The First Complete Mitochondrial Genome of Lachninae Species and Comparative Genomics Provide New Insights into the Evolution of Gene Rearrangement and the Repeat Region


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Preparation and DNA Extraction
2.2. Mitogenome Sequencing and Assembly
2.3. Annotation and Analysis of S. sinisalicis Mitogenome
2.4. Phylogenetic Analysis
2.5. Comparative Mitogenomic Analysis
3. Results and Discussion
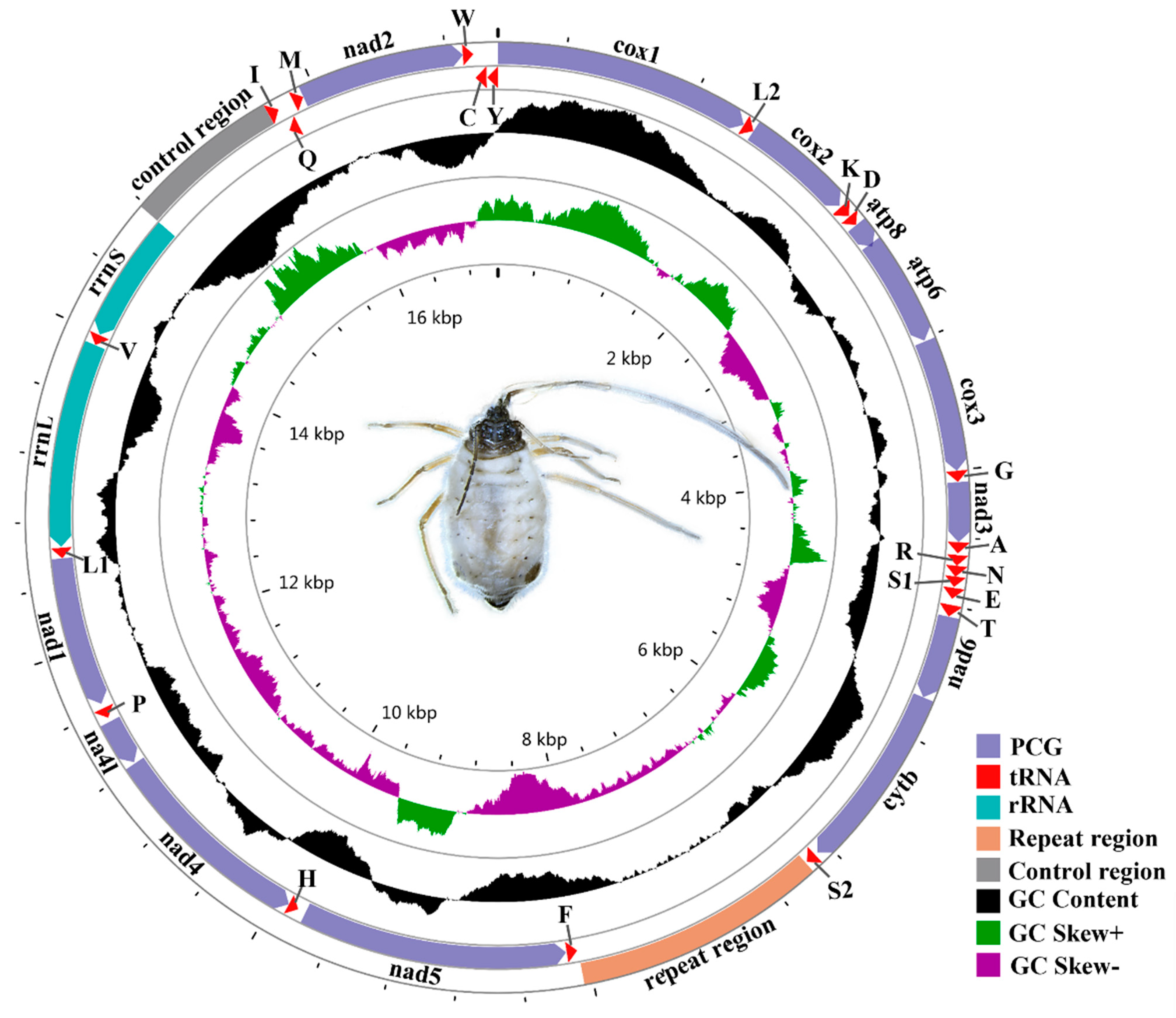
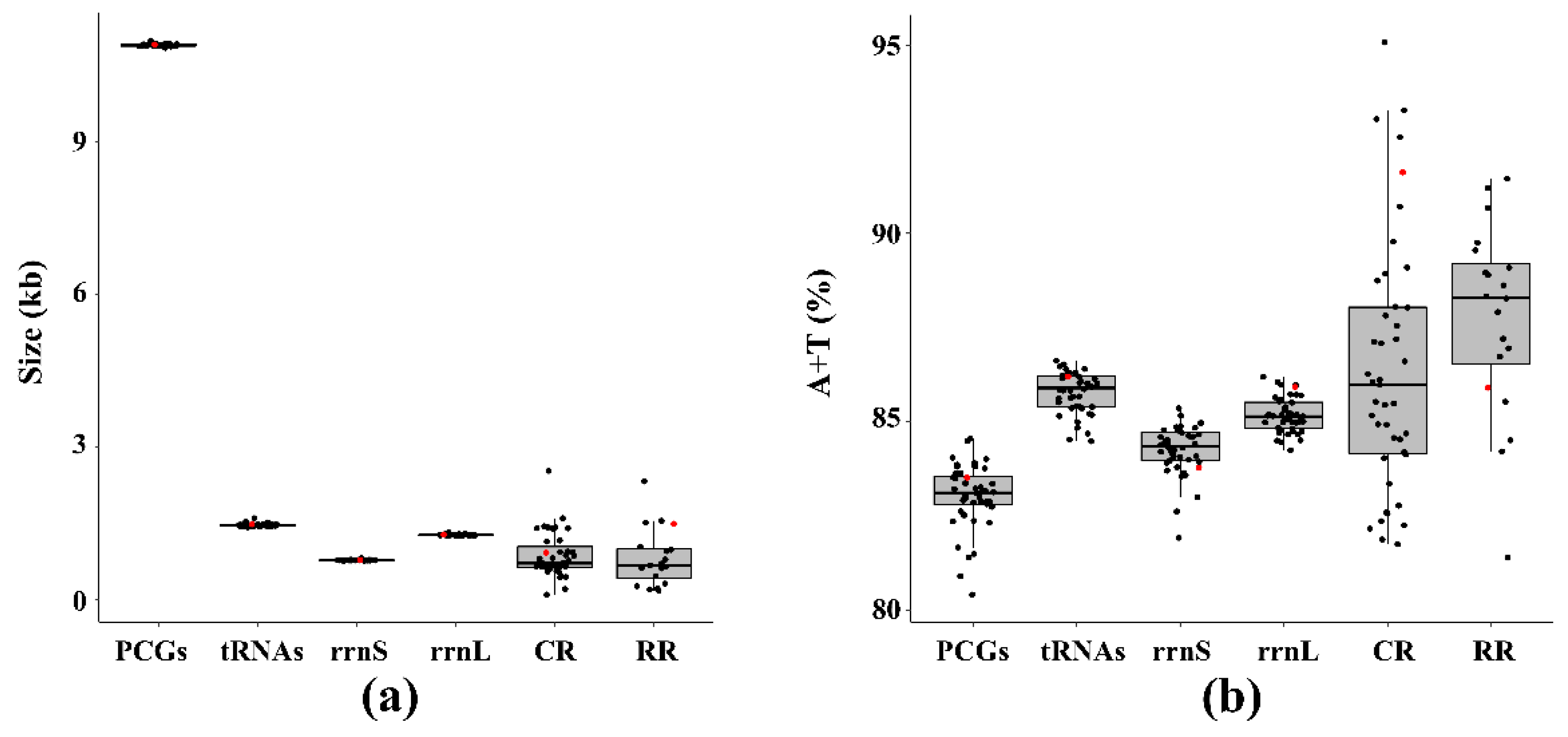
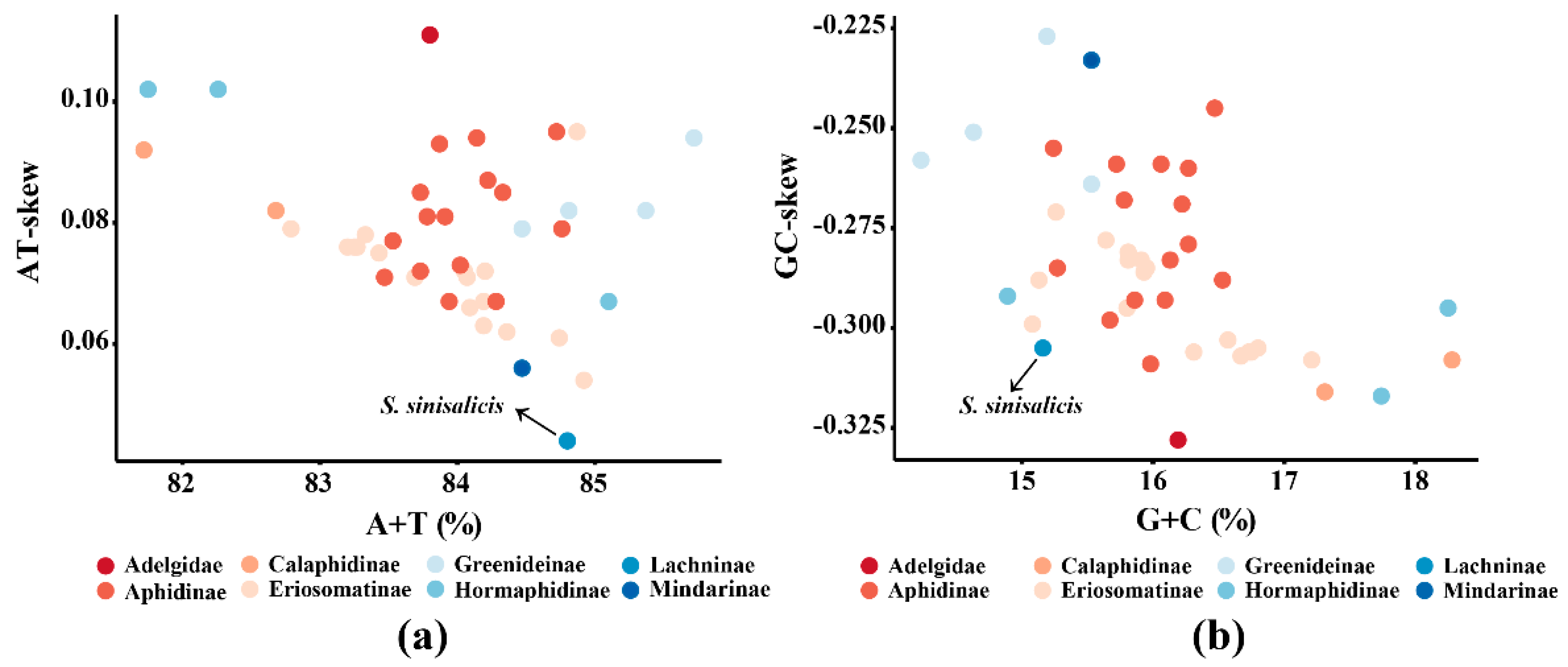
3.1. Mitogenome Features of S. sinisalicis and Other Aphids
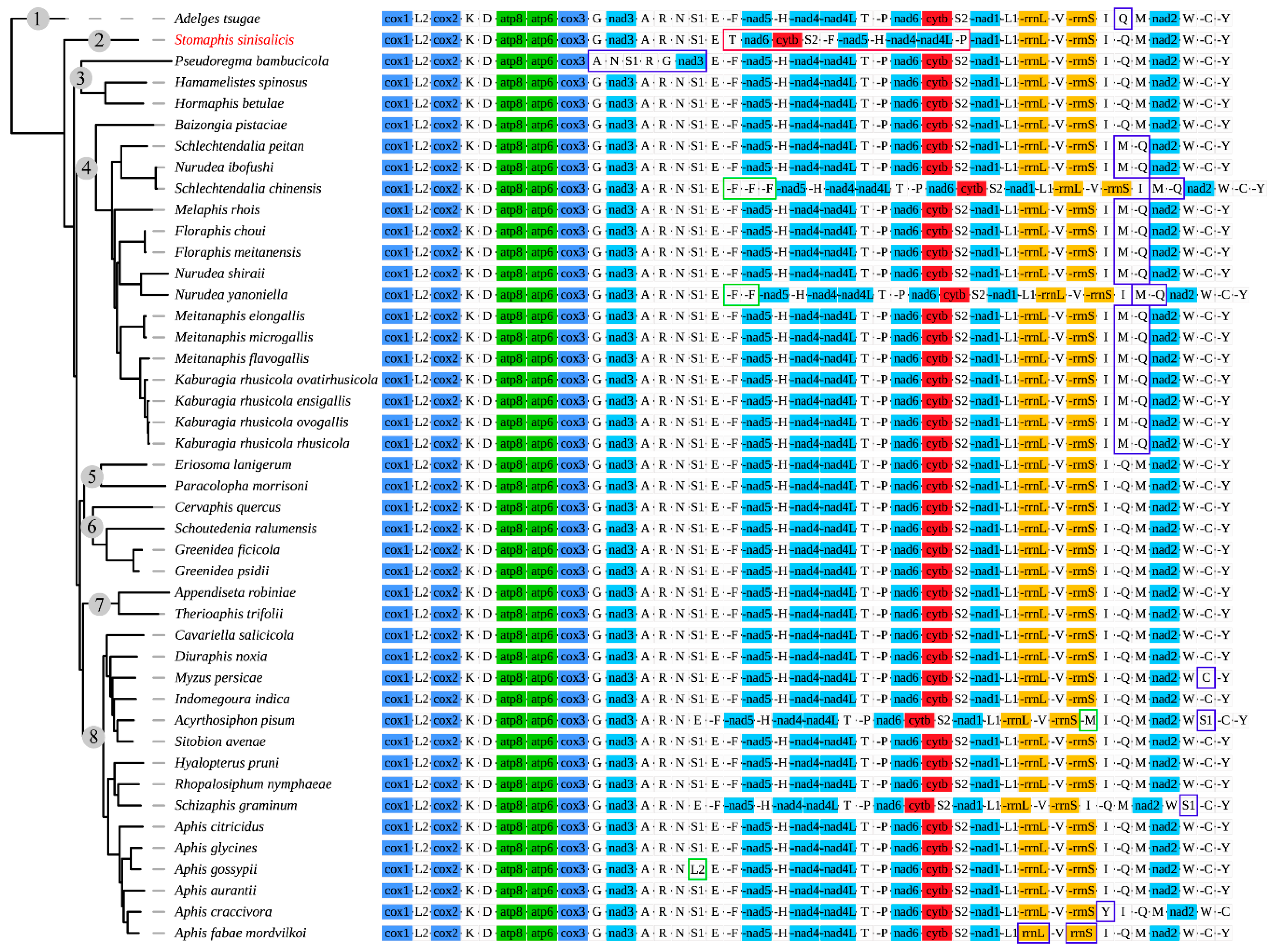
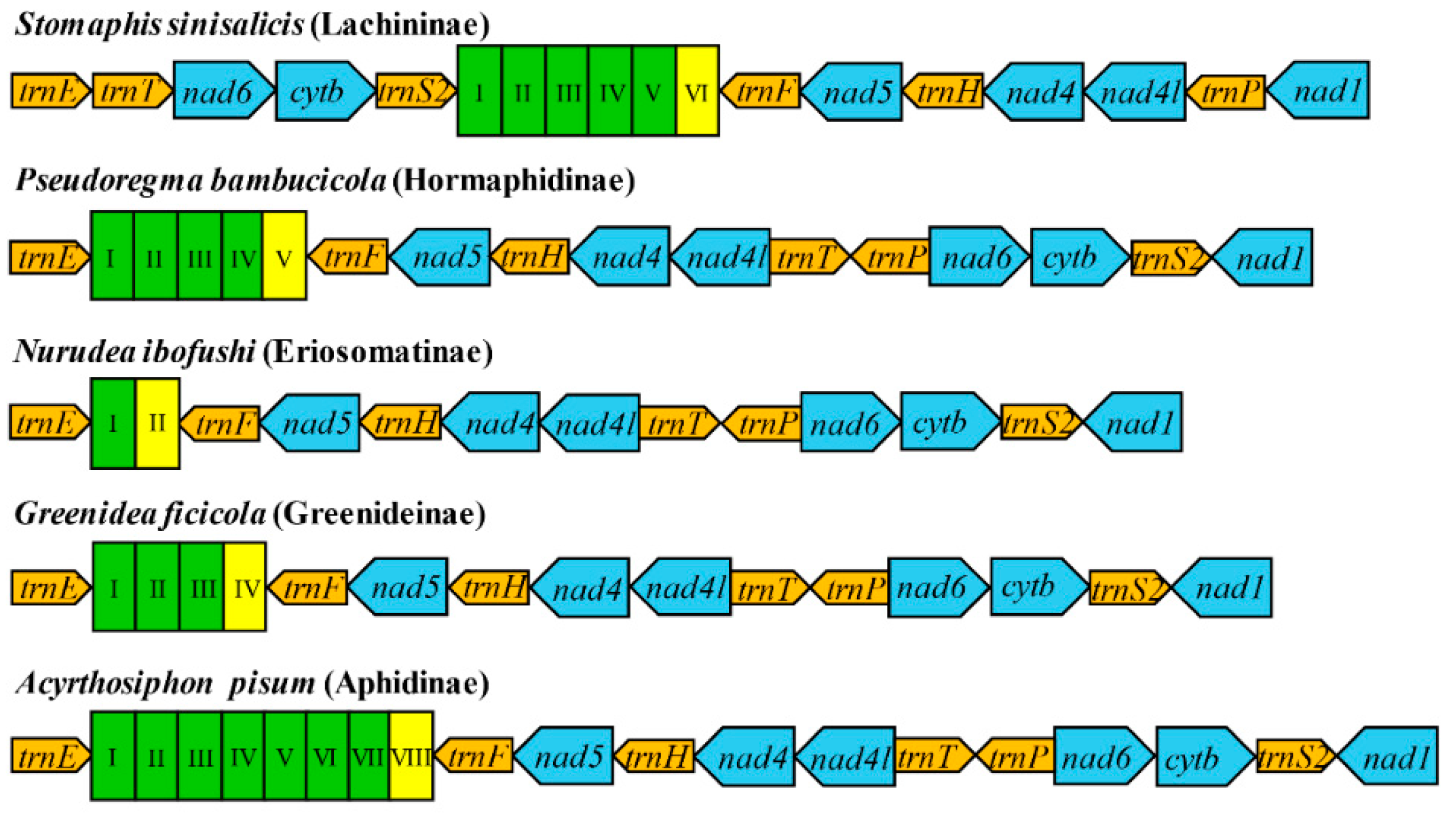
3.2. Gene Arrangement Patterns
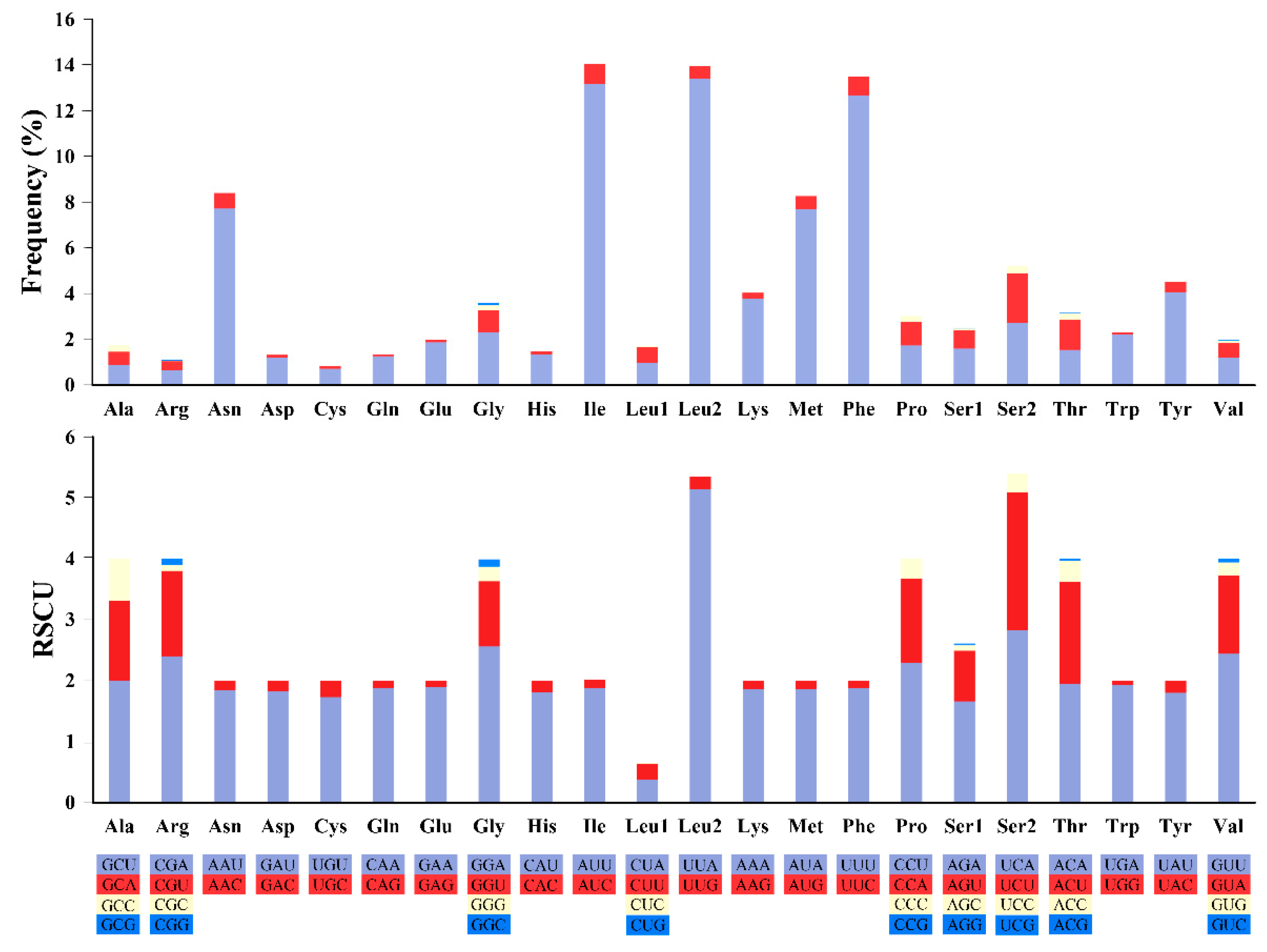
3.3. Protein Coding Genes and Codon Usage Patterns
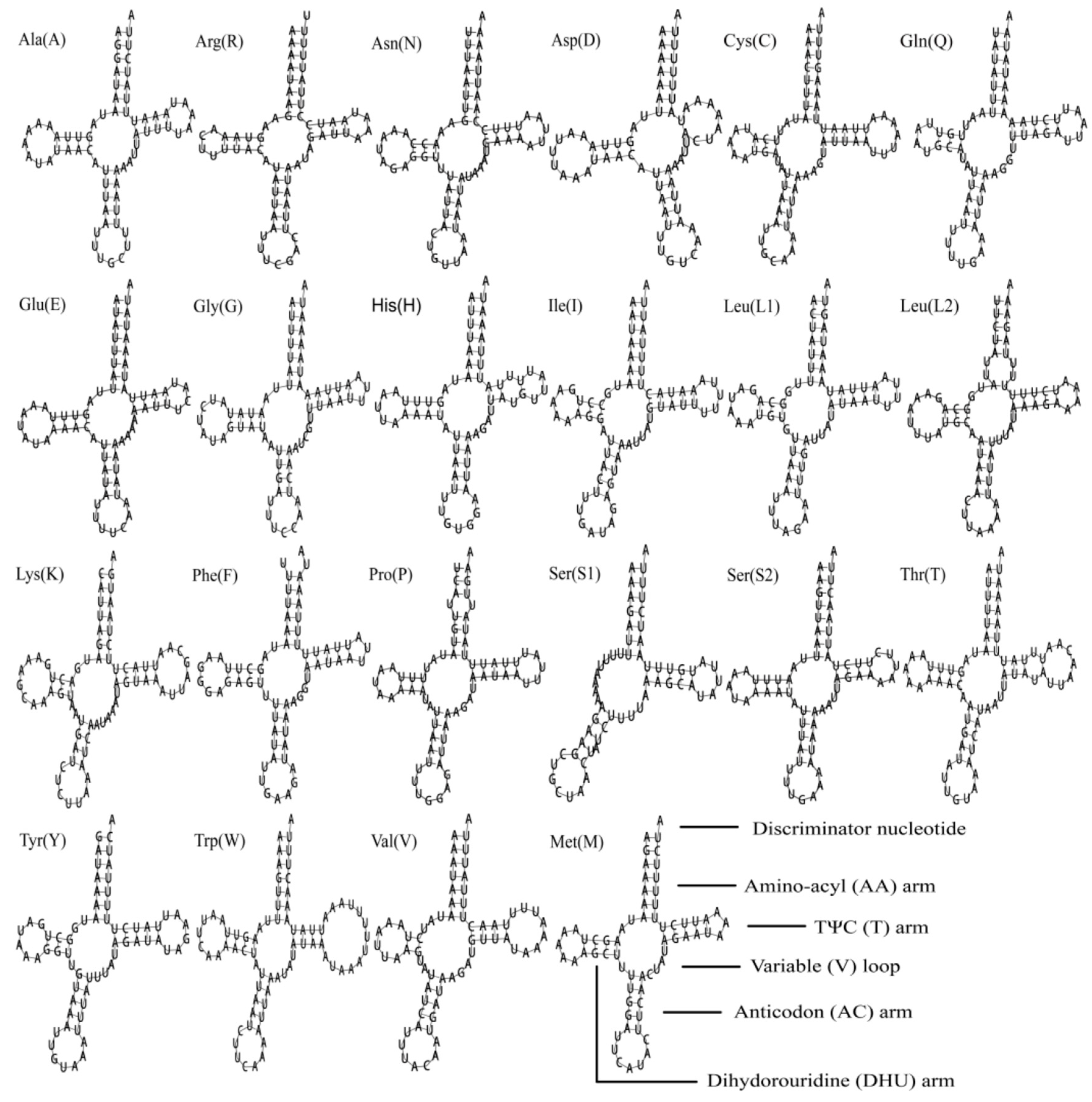
3.4. Transfer and Ribosomal RNA Genes
3.5. Non-Protein Coding Regions
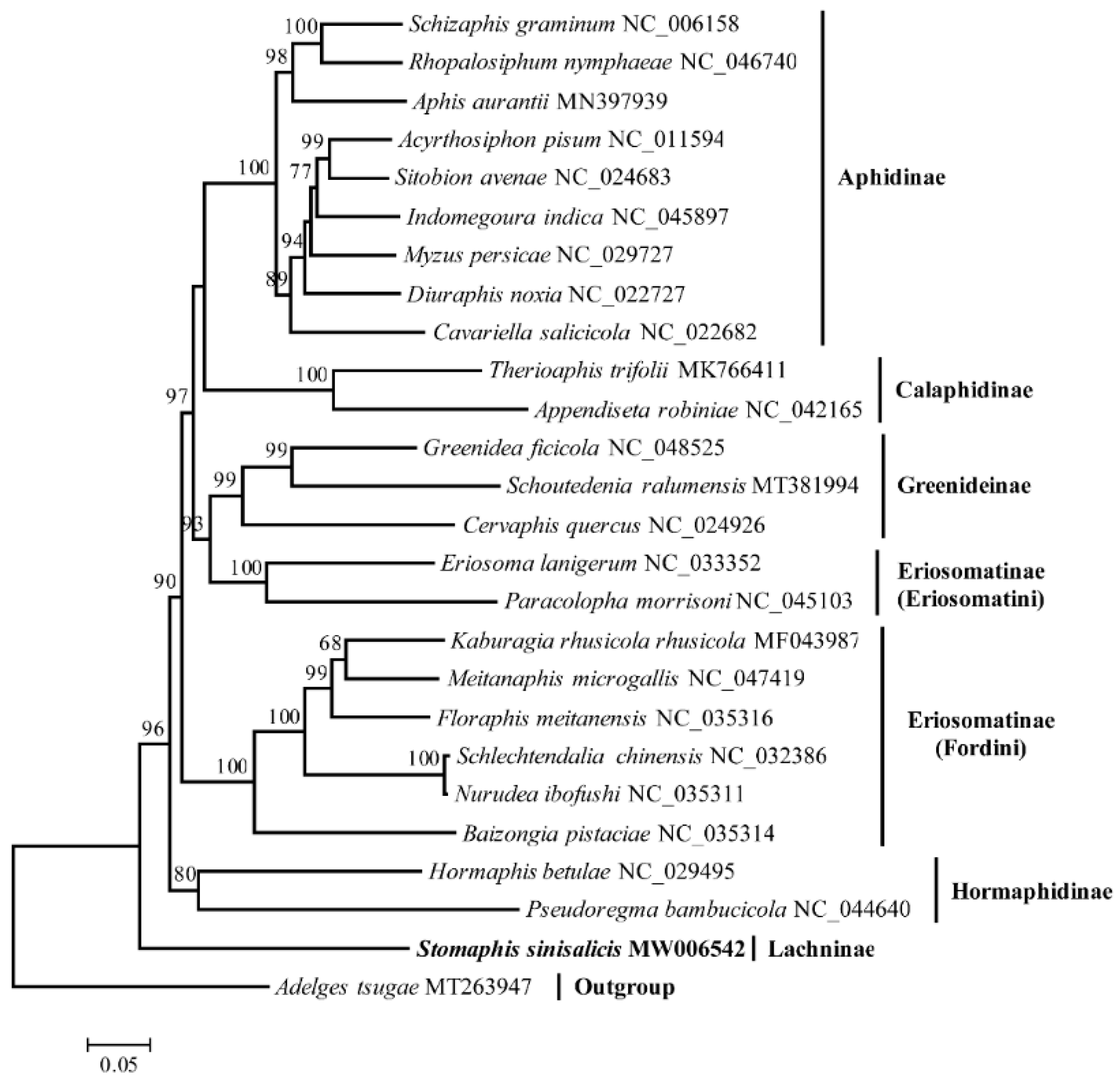
3.6. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [Green Version]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [Green Version]
- Boore, J.L.; Collins, T.M.; Stanton, D.; Daehler, L.L.; Brown, W.M. Deducing the pattern of arthropod phylogeny from mitochondrial DNA rearrangements. Nature 1995, 376, 163–165. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, X.-L.; Qiao, G.-X. Comparative analysis of mitochondrial genomes of five aphid species (Hemiptera: Aphididae) and phylogenetic implications. PLoS ONE 2013, 8, e77511. [Google Scholar] [CrossRef] [Green Version]
- Wei, D.D.; Lang, N.; Tao, Y.; He, W.; Wang, J.J. The mitochondrial genome of the brown citrus aphid Aphis (Toxoptera) citricidus: Insights into the repeat regions in aphids and phylogenetic implications. Int. J. Biol. Macromol. 2019, 136, 531–539. [Google Scholar] [CrossRef]
- Chen, J.; Wang, Y.; Qin, M.; Jiang, L.-Y.; Qiao, G.-X. The mitochondrial genome of Greenidea psidii van der Goot (Hemiptera: Aphididae: Greenideinae) and comparisons with other Aphididae aphids. Int. J. Biol. Macromol. 2019, 122, 824–832. [Google Scholar] [CrossRef]
- Zhang, B.; Ma, C.; Edwards, O.; Fuller, S.; Kang, L. The mitochondrial genome of the Russian wheat aphid Diuraphis noxia: Large repetitive sequences between trnE and trnF in aphids. Gene 2014, 533, 253–260. [Google Scholar] [CrossRef]
- Chen, L.; Chen, P.-Y.; Xue, X.-F.; Hua, H.-Q.; Li, Y.-X.; Zhang, F.; Wei, S.-J. Extensive gene rearrangements in the mitochondrial genomes of two egg parasitoids, Trichogramma japonicum and Trichogramma ostriniae (Hymenoptera: Chalcidoidea: Trichogrammatidae). Sci. Rep. 2018, 8, 7034. [Google Scholar] [CrossRef]
- Yoshizawa, K.; Johnsonb, K.P.; Sweet, A.D.; Yao, I.; Ferreira, R.L.; Cameron, S.L. Mitochondrial phylogenomics and genome rearrangements in the barklice (Insecta: Psocodea). Mol. Phylogenet. Evol. 2018, 119, 118–127. [Google Scholar] [CrossRef] [Green Version]
- Shao, R.; Barker, S.C. The highly rearranged mitochondrial genome of the plague thrips, Thrips imaginis (Insecta : Thysanoptera): Convergence of two novel gene boundaries and an extraordinary arrangement of rRNA genes. Mol. Biol. Evol. 2003, 20, 362–370. [Google Scholar] [CrossRef] [Green Version]
- Shao, R.; Campbell, N.J.H.; Barker, S.C. Numerous gene rearrangements in the mitochondrial genome of the wallaby louse, Heterodoxus macropus (Phthiraptera). Mol. Biol. Evol. 2001, 18, 858–865. [Google Scholar] [CrossRef] [Green Version]
- Boore, J.L.; Lavrov, D.V.; Brown, W.M. Gene translocation links insects and crustaceans. Nature 1998, 392, 667–668. [Google Scholar] [CrossRef]
- Shao, R.; Campbell, N.J.H.; Schmidt, E.R.; Barker, S.C. Increased rate of gene rearrangement in the mitochondrial genomes of three orders of hemipteroid insects. Mol. Biol. Evol. 2001, 18, 1828–1832. [Google Scholar] [CrossRef] [Green Version]
- Thao, M.L.; Baumann, L.; Baumann, P. Organization of the mitochondrial genomes of whiteflies, aphids, and psyllids (Hemiptera, Sternorrhyncha). BMC Evol. Biol. 2004, 4, 25. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Chen, J.; Jiang, L.-Y.; Qiao, G.-X. Hemipteran mitochondrial genomes: Features, structures and implications for phylogeny. Int. J. Mol. Sci. 2015, 16, 12382–12404. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Lu, C.; Huang, X. The first mitochondrial genome of scale insects (Hemiptera: Coccoidea). Mitochondrial DNA B 2019, 4, 2094–2095. [Google Scholar] [CrossRef]
- Liu, H.-L.; Chen, Q.-D.; Chen, S.; Pu, D.-Q.; Chen, Z.-T.; Liu, Y.-Y.; Liu, X. The highly rearranged mitochondrial genomes of three economically important scale insects and the mitochondrial phylogeny of Coccoidea (Hemiptera: Sternorrhyncha). PeerJ 2020, 8, e9932. [Google Scholar] [CrossRef]
- Lu, C.; Huang, X.; Deng, J. The challenge of Coccidae (Hemiptera: Coccoidea) mitochondrial genomes: The case of Saissetia coffeae with novel truncated tRNAs and gene rearrangements. Int. J. Biol. Macromol. 2020, 158, 854–864. [Google Scholar] [CrossRef]
- Favret, C. Aphid Species File. Version 5.0/5.0. Available online: http://Aphid.SpeciesFile.org (accessed on 7 November 2020).
- Wang, Y.; Chen, J.; Jiang, L.-Y.; Qiao, G.-X. The complete mitochondrial genome of Mindarus keteleerifoliae (Insecta: Hemiptera: Aphididae) and comparison with other Aphididae insects. Int. J. Mol. Sci. 2015, 16, 30091–30102. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Jiang, L.; Liu, Y.; Chen, J.; Qiao, G. General methods to obtain and analyze the complete mitochondrial genome of aphid species: Eriosoma lanigerum (Hemiptera: Aphididae) as an example. Zool. Syst. 2016, 41, 123–132. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, H.; Deng, J.; Lin, X.; Huang, X. The complete mitochondrial genome of Greenidea ficicola (Hemiptera: Aphididae: Greenideinae), a pest of Ficus. Mitochondrial DNA B 2020, 5, 254–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voronova, N.V.; Levykina, S.; Warner, D.; Shulinski, R.; Bandarenka, Y.; Zhorov, D. Characteristic and variability of five complete aphid mitochondrial genomes: Aphis fabae mordvilkoi, Aphis craccivora, Myzus persicae, Therioaphis tenera and Appendiseta robiniae (Hemiptera; Sternorrhyncha; Aphididae). Int. J. Biol. Macromol. 2020, 149, 187–206. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Deng, J.; Liu, Q.; Huang, X. The mitochondrial genome of a social aphid, Pseudoregma bambucicola (Hemiptera: Aphididae: Hormaphidinae). Mitochondrial DNA B 2019, 4, 2100–2101. [Google Scholar] [CrossRef]
- Song, H.; Donthu, R.K.; Hall, R.; Hon, L.; Weber, E.; Badger, J.H.; Giordano, R. Description of soybean aphid (Aphis glycines Matsumura) mitochondrial genome and comparative mitogenomics of Aphididae (Hemiptera: Sternorrhyncha). Insect Biochem. Mol. Biol. 2019, 113, 103208. [Google Scholar] [CrossRef]
- Chen, R.; Favret, C.; Jiang, L.; Wang, Z.; Qiao, G. An aphid lineage maintains a bark-feeding niche while switching to and diversifying on conifers. Cladistics 2016, 32, 555–572. [Google Scholar] [CrossRef]
- Ortiz-Rivas, B.; Moya, A.; Martínez-Torres, D. Molecular systematics of aphids (Homoptera: Aphididae): New insights from the long-wavelength opsin gene. Mol. Phylogenet. Evol. 2004, 30, 24–37. [Google Scholar] [CrossRef]
- Ortiz-Rivas, B.; Martínez-Torres, D. Combination of molecular data support the existence of three main lineages in the phylogeny of aphids (Hemiptera: Aphididae) and the basal position of the subfamily Lachninae. Mol. Phylogenet. Evol. 2010, 55, 305–317. [Google Scholar] [CrossRef]
- Qiao, G.; Zhang, G. A revision of Stomaphis Walker from China with descriptions of three new species (Homoptera: Lachnidae). Insect Sci. 1999, 6, 289–298. [Google Scholar] [CrossRef]
- Depa, Ł.; Mróz, E. Central European Acer- and salicaceae-feeding aphids of the genus Stomaphis (Insecta: Aphidoidea: Lachnidae) —Separate species or populations? Zool. Sci. 2013, 30, 509–518. [Google Scholar] [CrossRef]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2016, 45, e18. [Google Scholar] [CrossRef] [Green Version]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Laslett, D.; Canbäck, B. ARWEN: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenz, R.; Bernhart, S.H.; zu Siederdissen, C.H.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms. Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 37, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Grant, J.R.; Stothard, P. The CGView server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Yeh, H.-T.; Ko, C.-C.; Wu, L.-W. The first complete mitochondrial genome of Adelges tsugae Annand (Hemiptera: Adelgidae). Mitochondrial DNA B 2020, 5, 2288–2290. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.-H.; Liang, Y.-K.; Wen, J.; Ren, Z.-M. Complete mitochondrial genome of the witch-hazel leaf gall aphid Hamamelistes spinosus (Hemiptera: Aphididae: Hormaphidinae). Mitochondrial DNA B 2020, 5, 1388–1389. [Google Scholar] [CrossRef] [Green Version]
- Nong, X.; Liu, Y.; Wang, L.; Zhong, S.; Yu, X.; Xie, Y. Mitochondrial genome of Hormaphis betulae and its comparative analysis with Pseudoregma bambucicola (Hemiptera: Hormaphidinae). Mitochondrial DNA B 2020, 5, 906–907. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Park, J.; Lee, H.; Park, J.; Lee, W. The complete mitochondrial genome of Paracolopha morrisoni (Baker, 1919) (Hemiptera: Aphididae). Mitochondrial DNA B 2019, 4, 3037–3039. [Google Scholar] [CrossRef]
- Ren, Z.; Harris, A.J.; Dikow, R.B.; Ma, E.; Zhong, Y.; Wen, J. Another look at the phylogenetic relationships and intercontinental biogeography of eastern Asian - North American Rhus gall aphids (Hemiptera: Aphididae: Eriosomatinae): Evidence from mitogenome sequences via genome skimming. Mol. Phylogenet. Evol. 2017, 117, 102–110. [Google Scholar] [CrossRef]
- Ren, Z.; von Dohlen, C.D.; Harris, A.J.; Dikow, R.B.; Su, X.; Wen, J. Congruent phylogenetic relationships of Melaphidina aphids (Aphididae: Eriosomatinae: Fordini) according to nuclear and mitochondrial DNA data with taxonomic implications on generic limits. PLoS ONE 2019, 14, e0213181. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.-K.; Wen, J.; Ren, Z.-M. Complete mitochondrial genome of Rhus gall aphid Meitanaphis microgallis (Hemiptera: Aphididae: Eriosomatinae). Mitochondrial DNA B 2019, 4, 2363–2364. [Google Scholar] [CrossRef] [Green Version]
- Ren, Z.-M.; Wen, J. Complete mitochondrial genome of the North American Rhus gall aphid Melaphis rhois (Hemiptera: Aphididae: Eriosomatinae). Mitochondrial DNA B 2017, 2, 169–170. [Google Scholar] [CrossRef] [Green Version]
- Ren, Z.-M.; Bai, X.; Harris, A.J.; Wen, J. Complete mitochondrial genome of the Rhus gall aphid Schlechtendalia chinensis (Hemiptera: Aphididae: Eriosomatinae). Mitochondrial DNA B 2016, 1, 849–850. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, X.-L.; Qiao, G.-X. The complete mitochondrial genome of Cervaphis quercus (Insecta: Hemiptera: Aphididae: Greenideinae). Insect Sci. 2014, 21, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Jing, C.; Jiang, L.; Zhang, X.; Qiao, G. The complete mitochondrial genome of Schoutedenia ralumensis Rübsaamen, 1905 (Hemiptera: Aphididae: Greenideinae). Mitochondrial DNA B 2020, 5, 2217–2218. [Google Scholar] [CrossRef]
- Liu, X.; Wei, S.; He, J.; Song, F.; Cai, W. Complete mitochondrial genome of the spotted alfalfa aphid, Therioaphis trifolii (Hemipera: Aphididae). Mitochondrial DNA B 2019, 4, 3260–3261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pu, D.; Liu, C.; Liu, H.; Chen, Z.-T.; Wu, X.; Xiao, K.; Luo, X.; Mao, J.; Huang, Q. Complete mitochondrial genome of Sichuan’s population of Aphis aurantii (Hemiptera: Aphididae). Mitochondrial DNA B 2020, 5, 2119–2120. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Zhang, H.; Li, H.; Cai, W. All 37 mitochondrial genes of aphid Aphis craccivora obtained from transcriptome sequencing: Implications for the evolution of aphids. PLoS ONE 2016, 11, e0157857. [Google Scholar] [CrossRef]
- Zhang, S.; Luo, J.; Wang, C.; Lv, L.; Li, C.; Jiang, W.; Cui, J.; Rajput, L.B. Complete mitochondrial genome of Aphis gossypii Glover (Hemiptera: Aphididae). Mitochondrial DNA A 2016, 27, 854–855. [Google Scholar] [CrossRef]
- Hong, B.; Zhang, F.; Hu, Z.; Zhao, H. The complete mitochondrial genome of Indomegoura indica (Hemiptera: Aphididae). Mitochondrial DNA B 2019, 4, 882–883. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Kim, Y.; Xi, H.; Park, J.; Lee, W. The complete mitochondrial genome of Rhopalosiphum nymphaeae (Linnaeus, 1761) (Hemiptera: Aphididae). Mitochondrial DNA B 2020, 5, 1613–1615. [Google Scholar] [CrossRef]
- Zhang, B.; Zheng, J.; Liang, L.; Fuller, S.; Ma, C.-S. The complete mitochondrial genome of Sitobion avenae (Hemiptera: Aphididae). Mitochondrial DNA A 2016, 27, 945–946. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]
- Bernt, M.; Merkle, D.; Ramsch, K.; Fritzsch, G.; Perseke, M.; Bernhard, D.; Schlegel, M.; Stadler, P.F.; Middendorf, M. CREx: Inferring genomic rearrangements based on common intervals. Bioinformatics 2007, 23, 2957–2958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clary, D.O.; Wolstenholme, D.R. The mitochondrial DNA molecule of Drosophila yakuba: Nucleotide sequence, gene organization, and genetic code. J. Mol. Evol. 1985, 22, 252–271. [Google Scholar] [CrossRef] [PubMed]
- Lavrov, D.V.; Boore, J.L.; Brown, W.M. The complete mitochondrial DNA sequence of the horseshoe crab Limulus polyphemus. Mol. Biol. Evol. 2000, 17, 813–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, S.-J.; Shi, M.; Chen, X.-X.; Sharkey, M.J.; van Achterberg, C.; Ye, G.-Y.; He, J.-H. New views on strand asymmetry in insect mitochondrial genomes. PLoS ONE 2010, 5, e12708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowton, M.; Castro, L.R.; Austin, A.D. Mitochondrial gene rearrangements as phylogenetic characters in the invertebrates: The examination of genome ‘morphology’. Invertebr. Syst. 2002, 16, 345–356. [Google Scholar] [CrossRef]
- Boore, J.L. The duplication/random loss model for gene rearrangement exemplified by mitochondrial genomes of deuterostome animals. In Comparative Genomics; Springer: Dordrecht, The Netherlands, 2000; pp. 133–147. [Google Scholar] [CrossRef]
- Chaudhuri, K.; Chen, K.; Mihaescu, R.; Rao, S. On the tandem duplication-random loss model of genome rearrangement. Proc. Seventeenth Annu. ACM-SIAM Symp. 2006, 564–570. [Google Scholar] [CrossRef]
- Lavrov, D.V.; Boore, J.L.; Brown, W.M. Complete mtDNA sequences of two millipedes suggest a new model for mitochondrial gene rearrangements: Duplication and nonrandom loss. Mol. Biol. Evol. 2002, 19, 163–169. [Google Scholar] [CrossRef] [Green Version]
- Lunt, D.H.; Hyman, B.C. Animal mitochondrial DNA recombination. Nature 1997, 387, 247. [Google Scholar] [CrossRef]
- Cantatore, P.; Gadaleta, M.N.; Roberti, M.; Saccone, C.; Wilson, A.C. Duplication and remoulding of tRNA genes during the evolutionary rearrangement of mitochondrial genomes. Nature 1987, 329, 853–855. [Google Scholar] [CrossRef]
- Xu, S.-Y.; Long, J.-K.; Chen, X.-S. Comparative analysis of the complete mitochondrial genomes of five Achilidae species (Hemiptera: Fulgoroidea) and other Fulgoroidea reveals conserved mitochondrial genome organization. PeerJ 2019, 7, e6659. [Google Scholar] [CrossRef]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Lavrov, D.V.; Brown, W.M.; Boore, J.L. A novel type of RNA editing occurs in the mitochondrial tRNAs of the centipede Lithobius forficatus. Proc. Natl. Acad. Sci. USA 2000, 97, 13738–13742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masta, S.E.; Boore, J.L. The complete mitochondrial genome sequence of the spider Habronattus oregonensis reveals rearranged and extremely truncated tRNAs. Mol. Biol. Evol. 2004, 21, 893–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.-X.; Hewitt, G.M. Insect mitochondrial control region: A review of its structure, evolution and usefulness in evolutionary studies. Biochem. Syst. Ecol. 1997, 25, 99–120. [Google Scholar] [CrossRef]
- Crozier, R.H.; Crozier, Y.C. The mitochondrial genome of the honeybee Apis mellifera: Complete sequence and genome organization. Genetics 1993, 133, 97–117. [Google Scholar]
- Nováková, E.; Hypša, V.; Klein, J.; Foottit, R.G.; von Dohlen, C.D.; Moran, N.A. Reconstructing the phylogeny of aphids (Hemiptera: Aphididae) using DNA of the obligate symbiont Buchnera aphidicola. Mol. Phylogenet. Evol. 2013, 68, 42–54. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Subfamily | Species | Length (bp) | Accession Number | References |
|---|---|---|---|---|---|
| Adelgidae | Adelges tsugae | 16,056 | MT263947 | [43] | |
| Aphididae | Lachninae | Stomaphis sinisalicis | 17,109 | MW006542 | This study |
| Hormaphidinae | Hamamelistes spinosus | 15,089 | MT010853 | [44] | |
| Hormaphis betulae | 15,088 | NC_029495 | [45] | ||
| Pseudoregma bambucicola | 16,632 | NC_044640 | [24] | ||
| Mindarinae | Mindarus keteleerifoliae | 15,199 | NC_033410 | [20] | |
| Eriosomatinae | Eriosoma lanigerum | 15,640 | NC_033352 | [21] | |
| Paracolopha morrisoni | 16,330 | NC_045103 | [46] | ||
| Baizongia pistaciae | 15,602 | NC_035314 | [47] | ||
| Floraphis choui | 15,308 | NC_035310 | [48] | ||
| Floraphis meitanensis | 15,301 | NC_035316 | [48] | ||
| Kaburagia rhusicola ensigallis | 16,164 | MF043984 | [47] | ||
| Kaburagia rhusicola ovatirhusicola | 16,184 | MF043985 | [47] | ||
| Kaburagia rhusicola ovogallis | 16,164 | MF043986 | [47] | ||
| Kaburagia rhusicola rhusicola | 16,159 | MF043987 | [47] | ||
| Meitanaphis elongallis | 16,191 | NC_035315 | [48] | ||
| Meitanaphis flavogallis | 16,150 | NC_035312 | [48] | ||
| Meitanaphis microgallis | 16,191 | NC_047419 | [49] | ||
| Melaphis rhois | 15,436 | NC_036065 | [50] | ||
| Nurudea ibofushi | 16,054 | NC_035311 | [47] | ||
| Nurudea shiraii | 15,389 | NC_035301 | [47] | ||
| Nurudea yanoniella | 15,858 | NC_035313 | [48] | ||
| Schlechtendalia chinensis | 16,047 | NC_032386 | [51] | ||
| Schlechtendalia peitan | 15,609 | NC_035302 | [47] | ||
| Greenideinae | Cervaphis quercus | 15,272 | NC_024926 | [52] | |
| Greenidea ficicola | 17,361 | NC_048525 | [22] | ||
| Greenidea psidii | 16,202 | NC_041198 | [6] | ||
| Schoutedenia ralumensis | 16,051 | MT381994 | [53] | ||
| Calaphidinae | Appendiseta robiniae | 15,049 | NC_042165 | [23] | |
| Therioaphis trifolii | 16,068 | MK766411 | [54] | ||
| Aphidinae | Acyrthosiphon pisum | 16,971 | NC_011594 | NA | |
| Aphis aurantii | 15,469 | MN397939 | [55] | ||
| Aphis citricidus | 16,763 | NC_043903 | [5] | ||
| Aphis craccivora | 15,308 | NC_031387 | [56] | ||
| Aphis fabae mordvilkoi | 15,346 | NC_039988 | [23] | ||
| Aphis glycines | 17,954 | NC_045236 | [25] | ||
| Aphis gossypii | 15,869 | NC_024581 | [57] | ||
| Cavariella salicicola | 16,317 | NC_022682 | [4] | ||
| Diuraphis noxia | 15,784 | NC_022727 | [7] | ||
| Hyalopterus pruni | 15,410 | MT898422 | NA | ||
| Indomegoura indica | 15,220 | NC_045897 | [58] | ||
| Myzus persicae | 17,382 | NC_029727 | [23] | ||
| Rhopalosiphum nymphaeae | 15,594 | NC_046740 | [59] | ||
| Schizaphis graminum | 15,721 | NC_006158 | [14] | ||
| Sitobion avenae | 15,180 | NC_024683 | [60] |
| Name | Strand | Location | Length (bp) | Start Codons | Stop Codons | Intergenic Sequence (bp) |
|---|---|---|---|---|---|---|
| cox1 | J | 1–1531 | 1531 | ATA | T | −1 |
| trnL2 (taa) | J | 1531–1599 | 67 | −1 | ||
| cox2 | J | 1599–2273 | 675 | ATC | TAA | 9 |
| trnK (ctt) | J | 2283–2354 | 72 | 3 | ||
| trnD (gtc) | J | 2358–2421 | 64 | 0 | ||
| atp8 | J | 2422–2580 | 159 | ATT | TAA | −20 |
| atp6 | J | 2561–3214 | 654 | ATT | 4 | |
| cox3 | J | 3219–4004 | 786 | ATG | TAA | −1 |
| trnG (tcc) | J | 4004–4070 | 67 | 0 | ||
| nad3 | J | 4071–4424 | 354 | ATT | TAA | −1 |
| trnA (tgc) | J | 4424–4490 | 67 | 8 | ||
| trnR (tcg) | J | 4499–4564 | 64 | −3 | ||
| trnN (gtt) | J | 4562–4629 | 66 | −2 | ||
| trnS1 (gct) | J | 4628–4687 | 60 | 5 | ||
| trnE (ttc) | J | 4693–4760 | 66 | 34 | ||
| trnT (tgt) | J | 4795–4863 | 69 | 0 | ||
| nad6 | J | 4864–5361 | 498 | ATT | −1 | |
| cytb | J | 5361–6473 | 1113 | ATG | TAA | 17 |
| trnS2 (tga) | J | 6491–6557 | 65 | 7 | ||
| repeat region | J | 6565–8053 | 1489 | 6 | ||
| trnF (gaa) | N | 8060–8125 | 66 | 3 | ||
| nad5 | N | 8129–9796 | 1668 | ATA | TAA | 50 |
| trnH (gtg) | N | 9847–9910 | 64 | 0 | ||
| nad4 | N | 9911–11,219 | 1309 | ATA | T | 8 |
| nad4l | N | 11,228–11,521 | 294 | ATG | TAA | 24 |
| trnP (tgg) | N | 11,546–11,614 | 67 | 28 | ||
| nad1 | N | 11,643–12,578 | 936 | ATT | TAA | 0 |
| trnL1 (tag) | N | 12,579–12,643 | 65 | 6 | ||
| rrnL | N | 12,650–13,927 | 1278 | 0 | ||
| trnV (tac) | N | 13,928–13,994 | 67 | 3 | ||
| rrnS | N | 13,998–14,774 | 777 | 0 | ||
| control region | J | 14,775–15,693 | 919 | 0 | ||
| trnI (gat) | J | 15,694–15,759 | 66 | −3 | ||
| trnQ (ttg) | N | 15,757–15,822 | 66 | 35 | ||
| trnM (cat) | J | 15,858–15,922 | 65 | 0 | ||
| nad2 | J | 15,923–16,903 | 981 | ATA | TAA | −1 |
| trnW (tca) | J | 16,903–16,977 | 60 | −9 | ||
| trnC (gca) | N | 16,969–17,039 | 69 | 2 | ||
| trnY (gta) | N | 17,042–17,108 | 67 | 1 |
| Regions | Size (bp) | T(U) | C | A | G | A+T (%) | G+C (%) | AT-Skew | GC-Skew |
|---|---|---|---|---|---|---|---|---|---|
| Whole genome | 17,109 | 40.5 | 9.9 | 44.3 | 5.3 | 84.8 | 15.2 | 0.044 | −0.305 |
| PCGs * | 10,923 | 48.3 | 8.8 | 35.2 | 7.7 | 83.5 | 16.5 | −0.156 | −0.067 |
| 1st codon | 3641 | 40.3 | 8.7 | 40.4 | 10.6 | 80.7 | 19.3 | 0.001 | 0.098 |
| 2nd codon | 3641 | 53.5 | 13.1 | 23.1 | 10.3 | 76.6 | 23.4 | −0.397 | −0.120 |
| 3rd codon | 3641 | 51.1 | 4.6 | 42.3 | 2.1 | 93.4 | 6.7 | −0.094 | −0.373 |
| tRNAs | 1478 | 41.5 | 5.7 | 44.9 | 7.9 | 86.4 | 13.6 | 0.038 | 0.164 |
| rRNAs | 2055 | 42.9 | 4.9 | 42.2 | 10.0 | 85.1 | 14.9 | −0.007 | 0.340 |
| Repeat region | 1489 | 41.5 | 9.7 | 44.4 | 4.3 | 85.9 | 14.0 | 0.034 | −0.385 |
| Control region | 919 | 46.8 | 4.6 | 44.7 | 3.5 | 91.5 | 8.1 | −0.023 | −0.135 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Liu, Q.; Lu, C.; Deng, J.; Huang, X. The First Complete Mitochondrial Genome of Lachninae Species and Comparative Genomics Provide New Insights into the Evolution of Gene Rearrangement and the Repeat Region. Insects 2021, 12, 55. https://doi.org/10.3390/insects12010055
Zhang H, Liu Q, Lu C, Deng J, Huang X. The First Complete Mitochondrial Genome of Lachninae Species and Comparative Genomics Provide New Insights into the Evolution of Gene Rearrangement and the Repeat Region. Insects. 2021; 12(1):55. https://doi.org/10.3390/insects12010055
Chicago/Turabian StyleZhang, Hui, Qian Liu, Congcong Lu, Jun Deng, and Xiaolei Huang. 2021. "The First Complete Mitochondrial Genome of Lachninae Species and Comparative Genomics Provide New Insights into the Evolution of Gene Rearrangement and the Repeat Region" Insects 12, no. 1: 55. https://doi.org/10.3390/insects12010055