEL staining and counterstained with DAPI. Treatment with CCl4 (24 h) induced a significant apoptotic response, consistent with the above results (Fig. 3G). Apoptosis was significantly less frequent in the liver of Per1−/− mice as compared with WT mice, as seen by TUNEL (Fig. 3G). Histological examination of the liver tissues revealed that hepatic damage was limited to pericentral areas in mice at 24 h (Fig. 3H). The necrosis was less severe in the Per1−/− mice compared with WT mice during 24–72 h after CCl4 administration. These observations suggested that the elevation of Per1 expression in toxin CCl4 exposure was associated with the enhancement of liver metabolism of toxins capability.
Per1 deficiency alleviated chronic CCl4-induced liver injury and fibrosis in mice
To expand our observation that loss of Per1 reduced CCl4-induced liver acute injury, we performed a series of experiments to assess CCl4-induced chronic liver injury and fibrosis in WT and Per1−/− mice. Mice were injected with 0.6 mL/kg body weight CCl4 twice weekly for 4 weeks and killed 48 h after the last injection. As expected, histological examination of the liver tissues after CCl4 administration also revealed significantly less severe necrosis in Per1−/− mice compared with WT mice (Fig. 4A, B). Chronic liver injury was alleviated in Per1−/− mice as evidenced by lower serum ALT and AST levels after CCl4 administration (Fig. 4G, H). After chronic treatment of CCl4 for 4 weeks, WT mice had significant hepatic fibrosis, as demonstrated by Masson’s trichrome staining. In contrast, Per1−/− mice had less fibrosis as demonstrated by reduced collagen deposition (Fig. 4C, D). Expression of α-SMA, a marker of hepatic stellate cell (HSC) activation, was increased in both genotypes in response to chronic CCl4 treatment. Per1 deficiency significantly reduced α-SMA expression after CCl4 administration as assessed by immunohistochemistry (Fig. 4E, F). CCl4 administration elevated mRNA levels of markers for fibrogenesis including Col1α1, Col3α1, and Acta2. The expression of all these genes after CCl4 treatment was significantly suppressed in Per1−/− mice compared with WT mice (Fig. 4I–K). These results indicated that Per1 deficiency alleviated chronic CCl4-induced liver injury and fibrosis in mice.

A H&E staining of livers from each treatment group. B The percentage of necrotic area was quantified using Image J software. C Masson’s trichrome of livers from each treatment group for the visualization of hepatic collagen deposition. D The percentage of collagen volume fraction was quantified using Image J software. E Expression of α-SMA was determined by immunohistochemistry. F Relative α-SMA Protein level was quantified using Image J software. Bar = 100 μm. Representative images from N = 3 biological replicates. Serum activities of G ALT and H AST were measured after chronic CCl4 administration. Relative mRNA levels of I Col1α1 and J Col3α1 were measured in the liver of WT and Per1−/− mice. K Relative mRNA levels of Atca2 were measured in liver of WT and Per1−/− mice. CK: control. Data were shown as means ± S.E.M. N = 5 independent biological replicates per group. * indicates P value <0.05, ** indicates P value <0.01, Per1−/− group versus WT group; # indicates P value < 0.05, ## indicates P value <0.01, CCl4 group versus control group.
Per1 deficiency declined the level of Cyp2e1 expression in vivo
As circadian genes can influence hepatic oxidative stress by mediating the expression of antioxidative enzymes in mice liver27,28, we examined the effect of Per1 deletion on hepatic expression of these enzymes, including Gclc, Ho-1, Cat, Sod1, and Sod2. CCl4 treatment significantly elevated the expression of Gclc and Ho-1. Cat, Sod1, and Sod2 were downregulated in response to CCl4. These antioxidative enzymes showed no significant changes between WT and Per1−/− mice (Fig. 5A–E). The injurious effects of a few toxins are results of their intermediary metabolites by CYP2E1, some highly toxic molecules that can lead to oxidative injury29,30. CCl4 is metabolized by CYP2E1 to trichloromethyl peroxyl radical that induces oxidative stress cell damage and cell death31. The change of Cyp2e1 expression by qRT-PCR analysis was in agreement with RNA-seq data (Fig. S2). Intraperitoneal injection of mice with CCl4 decreased Cyp2e1 expression as previously described31. Analysis of mRNA levels in the liver revealed that Cyp2e1 expression was significantly downregulated by Per1 deletion in both control and CCl4 group (Fig. 5F), indicating a possibility of decreased hepatic injury after CCl4 was primarily owing to downregulation of Cyp2e1 in Per1−/− mice. Then we investigated whether the expression of Cyp2e1 was circadian controlled. The results showed the mRNA expression of Cyp2e1 was maximal at the beginning of the dark phase of the entraining photocycle at ZT13 and minimal at ZT1, the onset of the light phase (Fig. 5G). The knockdown of Per1 resulted in an approximately twofold diminution of Cyp2e1 expression at the mRNA level (Fig. 5G). Western blot analysis revealed that CYP2E1 protein levels were higher during the dark phase (Fig. 5H). The CYP2E1 protein was also reduced in Per1−/− mice, and the circadian rhythm of CYP2E1 was impaired (Fig. 5H). Analysis of protein levels in the liver revealed that CYP2E1 expression was significantly downregulated by Per1 deletion in both control and CCl4 group (Fig. 5I). Next, we performed immunoblotting analysis to examine putative changes in CYP2E1 protein levels in mouse liver tissues, resulting from CCl4 treatment. Parallel immunohistochemical examination revealed that CYP2E1-positive staining was decreased in Per1−/− mice exposed CCl4 compared with WT mice (Fig. 5J). Taken together, these findings demonstrated that Per1 participated in the regulation of transcriptional activation of Cyp2e1 and, in turn, played a critical role in mediating CCl4-induced hepatotoxicity.

Liver tissues were collected 24 h after single CCl4 administration. Hepatic mRNA levels of A Gclc, B Ho-1, C Cat, D Sod1, E Sod2, and F Cyp2e1 were measured by real-time PCR. CK: control. Data were shown as means ± S.E.M. N = 5 independent biological replicates per group. * indicates P value < 0.05, ** indicates P value <0.01, Per1−/−group versus WT group; # indicates P value <0.05, ## indicates P value <0.01, CCl4 group versus control group. G Cyp2e1 expression was measured by real-time PCR in the mouse liver at ZT1 and ZT13. H Representative western blots for rhythm of CYP2E1 protein expression in the mouse liver. Extracts were measured via western blot analysis with anti-CYP2E1 or anti–β-ACTIN antibody. CK: control. Data were shown as means ± S.E.M. N = 3 independent biological replicates per group. * indicates P value <0.05, ** indicates P value <0.01, Per1−/− group versus WT group; # indicates P value <0.05, ## indicates P value <0.01, ZT1 group versus ZT13 group. I Western blot assay of CYP2E1 protein after CCl4 treatment in vivo. J Representative images of immunohistochemical staining were shown for liver sections stained with anti-CYP2E1 antibody. This is in agreement with the RT-PCR and western blot data. Bar = 100 μm. Representative images from N = 3 biological replicates. CK: control. Data were shown as means ± S.E.M. N = 3 independent biological replicates per group. * indicates P value <0.05, ** indicates P value <0.01, Per1−/− group versus WT group; # indicates P value <0.05, ## indicates P value <0.01, CCl4 group versus control group. Data represent cumulative results from three independent experiments.
Effect of Per1 on CCl4-induced oxidative injury and Cyp2e1 expression in hepatocytes
To investigate whether Per1 directly regulated Cyp2e1 expression in hepatocytes, we analyzed CCl4-induced oxidative injury in primary hepatocytes and HepG2 E47 cells. The primary hepatocytes were transfected with control shRNA or Per1-specific shRNA, and the cells were challenged with CCl4. shRNA-mediated knockdown of Per1 led to a 70–80% decrease in mRNA levels (Fig. 6A). Next, we created stable HepG2 E47 cell lines expressing shPer1 through retroviral infection and assessed the effect of Per1 suppression on the expression of Cyp2e1. As expected, real-time PCR experiments showed that cells treated with Per1 shRNA decreased the expression level of Per1 (Fig. 6A). CCl4 exposure increased intracellular MDA levels, and suppression of Per1 expression significantly impaired the elevation of MDA in primary hepatocytes (Fig. 6B). The contents of GSH in cells also decreased after CCl4, which could be alleviated by knockdown of Per1 in primary hepatocytes (Fig. 6C). Per1 inhibition improved the increase in MDA levels and the decrease in GSH levels in HepG2 E47 cells (Fig. 6B, C). Per1 inhibition also effectively reduced mRNA levels of Cyp2e1 in primary hepatocytes (Fig. 6D). Per1 inhibition reduced Cyp2e1 mRNA levels in HepG2 E47 cells, consistent with the results of primary hepatocytes (Fig. 6D). Apoptosis was measured by annexin V/PI staining followed by flow cytometry. Compared with control shRNA, the cells treated with Per1 shRNA showed less apoptosis in vitro in primary cells (Fig. 6E). Consistent with the above observation, inhibition of Per1 expression resulted in the reduction of apoptosis in HepG2 E47 cells (Fig. 6E). To further study the function of Per1, HepG2 E47 cells expressing Cyp2e1 were transfected with Per1-cDNA plasmid and then treated with CCl4 for 24 h. Overexpression was verified by real-time PCR (Fig. 6F). Overexpression of Per1 in HepG2 E47 increased CCl4 toxicity. Moreover, MDA induced by CCl4 was increased to a greater extent, and GSH was further decreased (Fig. 6G, H).The Per1 transfection significantly increased Cyp2e1 expression in hepatocytes both in control and CCl4 group (Fig. 6I), which was consistent with that observed in vivo. We observed a significant increase in apoptosis after CCl4 in cells of Per1 cDNA transfected (Fig. 6J).

A Knockdown of Per1 was verified by real-time PCR after infection with shPer1 or shRNA control (shCont) in primary cells and HepG2 E47 cells. Cells were harvested after treatment with 0.5% CCl4 for 24 h. Levels of B MDA and C GSH in cell lysates were determined. D Cyp2e1 expression was measured by real-time PCR. E Representative images of flow cytometry analysis of apoptosis by annexin V/PI staining in primary cells and HepG2 E47 cells infected with shPer1 or shCont. CK: control. Data were shown as means ± S.E.M. N = 3–5 independent biological replicates per group. * indicates P value <0.05, ** indicates P value <0.01, shPer1 group versus shCont group; # indicates P value <0.05, ## indicates P value < 0.01, CCl4 group versus control group. HepG2 cells were transfected with Per1 cDNA or vector as control. F Real-time PCR was used to analyze changes in gene expression of Per1. Levels of G MDA and H GSH in cell lysates were determined. I Cyp2e1 expression was measured by real-time PCR. J Representative flow profile of annexin V/PI staining. CK: control. Data were shown as means ± S.E.M. N = 3–5 independent biological replicates per group. * indicates P value <0.05, ** indicates P value <0.01, Per1 cDNA group versus vector group; # indicates P value <0.05, ## indicates P value <0.01, CCl4 group versus control group. Data represent cumulative results from three independent experiments.
The Per1 increased Cyp2e1 expression by interacting with HNF-1α on Cyp2e1 promoter
Next, we investigated how Per1 regulates Cyp2e1 expression. Cyp2e1 exhibits 24-hour periodicity in its hepatic mRNA levels, and this circadian rhythm is regulated by HNF-1α and the circadian organization of molecular clocks12. Both hepatic mRNA and protein levels of HNF-1α were similar between WT and Per1−/− mice (Fig. 7A–C). The role of PER1 in the transcriptional activation of HNF-1α was confirmed by ChIP-qPCR, which showed a marked reduction (about twofold) in HNF-1α binding to Cyp2e1 promoter regions in Per1-deficient liver tissues (Fig. 7D, E). PER1 protein also occurred in the HNF-1α-binding site in the Cyp2e1 promoter (Fig. 7F), implying a possible interaction between PER1 and HNF-1α. Co-immunoprecipitation and western blot analysis of the liver lysates showed that HNF-1α interacted with PER1 and forms a protein complex (Fig. 7G). Interaction of CREB binding protein (CBP) with the N-terminal domain of HNF-1 greatly increased the binding affinity32. To further verify the specificity of HNF-1α to the PER1, we tested the binding of CBP to PER1. CBP was also co-immunoprecipitated by the anti-PER1 (Fig. 7G). Not surprisingly, we also observed strongly synergy between PER1 and HNF-1α, whereas CBP specifically interacted with HNF-1α in the whole-liver homogenates immunoprecipitated with anti-HNF-1α antibody (Fig. 7H). These results indicated that PER1 protein interacted with the HNF-1α–CBP complex and increased the binding activity of HNF-1α on the Cyp2e1 promoter, enhancing the transcription of Cyp2e1 in mice liver.

A Hepatic mRNA levels of HNF-1α were measured in Per1−/− and WT mice. B–C Hepatic protein levels of HNF-1α were analyzed by western blot and β-actin was used as internal control. D Recruitments of HNF-1α to Cyp2e1 promoter were detected by ChIP assays. E ChIP assays were performed followed by real-time PCR. F Recruitments of PER1 to Cyp2e1 promoters were detected by ChIP assays. G–H Representative immunoblots showing recovery of the indicated proteins from mouse liver after immunoprecipitation with the indicated antibodies. I A hypothetical model of how PER1 interacted with HNF-1α on Cyp2e1 promoter and target gene transcription. Data were shown as means ± S.E.M. N = 5 independent biological replicates per group; *P < 0.05, Per1−/− group versus WT group.
Discussion
Our observations that toxins specifically activate Per1 expression illustrate the central role played by the circadian clock in mediating the metabolism of toxins physiology in mice. Our previous study showed that Per2 plays a protective role in CCl4-induced hepatotoxicity33. The opposite gene expression of Per1 and Per2 in mice exposed to CCl4 suggested different roles of core circadian genes. Per2 gene functions in hepatocyte protection from chemical toxicants via the regulation of hepatic Ucp2 gene expression levels33. Loss of Per1 reduced metabolism of toxins capability, and eliminated the difference of toxin residues in plasma between day and night, suggesting Per1 is required for maintaining daily xenobiotic-metabolizing rhythms. It is well known that Cyp2e1 is responsible for the metabolism of most xenobiotics and is responsible for the efficient elimination of foreign chemicals from the body34. Our results supported that the metabolizing capacity of Per1 is through regulating Cyp2e1 expression. Paradoxically, Cyp2e1 metabolically activates biologically inert compounds to electrophilic derivatives that can cause toxicity and cell death10,11. CYP2E1 catalyzes CCl4 molecule to the trichloromethyl free radical and induces hepatotoxicity. Elevation of CYP2E1 activity with phenobarbital, dichlorodiphenyltrichloroethane, or Arochlor 1254 R increases the hepatotoxic effect of CCl435. Mice lacking Cyp2e1 are resistant to CCl4 hepatotoxicity36. Therefore, it is reasonable that mice lacking Per1 alleviate CCl4-induced liver injury and fibrosis due to the low level of hepatic Cyp2e1.
The Cyp2e1 expression and activities are circadian controlled with higher levels in the dark phase in mice liver12,37, and this rhythm was impaired in Per1−/− mice. Previous study has demonstrated transcription of the Cyp2e1 is rhythmically enhanced by HNF-1α, and circadian gene CRY1 rhythmically inhibits the binding activity of HNF-1α on Cyp2e1 promoter12,38,39. PER and CRY play a central role in negative-feedback loop in the core clock system. The circadian pattern of Per1 mRNA in the liver is consistent with the rhythmic expression of Cyp2e1, which peaks at ZT12 as previously described40. These evidences implied a possible role of Per1 in regulating the circadian transcription of Cyp2e1. In the present study, we showed that Per1 directly enhanced mRNA levels of Cyp2e1. The expression of HNF-1α was not affected by Per1. However, the recruitment of HNF-1α to Cyp2e1 promoter was significantly weakened in Per1−/− mice. ChIP assays also revealed the recruitment of PER1 protein to the binding site of HNF-1α in the Cyp2e1 promoter. Previous study of the crystal structure of PER1 protein demonstrated that PER1 cannot directly bind to DNA41, implying that PER1 may associate with other proteins and indirectly bind to the Cyp2e1 promoter. Results of co-immunoprecipitated assays revealed a novel interaction between PER1 and HNF-1α. The present study suggests that the Per1 maintains the daily metabolism of toxin rhythm through regulation of Cyp2e1 expression (Fig. 7I). Our findings presented a control system that causes alternating turn-off/turn-on of transcription on Cyp2e1 by endogenous circadian PER1 interaction.
The overarching evidences have already showed a reciprocal regulation of circadian rhythms and metabolic states. Disrupted rhythms can lead to metabolic impairments, the reverse is also true that disrupting metabolism can alter circadian rhythms3. Patients with cirrhosis have liver portal hypertension and dysfunctional circadian clock systems5. Hepatic fibrosis induced by CCl4 leads to alterations in the rhythms of hepatic clock genes6, and the patients with hepatic fibrosis suffer abnormal sleeping cycles42, indicating possible side-effects of clock responding to liver injury and metabolic diseases. Per1 is the unique clock gene that responds rapidly to toxin exposure, and this response inevitably influences the normal circadian system.
There are multiple CRE binding sites in the promoter of Per1 gene, which may be caused by cAMP signal (enriched in KEGG). Loss of Per1 reduced significantly CCl4-induced acute and chronic liver injury. Thus, appropriate suppression of Per1 response is a potential therapeutic target for toxin-induced hepatotoxicity, also is a promising method to improve the dysfunctional circadian clock system caused by toxin metabolism.
Data availability
Data are available upon request from the corresponding author.
References
Reppert, S. M. & Weaver, D. R. Coordination of circadian timing in mammals. Nature 418, 935–941 (2002).
Fu, L., Patel, M. S., Bradley, A., Wagner, E. F. & Karsenty, G. The molecular clock mediates leptin-regulated bone formation. Cell 122, 803–815 (2005).
Arble, D. M., Ramsey, K. M., Bass, J. & Turek, F. W. Circadian disruption and metabolic disease: findings from animal models. Best. Pract. Res., Clin. Endocrinol. Metab. 24, 785–800 (2010).
Tahara, Y. & Shibata, S. Circadian rhythms of liver physiology and disease: experimental and clinical evidence. Nat. Rev. Gastroenterol. Hepatol. 2016 13, 217–226 (2016).
Montagnese, S. et al. Sleep-wake abnormalities in patients with cirrhosis. Hepatology 59, 705–712 (2014).
Chen, P., Kakan, X. & Zhang, J. Altered circadian rhythm of the clock genes in fibrotic livers induced by carbon tetrachloride. FEBS Lett. 584, 1597–1601 (2010).
Kleiner, D. E. et al. Hepatic histological findings in suspected drug-induced liver injury: systematic evaluation and clinical associations. Hepatology 59, 661–670 (2014).
Malhi, H. & Gores, G. J. Cellular and molecular mechanisms of liver injury. Gastroenterology 134, 1641–1654 (2008).
Bataller, R. & Brenner, D. A. Liver fibrosis. J. Clin. Invest. 115, 209 (2005).
Gong, P. & Cederbaum, A. I. Nrf2 is increased by CYP2E1 in rodent liver and HepG2 cells and protects against oxidative stress caused by CYP2E1. Hepatology 43, 144–153 (2006).
Dey, A., Caro, A. A. & Cederbaum, A. I. S-adenosyl methionine protects ob/ob mice from CYP2E1-mediated liver injury. Am. J. Physiol. Gastrointest. Liver Physiol. 293, G91–G103 (2007).
Matsunaga, N., Ikeda, M., Takiguchi, T., Koyanagi, S. & Ohdo, S. The molecular mechanism regulating 24-hour rhythm of CYP2E1 expression in the mouse liver. Hepatology 48, 240–251 (2008).
Beauchamp, D. & Labrecque, G. Chronobiology and chronotoxicology of antibiotics and aminoglycosides. Adv. Drug Deliv. Rev. 59, 896–903 (2007).
Krista, K. & Lee, C. C. Reciprocal regulation of haem biosynthesis and the circadian clock in mammals. Nature 430, 467–471 (2004).
Hjelle, J. J. & Petersen, D. R. Decreased in vivo acetaldehyde oxidation and hepatic aldehyde dehydrogenase inhibition in C57BL and DBA mice treated with carbon tetrachloride. Toxicol. Appl. Pharmacol. 59, 15–24 (1981).
Cong, M. et al. Antifibrotic effects of a recombinant adeno-associated virus carrying small interfering RNA targeting TIMP-1 in rat liver fibrosis. Am. J. Pathol. 182, 1607–1616 (2013).
Seglen, P. O. Preparation of isolated rat liver cells. Methods Cell Biol. 13, 29–83 (1976).
Zheng, B. et al. Nonredundant roles of the mPer1 and mPer2 genes in the mammalian circadian clock. Cell 105, 683–694 (2001).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Tarazona, S., García-Alcalde, F., Dopazo, J., Ferrer, A. & Conesa, A. Differential expression in RNA-seq: a matter of depth. Genome Res 21, 2213–2223 (2011).
Yang, P. et al. Loss of A(1) adenosine receptor attenuates alpha-naphthylisothiocyanate-induced cholestatic liver injury in mice. Toxicol. Sci. 131, 128–138 (2013).
Tang, X. et al. Assessment of Brd4 inhibition in idiopathic pulmonary fibrosis lung fibroblasts and in vivo models of lung fibrosis. Am. J. Pathol. 183, 470–479 (2013).
Kong, X. et al. Interleukin-22 induces hepatic stellate cell senescence and restricts liver fibrosis in mice. Hepatology 56, 1150–1159 (2012).
Zhao, Y. et al. Preparation, characterization, and pharmacokinetics study of capsaicin via hydroxypropyl-beta-cyclodextrin encapsulation. Pharm. Biol. 54, 130–138 (2016).
Esteban, A., Graells, M., Satorre, J. & Pérez-Mateo, M. Determination of paracetamol and its four major metabolites in mouse plasma by reversed-phase ion-pair high-performance liquid chromatography. J. Chromatogr. 573, 121–126 (1992).
Mohri, T. et al. Alterations of circadian expressions of clock genes in Dahl salt-sensitive rats fed a high-salt diet. Hypertension 42, 189–194 (2003).
Hardeland, R., Coto-Montes, A. & Poeggeler, B. Circadian rhythms, oxidative stress, and antioxidative defense mechanisms. Chronobiol. Int. 20, 921–962 (2003).
Xu, Y. Q. et al. Diurnal variation of hepatic antioxidant gene expression in mice. PLoS ONE 7, e44237 (2012).
Tsujimoto, I., Moriya, K., Sakai, K., Dickneite, G. & Saka, T. Critical role of factor XIII in the initial stages of carbon tetrachloride-induced adult liver remodeling. Am. J. Pathol. 179, 3011–3019 (2011).
Weber, L. W. D., Boll, M. & Stampfl, A. Hepatotoxicity and mechanism of action of haloalkanes: carbon tetrachloride as a toxicological model. Crit. Rev. Toxicol. 33, 105–136 (2003).
Knockaert, L. et al. Carbon tetrachloride-mediated lipid peroxidation induces early mitochondrial alterations in mouse liver. Lab. Invest. 92, 396–410 (2012).
Soutoglou, E., Papafotiou, G., Katrakili, N. & Talianidis, I. Transcriptional activation by hepatocyte nuclear factor-1 requires synergism between multiple coactivator proteins. J. Biol. Chem. 275, 12515–12520 (2000).
Chen, P. et al. The protective role of Per2 against carbon tetrachloride-induced hepatotoxicity. Am. J. Pathol. 174, 63–70 (2009).
Chen, J. et al. A comprehensive review of cytochrome P450 2E1 for xenobiotic metabolism. Drug Metab. Rev. 51, 178–195 (2019).
Rechnagel, R. O., Glende, E. A. & Plaa, G. L. Carbon tetrachloride hepatotoxicity: an example of lethal cleavage. Crit. Rev. Toxicol. 2, 263–297 (1973).
Wong, W. Y., Chan, W. Y. & Lee, S. T. Resistance to carbon tetrachloride-induced hepatotoxicity in mice which lack CYP2E1 expression. Toxicol. Appl. Pharmacol. 153, 109–118 (1998).
Matsunaga, N. et al. Influence of feeding schedule on 24-h rhythm of hepatotoxicity induced by acetaminophen in mice. J. Pharmacol. Exp. Ther. 311, 594–600 (2004).
Cheung, C., Akiyama, T. E., Kudo, G. & Gonzalez, F. J. Hepatic expression of cytochrome P450s in hepatocyte nuclear factor 1-alpha (HNF1α)-deficient mice. Biochem. Pharmacol. 66, 2011–2020 (2003).
Ktistaki, E. & Talianidis, I. Modulation of hepatic gene expression by hepatocyte nuclear factor 1. Science 277, 109–112 (1997).
Wang, T. et al. Deletion of circadian gene Per1 alleviates acute ethanol-induced hepatotoxicity in mice. Toxicology 314, 193–201 (2013).
Kucera, N. et al. Unwinding the differences of the mammalian PERIOD clock proteins from crystal structure to cellular function. Proc. Natl. Acad. Sci. USA 109, 3311–3316 (2012).
Montagnese, S. et al. Sleep‐wake abnormalities in patients with cirrhosis. Hepatology 59, 705–712 (2014).
Acknowledgements
This work was supported by the grant from the National Natural Science Foundation of China (31671220, 31871178) and the Fundamental Research Funds for the Central Universities (30920031102).
Author information
Authors and Affiliations
Contributions
W.H.G. and T.W. performed the experiments. Y.Z., Y.X.Y., Q.S., X.Y., G.Y., and X.X. contributed reagents and analytical tools. J.F.Z. supervised the project and conceived the study and designed the experiments. W.H.G. and J.F.Z. wrote the manuscript with input from other authors, and all authors revised it.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval and consent to participate
All procedures were approved by the Animal Care and Use Committee at Nanjing University of Science and Technology (ACUC-NUST-20160016).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by M. Agostini
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ge, W., Wang, T., Zhao, Y. et al. Period1 mediates rhythmic metabolism of toxins by interacting with CYP2E1. Cell Death Dis 12, 76 (2021). https://doi.org/10.1038/s41419-020-03343-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-020-03343-7

{kind=link}
{kind=link}