
Abstract
We report herein the synthesis and evaluation of phenyl ureas derived from 4-oxotetrahydropyrimidine as novel capsid assembly modulators of hepatitis B virus (HBV). Among the derivatives, compound 27 (58031) and several analogs showed an activity of submicromolar EC50 against HBV and low cytotoxicities (>50 μM). Structure–activity relationship studies revealed a tolerance for an additional group at position 5 of 4-oxotetrahydropyrimidine. The mechanism study indicates that compound 27 (58031) is a type II core protein allosteric modulator (CpAMs), which induces core protein dimers to assemble empty capsids with fast electrophoresis mobility in native agarose gel. These compounds may thus serve as leads for future developments of novel antivirals against HBV.
Similar content being viewed by others


Introduction
Hepatitis B virus (HBV) chronically infects 258 million people worldwide and causes 880 thousand deaths annually due to cirrhosis, hepatocellular carcinoma, and liver failure [1]. The current standard of care medications, including pegylated interferon alpha that regulates host antiviral immune response and nucleos(t)ide analogues (NUCs) that inhibit viral DNA polymerase, can potently suppress viral replication, but fail to induce the loss of HBV surface antigen (HBsAg), an indication of successful immune control or the functional cure of chronic hepatitis B, in the vast majority of the treated patients [2, 3]. Therefore, development of novel antivirals targeting other steps of HBV replication as well as drugs that can activate host antiviral immune response is required to achieve the functional cure of chronic hepatitis B [4, 5]. Particularly, selective packaging of viral pregenomic (pg) RNA–DNA polymerase complex by 120 core protein (Cp) dimers into a nucelocapsid for viral DNA synthesis to take place is a key step of HBV replication and thus an ideal target for novel antiviral development [6]. In the last two decades, multiple small molecule inhibitors of HBV pgRNA encapsidation have been discovered and several leads from three chemotypes—heteroaryldihydropyrimidine (HAPs, 1), dibenzothiazepine derivatives (DBTs, 2), and sulfamoylbenzamides (SBAs, 3)—have been extensively developed and are currently in clinical trials for the treatment of chronic hepatitis B (Figs 1 and 2) [6, 7]. Mechanistically, all the structurally diversified capsid assembly modulators or core protein allosteric modulators (CpAMs) bind to a hydrophobic pocket (HAP pocket) between Cp dimer–dimer interfaces to misdirect the assembly of Cp dimers into non-capsid polymers (type I CpAM) or morphologically “normal” capsids devoid of pgRNA and viral DNA polymerase (type II CpAM) [6, 8]. Of over a dozen families of CpAMs discovered thus far, SBAs have received much attention due to their structural simplicity, availability of the cocrystal structures with capsids or Y132A mutant Cp heximers, and potential for structural modifications [9, 10].

Representative capsid modulators from the three major chemotype families, and modulators with the feature of two side chains meta to each other, which is similar to SBAs

Rationale of the design of 4-oxo-tetrahydropyrimidine-derived phenyl ureas
Extensive SARs have been reported for SBAs [7]. Early works on SBAs focused on the optimization of the two side chains out of the central phenyl ring, sulfonamide and benzamide, that are meta to each other, and two clinical candidates were produced (4 and 5, Fig. 1) [11,12,13,14]. Further investigation has turned the attention to modifying the central core, because the angles and trajectories of the two side chains partially depend on how they are connected to and the shape of the central cores, and the trajectory and conformation of the side chains are important for directing their interactions with the target amino acid units, even at the same HAP pocket between Cp dimer–dimer interfaces. Several new central cores ranging from 7-membered to 5-membered rings, saturated and unsaturated, have been reported including aniline and pyrrole, which has led to more potent lead compounds, such as 14 and 15 (Fig. 1) [15,16,17,18,19,20,21,22,23,24]. Although they are promising, considering different scaffolds may confer different resistance profiles due to the interaction with different amino acid residues of Cp at HAP pocket [10, 25], so the identification of new central cores that can serve as bases for directing new interactions remains necessary. Here we report the design, synthesis, and characterization of phenyl ureas based on a new central core, 4-oxo-tetrahydropyrimidine, as nevol CpAMs.
4-Oxo-tetrahydropyrimidine is a pharmacophore that can be directly derivatized to put forward two branch side chains from the two nitrogen atoms separated by a methylene. The introduction of other groups at the 5-,6- carbons is also achievable. This moiety has been used in the preparation of other biologically active compounds, such as p38 MAP kinase inhibitors [26], cell adhesion inhibitors [27], metalloprotease inhibitors [28], MMP2, MMP3, and MMP9 inhibitors [29], and aspartyl protease inhibitors [30]. We envisioned that we could introduce an aryl group at N1 through a urea linker to mimic the benzamide side chain in some of the capsid modulators and another group at N3 to explore additional bindings with the target. This structure can also be viewed as a combination of the partial pyrazine compound 11 with partial pyridazinone 7.
Results and discussion
Synthesis
A general synthetic route for the compounds is illustrated in Scheme 1. A Boc-protected β-amino acid was coupled with an amine to form an amide 18. Next, the Boc was removed with HCl to afford intermediate 19, followed by cyclization with paraformaldehyde in the presence of NaOH or cyanuric chloride to generate the 4-oxo-tetrahydropyrimidine core 20 [31, 32], which could be reacted with phenyl carbamates to afford the desired phenyl ureas for evaluation.

General route for the synthesis of phenyl ureas derived from 4-oxotetrahydropyrimidine
Biological evaluation of new compounds. The antiviral activity of compounds was tested in an immortalized mouse hepatocyte (AML12)-derived stable cell line (AML12HBV10) that supports a high level of HBV replication. The effect of compounds on HBV-DNA replication in this cell line was determined by a dot-blot hybridization assay, which is the platform for our initial high throughput screening of HBV replication inhibitors, which resulted in the discovery of three chemotypes of CpAMs. Taking the advantage of its high throughput property, AML12HBV10-based antiviral and cytotoxicity assays were used to determine EC50 and CC50 of new compounds and direct the structure–activity relationship (SAR) study [9]. The antiviral activity of selected compounds was further confirmed in a human hepatoma-derived stable cell line supporting HBV replication (HepDES19). The mode of action of representative compounds on capsid assembly and pgRNA encapsidation was also investigated in hepatocytes by examination of capsid electrophoresis mobility and capsid-associated viral DNA as well as Cp dephosphorylation, a process-associated with pgRNA encapsidation.
In our previous work on benzamide CpAMs, we identified 3-chloro-4-fluoroaniline as a suitable fragment for the amine part of the amide [33]. We applied this aniline at the 1-position of the 4-oxo-tetrahydropyrimidine via a urea linker, and explored the effect of R1 substitution at the 3-position first (Table 1). A para-fluorobenzene connected to the benzene through one methyl is slightly more potent than one connected through a two-methylene linker (EC50, 2.02 μM of 21 vs. EC50, 5.08 μM of 22). Fluorine scan and walking based on 21 provided compound 27 (58031), with a fourfold potency increase (EC50, 0.52 μM) and no cytotoxicity at 50 μM. Interestingly, this 2,4-disubstitution pattern worked well when dimethoxy was evaluated in 28, albeit with a slightly weaker potency observed compared to 27 (58031). The basic and more hydrophilic pyridine was also evaluated at that position, but rendered compounds 29 and 30 less active. An attempt to extend the side chain through five-membered rings as used in the optimization of compound 11, resulting in compound 32 and 33, failed to improve the potencies. The (2,4-difluorophenyl)methylene group at position 3 in 27 (58031) was thus considered a proper starting point for SAR at other positions.
3-Chloro-4-fluoroaniline was found optimal in our previous benzamide optimization [33], but more anilines have been used and have shown unique physiochemical properties since then [22]. The variation of R4 was therefore investigated (Table 2). 3-chloroaniline in 35 and 3-(difluoromethyl)-4-fluoroaniline in 36 were found to have comparable activities to 27 (58031), while the tri-halogen substitution in 40, 41, and 42 reduced the activities. The replacement of the aniline with an ortho-fluorobenzylamine in 43 was not tolerated.
The effect of adding functional groups at the 5- and 6- positions of the central 4-oxo-tetrahydropyrimidine core was also explored. The assessment of substitutions at the 5- and 6- positions was performed based on 27 (58031) (Table 3). Among the small number of groups tested at the 6-position (R3), the methyl group resulted in reduced activity, suggesting that compound 44 does not have the same SAR as compound 7, in which a methyl substitution next to the endocyclic carbonyl group has a beneficial impact to the activity [16]. The introduction of a benzyl group, or 4,4-difluoropiperidine or 3,3-difluoropyrrolidine through a methylene linker, deteriorated the potency of compounds 45, 46, and 47. In contrast, substitutions at the 5-position were shown to be more tolerable. The compounds with substituents like phenyl in 50, triazole in 52, benzamide in 53, sulfonamide in 54, and acrylamide in 55 displayed comparable or slightly better potencies than 27 (58031), while the introduction of a dimethyl group, a fused cyclopentane, and a benzyl group diminished the activities only by two to threefold, as observed in 48, 49, and 51. The compounds 52, 53, 54, and 55 were prepared from 2-azido-3-((tert-butoxycarbonyl)amino)propanoic acid 56 according to the Scheme 1 to form the azide intermediate, 5-azido-N-(3-chloro-4-fluorophenyl)-3-(2,4-difluorobenzyl)-4-oxotetrahydropyrimidine-1(2H)-carboxamide 60. This azide 60 underwent cyclization with ethynylcyclopropane to form 52, or was reduced to 5-amino-N-(3-chloro-4-fluorophenyl)-3-(2,4-difluorobenzyl)-4-oxotetrahydropyrimidine-1(2H)-carboxamide 61, from which benzamide in 53 and sulfonamide in 54 were introduced (Scheme 2). The acceptance for the addition of functional groups at the 5-position (R2) indicated that this family of compounds does not have the same SAR as the structurally similar compounds 7 and 11, and that it can be further explored for better leads.

Synthesis of analogs of 27 derived from 5-amine
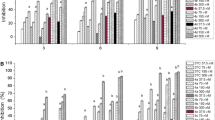
The anti-HBV activity of 27 (58031) was further assessed in a human hepatoma-derived cell line HepDES19 and demonstrated to inhibit HBV-DNA replication in a concentration-dependent manner with EC50 value of 0.84 (Fig. 3), which is slightly higher that its EC50 value in AML12HBV10 cells. Moreover, similar to ENAN-342017, a SBA chemotype of type II CpAM [34], treatment of AML12HBV_DE11 cells with 27 (58031) induced the assembly capsids with faster electrophoresis mobility in a native agarose gel and drastically reduced the amount of capsid-associated viral DNA (Fig. 4A) [35]. As anticipated, Bay 41-4109, a type I CpAM, inhibited capsid assembly and subsequent HBV-DNA synthesis. Also as expected, treatment of AML12HBV_DE11 cells with HBV-DNA polymerase inhibitor entecavir (ETV) did not alter capsid electrophoresis mobility, but significantly inhibited the synthesis of HBV DNA. These results indicate that 27 (58031) is a typical type II CpAM that misdirects the assembly of Cp dimers into empty capsids and thus precludes the synthesis of viral DNA. In support of this notion, similar to ENAN-34017 or Bay 41-4109, 27 (58031) treatment inhibited Cp dephosphorylation (Fig. 4B), which is catalyzed by cellular protein phosphatase 1 during pgRNA encapsidation and essential for the assembly of pgRNA-containing nucelocapsids, but not empty capsids. As anticipated, ETV treatment did not affect Cp dephosphorylation [36, 37].

Compound 58031 inhibits HBV replication in HepDES19 cells. The cells were treated with a serial twofold dilution of 58031 for 4 days. Intracellular HBV DNA were quantified by a qPCR assay. EC50 was calculated from two independent biologically triplicate experiments. Cell viability was determined by MTT assay and CC50 was calculated from a biologically triplicate experiment

Compound 58031 is a type II CpAM. AML12HBV_DE11 cells were cultured in the presence of tet (tet+) or cultured in the absence of tet and mock treated (tet-) or treated with the indicated concentrations of 27 (58031), 2 µM of ENAN-34017, 2 µM of Bay 41-4109 or 1 µM of entecavir (ETV) for 30 h. A HBV capsids and capsid-associated viral DNA were detected. Slow and fast migrating capsids were indicated. B Hyper-phopshorylated and dephosphorylated (or hypophosphorylated) HBV core protein were detected by a western blot assay, with β-actin as a loading control
Conclusions
In summary, the authors designed and synthesized 4-oxotetrahydropyrimidine-derived phenyl ureas as a new chemotype of CpAMs. SAR studies at four positions of the central core resulted in the discovery of compound 27 (58031) and several other analogs with submicromolar activities. Compound 27 (58031) was found to inhibit HBV in mouse and human hepatocytes and its mode of action is consistent with typical type II CpAM, i.e., misdirect the Cp dimers to assembly empty capsids devoid of pgRNA and thus precludes the synthesis of viral DNA.
Materials and methods
Chemistry
All reagents and solvents were used as purchased from commercial sources. Reactions were carried out under argon atmosphere. Flash column chromatography was performed on either CombiFlash Rf+ or CombiFlash Companion using the appropriate size Teledyne ISCO columns (20–40 µm or 40–60 µm) and prepacked silica filled cartridges. Preparative high-performance liquid chromatography (HPLC) was performed using a Gilson 331 and 332 pumps with a UV/VIS-155 detector and GX-271 liquid handler. Column was Phenomenex Luna LC Column (5 µm C18 100 Å, 150 × 21.2 mm). 1H NMR spectra were recorded on a 300 MHz INOVA VARIAN spectrometer. Chemical shifts values are given in ppm and referred against the internal standard of TMS (tetramethylsilane). The peak patterns are indicated as follows: s, singlet; d, doublet; t, triplet; q, quadruplet; m, multiplet and dd, doublet of doublets. The coupling constants (J) are reported in Hertz (Hz). Mass Spectra were obtained on an Agilent 6120 mass spectrometer with electrospray ionization source (1200 Aligent LC-MS spectrometer, Positive). Mobile phase flow was 1.0 mL/min with a 3.0 min gradient from 20% aqueous media (0.1% formic acid) to 95% CH3CN (0.1% formic acid) and a 9.0 min total acquisition time. All the tested compounds possess a purity of at least 95%, which was determined by LC/MS Data recorded using an Agilent 1200 liquid chromatography and Agilent 6120 mass spectrometer, and further supported by clean NMR spectra.
N-(3-chloro-4-fluorophenyl)-3-(4-fluorobenzyl)-4-oxotetrahydropyrimidine-1(2H)-carboxamide (21)
4-fluorobenzylamine (0.264 mmol), EDC·HCl (51 mg, 0.264 mmol), HOBt·H2O (40 mg, 0.264 mmol), and excess triethylamine was added to Boc-beta-Ala-OH (50 mg, 0.264 mmol) in 2 mL DCM. The reaction was stirred overnight. After diluting with EtOAc, the reaction mixture was washed with saturated aqueous NaHCO3 and then brine. The organic phase was dissolved in 1:1 MeOH to 4 M HCl in dioxane for several hours and then concentrated down. After drying on high vacuum overnight, the intermediate was refluxed in EtOH at 70 °C overnight with paraformaldehyde (10 mg, 0.343 mmol) and 10 N aqueous NaOH (42 μL, 0.422 mmol). The finished reaction was diluted with EtOAc and washed with brine twice. The organic phase was redissolved in DCM with excess Et3N to react with phenyl (3-chloro-4-fluorophenyl)carbamate, which was synthesized by reacting 3-chloro-4-fluoroaniline with 1.2 eq of phenyl chloroformate in 1:1 EtOAc to saturate aqueous NaHCO3 overnight and then purifying the concentrated organic phase with CombiFlash or HPLC. The reaction was diluted with EtOAc and washed with 2 M HCl thrice, saturated aqueous NaHCO3 once, and brine once. The desired product 21 (15.7 mg, 16%) was obtained after HPLC separation. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.49–7.40 (m, 1H), 7.29–7.25 (m, 1H), 7.24–7.19 (m, 1H), 7.19–7.10 (m, 1H), 7.06–6.93 (m, 3H), 4.82–4.76 (m, 2H), 4.60–4.54 (m, 2H), 3.73–3.63 (m, 2H), 2.70–2.60 (m, 2H); Calculated for C18H16ClF2N3O2, 379.1; observed (M + H)+ 380.5.
N-(3-chloro-4-fluorophenyl)-3-(4-fluorophenethyl)-4-oxotetrahydropyrimidine-1(2H)-carboxamide (22)
According to the procedure for preparation of compound 21, Boc-beta-Ala-OH (50 mg, 0.264 mmol) was treated with 2-(4-fluorophenyl)ethanamine (0.264 mmol), EDC·HCl (51 mg, 0.264 mmol), triethylamine, and HOBt·H2O (40 mg, 0.264 mmol), and then continued the same procedure to afford 22 (3.2 mg, 3%). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.53–7.46 (m, 1H), 7.23–7.16 (m, 1H), 7.16–7.08 (m, 2H), 7.08–6.98 (m, 1H), 6.98–6.87 (m, 1H), 4.71 (s, 2H), 3.64–3.50 (m, 4H), 2.83 (t, J = 7.2 Hz, 2H), 2.54–2.49 (m, 2H); Calculated for C19H18ClF2N3O2, 393.1; observed (M + H)+ 394.5.
N-(3-chloro-4-fluorophenyl)-3-(3-fluorophenethyl)-4-oxotetrahydropyrimidine-1(2H)-carboxamide (23)
According to the procedure for preparation of compound 21, Boc-beta-Ala-OH (50 mg, 0.264 mmol) was treated with 2-(3-fluorophenyl)ethanamine (0.264 mmol), EDC·HCl (51 mg, 0.264 mmol), triethylamine, and HOBt·H2O (40 mg, 0.264 mmol), and then continued the same procedure to afford 23 (3.7 mg, 4%). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.52–7.44 (m, 1H), 7.24–7.14 (m, 2H), 7.06–7.00 (m, 1H), 7.00–6.91 (m, 1H), 6.91–6.78 (m, 2H), 4.69 (s, 2H), 3.65–3.50 (m, 4H), 2.90–2.83 (m, 2H), 2.58–2.48 (m, 2H); Calculated for C19H18ClF2N3O2, 393.1; observed (M + H)+ 394.5.
N-(3-chloro-4-fluorophenyl)-3-(2-morpholinoethyl)-4-oxotetrahydropyrimidine-1(2H)-carboxamide (24)
According to the procedure for preparation of compound 21, Boc-beta-Ala-OH (100 mg, 0.529 mmol) was treated with 2-morpholinoethanamine (76 μL, 0.581), DCC (120 mg, 0.581 mmol), and HOBt·H2O (89 mg, 0.581 mmol), and then continued the same procedure to afford 24 (1.7 mg, 2%). 1H NMR (300 MHz, CD3OD): δ (ppm) 7.72–7.66 (m, 1H), 7.28–7.20 (m, 1H), 7.20–7.10 (m, 1H), 4.56 (s, 2H), 4.13–3.95 (m, 2H), 3.95–3.70 (m, 4H), 3.70–3.35 (m, 4H), 3.28–3.04 (m, 4H), 2.68–2.58 (m, 1H), 2.45–2.35 (m, 1H); Calculated for C12H22ClFN4O3, 384.1; observed (M + H)+ 385.5.
N-(3-chloro-4-fluorophenyl)-3-(3,4-difluorobenzyl)-4-oxotetrahydropyrimidine-1(2H)-carboxamide (25)
According to the procedure for preparation of compound 21, Boc-beta-Ala-OH (50 mg, 0.264 mmol) was treated with 3,4-difluorobenzylamine (0.264 mmol), EDC·HCl (51 mg, 0.264 mmol), triethylamine, and HOBt·H2O (40 mg, 0.264 mmol), and then continued the same procedure to afford 25 (8.0 mg, 8%). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.49–7.44 (m, 1H), 7.21–7.13 (m, 2H), 7.13–7.00 (m, 3H), 4.83 (s, 2H), 4.56 (s, 2H), 3.70 (t, J = 6.6 Hz, 2H), 2.68 (t, J = 6.6 Hz, 2H); Calculated for C18H15ClF3N3O2, 397.1; observed (M + H)+ 398.5.
N-(3-chloro-4-fluorophenyl)-3-(2,5-difluorobenzyl)-4-oxotetrahydropyrimidine-1(2H)-carboxamide (26)
According to the procedure for preparation of compound 21, Boc-beta-Ala-OH (50 mg, 0.264 mmol) was treated with 2,5-difluorobenzylamine (0.264 mmol), EDC·HCl (51 mg, 0.264 mmol), triethylamine, and HOBt·H2O (40 mg, 0.264 mmol), and then continued the same procedure to afford 26 (13.7 mg, 13%). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.48–7.40 (m, 1H), 7.21–7.12 (m, 1H), 7.04–6.84 (m, 4H), 4.85 (s, 2H), 2.59 (s, 2H), 3.72–3.60 (m, 2H), 2.65–2.54 (m, 2H); Calculated for C18H15ClF3N3O2, 397.1; observed (M + H)+ 398.5.
N-(3-chloro-4-fluorophenyl)-3-(2,4-difluorobenzyl)-4-oxotetrahydropyrimidine-1(2H)-carboxamide (27, 58031)
According to the procedure for preparation of compound 21, Boc-beta-Ala-OH (200 mg, 1.057 mmol) was treated with 2,4-difluorobenzylamine (0.14 mL, 1.163 mmol), DCC (240 mg, 1.163 mmol), and HOBt·H2O (178 mg, 1.163 mmol), and then continued the same procedure to afford 27 (58031) (159.4 mg, 38%). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.49–7.43 (m, 1H), 7.41–7.31 (m, 1H), 7.18–7.11 (m, 1H), 7.10–7.01 (m, 1H), 6.89–6.76 (m, 2H), 6.50 (s, 1H, NH), 4.91 (s, 2H), 4.63 (s, 2H), 3.70 (t, J = 6.6 Hz, 2H), 2.68 (t, J = 6.6 Hz, 2H); 13C NMR (100 MHz) showed a conformer mixture: δ (ppm) 169.8, 162.8 (dd, J = 249, 12 Hz), 161.1 (dd, J = 247, 12 Hz), 159.0 (d, J = 41 Hz), 154.9 (d, J = 245 Hz), 154.0, 134.4 (d, J = 3 Hz), 132.0 (dd, J = 10.0, 5.4 Hz), 123.1, 121.1 (d, J = 18 Hz), 120.5 (d, J = 8 Hz), 118.7 (dd, J = 16.4 Hz), 116.6 (d, J = 23 Hz), 111.9 (dd, J = 22, 4 Hz), 104.1 (t, J = 26 Hz), 58.2, 58.1, 42.7, 42.7, 41.0, 31.3; HRMS, Calculated for C18H15ClF3N3NaO2, 420.0697; observed (M + Na)+ 420.0687.
N-(3-chloro-4-fluorophenyl)-3-(2,4-dimethoxybenzyl)-4-oxotetrahydropyrimidine-1(2H)-carboxamide (28)
According to the procedure for preparation of compound 21, Boc-beta-Ala-OH (100 mg, 0.529 mmol) was treated with 2,4-dimethoxybenzylamine (76 μL, 0.581 mmol), DCC (120 mg, 0.581 mmol), and HOBt·H2O (89 mg, 0.581 mmol), and then continued the same procedure to afford 28 (62.5 mg, 28%). 1H NMR (300 MHz, CD3OD): δ (ppm) 7.52–7.46 (m, 1H), 7.25–7.18 (m, 1H), 7.16–7.08 (m, 2H), 6.49–6.45 (m, 1H), 6.45–6.39 (m, 1H), 4.88 (s, 2H), 4.54 (s, 2H), 3.80 (s, 3H), 3.74–3.68 (m, 5H), 2.64 (t, J = 6.6 Hz, 2H); Calculated for C20H21ClFN3O4, 421.1; observed (M + H)+ 422.5.
N-(3-chloro-4-fluorophenyl)-4-oxo-3-(pyridin-4-ylmethyl)tetrahydropyrimidine-1(2H)-carboxamide (29)
According to the procedure for preparation of compound 21, Boc-beta-Ala-OH (100 mg, 0.529 mmol) was treated with pyridin-4-ylmethanamine (58 μL, 0.581 mmol), DCC (120 mg, 0.581 mmol), and HOBt·H2O (89 mg, 0.581 mmol), and then continued the same procedure to afford 29 (5.0 mg, 3%). 1H NMR (300 MHz, CD3OD): δ (ppm) 8.79–8.69 (m, 2H), 7.96–7.88 (m, 2H), 7.67–7.59 (m, 1H), 7.36–7.27 (m, 1H), 7.22–7.13 (m, 1H), 5.03 (s, 2H), 4.92–4.84 (m, 2H), 3.97–3.87 (m, 2H), 2.76–2.66 (m, 2H); Calculated for C17H16ClFN4O2, 362.1; observed (M + H)+ 363.4.
N-(3-chloro-4-fluorophenyl)-4-oxo-3-(pyridin-2-ylmethyl)tetrahydropyrimidine-1(2H)-carboxamide (30)
According to the procedure for preparation of compound 21, Boc-beta-Ala-OH (100 mg, 0.529 mmol) was treated with pyridin-2-ylmethanamine (60 μL, 0.581 mmol), DCC (120 mg, 0.581 mmol), and HOBt·H2O (89 mg, 0.581 mmol), and then continued the same procedure to afford 30 (8.9 mg, 5%). 1H NMR (300 MHz, CD3OD): δ (ppm) 8.70–8.66 (m, 1H), 8.35–8.26 (m, 1H), 7.86–7.80 (m, 1H), 7.78–7.71 (m, 1H), 7.64–7.59 (m, 1H), 7.36–7.28 (m, 1H), 7.21–7.12 (m, 1H), 5.07 (s, 2H), 4.89 (s, 2H), 3.90 (t, J = 6.4 Hz, 2H), 2.69 (t, J = 6.4 Hz, 2H); Calculated for C17H16ClFN4O2, 362.1; observed (M + H)+ 363.5.
N-(3-chloro-4-fluorophenyl)-3-(cyclopropylmethyl)-4-oxotetrahydropyrimidine-1(2H)-carboxamide (31)
According to the procedure for preparation of compound 21, Boc-beta-Ala-OH (100 mg, 0.529 mmol) was treated with cyclopropylmethanamine (50 μL, 0.581 mmol), DCC (120 mg, 0.581 mmol), and HOBt·H2O (89 mg, 0.581 mmol), and then continued the same procedure to afford 31 (12.4 mg, 7%). 1H NMR (300 MHz, CD3OD): δ (ppm) 7.65–7.59 (m, 1H), 7.36–7.28 (m, 1H), 7.20–7.12 (m, 1H), 5.03 (s, 2H), 4.93–4.87 (m, 2H), 3.78 (t, J = 6.6 Hz, 2H), 2.61 (t, J = 6.6 Hz, 2H), 1.10–0.97 (m, 1H), 0.58–0.49 (m, 2H), 0.33–0.25 (m, 2H); Calculated for C15H17ClFN3O2, 325.1; observed (M + H)+ 326.4.
N-(3-chloro-4-fluorophenyl)-4-oxo-3-((1-phenylpyrrolidin-3-yl)methyl)tetrahydropyrimidine-1(2H)-carboxamide (32)
According to the procedure for preparation of compound 21, Boc-beta-Ala-OH (60 mg, 0.317 mmol) was treated with (1-phenylpyrrolidin-3-yl)methanamine (62 mg, 0.349 mmol), DCC (72 mg, 0.349 mmol), and HOBt·H2O (53 mg, 0.349 mmol), and then continued the same procedure to afford 32 (85.7 mg, 63%). 1H NMR (300 MHz, CD3OD): δ (ppm) 7.66–7.60 (m, 1H), 7.35–7.23 (m, 3H), 7.20–7.10 (m, 1H), 6.95–6.85 (m, 3H), 4.50 (s, 2H), 3.80 (t, J = 6.5 Hz, 2H), 3.61–3.41 (m, 6H), 2.92–2.78 (m, 1H), 2.65 (t, J = 6.5 Hz, 2H), 2.30–2.16 (m, 1H), 1.96–1.80 (m, 1H); Calculated for C22H24ClFN4O2, 430.2; observed (M + H)+ 431.6.
N-(3-chloro-4-fluorophenyl)-4-oxo-3-((2-oxo-3-phenyloxazolidin-5-yl)methyl)tetrahydropyrimidine-1(2H)-carboxamide (33)
According to the procedure for preparation of compound 21, Boc-beta-Ala-OH (60 mg, 0.317 mmol) was treated with 5-(aminomethyl)-3-phenyloxazolidin-2-one (80 mg, 0.349 mmol), DCC (72 mg, 0.349 mmol), and HOBt·H2O (53 mg, 0.349 mmol), and then continued the same procedure to afford 33 (13.2 mg, 9%). 1H NMR (300 MHz, CD3OD): δ (ppm) 7.65–7.59 (m, 1H), 7.57–7.50 (m, 2H), 7.40–7.26 (m, 3H), 7.18–7.08 (m, 2H), 5.16–5.00 (m, 2H), 5.00–4.90 (m, 1H), 4.22–4.13 (m, 1H), 3.93–3.71 (m, 5H), 2.68–2.60 (m, 2H); Calculated for C21H20ClFN4O4, 446.1; observed (M + H)+ 447.6.
3-(2,4-difluorobenzyl)-N-(4-fluoro-3-(trifluoromethyl)phenyl)-4-oxotetrahydropyrimidine-1(2H)-carboxamide (34)
2,4-difluorobenzylamine (378 mg, 2.64 mmol), EDC·HCl (557 mg, 2.91 mmol), HOBt·H2O (444 mg, 2.91 mmol), and excess triethylamine was added to Boc-beta-Ala-OH (500 mg, 2.64 mmol) in 10 mL DCM. The reaction was stirred overnight. After diluting with EtOAc, the reaction mixture was washed with saturated aqueous NH4Cl and brine. The organic phase was dissolved in 1:1 MeOH to 4 M HCl in dioxane for several hours and then dried overnight on high vacuum. Then, it was refluxed in acetonitrile at 50 °C overnight with paraformaldehyde (1.2 eq.) and cyanuric chloride (0.1 eq.). The reaction was concentrated down and purified with HPLC to afford the cyclized intermediate, 3-(2,4-difluorobenzyl)tetrahydropyrimidin-4(1H)-one. 3-(difluoromethyl)-4-fluoroaniline was reacted with 1.2 eq of phenyl chloroformate in 1:1 EtOAc to saturate aqueous NaHCO3 overnight. The organic phase was concentrated down and reacted with 3-(2,4-difluorobenzyl)tetrahydropyrimidin-4(1H)-one with excess DIPEA in DCM overnight. The desired product 34 was obtained after HPLC separation. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.61–7.54 (m, 2H), 7.39–7.28 (m, 1H), 7.15–7.06 (m, 1H), 6.86–6.74 (m, 2H), 4.88 (s, 2H), 4.61 (s, 2H), 3.70 (t, J = 6.8 Hz, 2H), 2.63 (t, J = 6.8 Hz, 2H); Calculated for C19H15F6N3O2, 431.1; observed (M + H)+ 432.6.
N-(3-chlorophenyl)-3-(2,4-difluorobenzyl)-4-oxotetrahydropyrimidine-1(2H)-carboxamide (35)
According to the procedure for preparation of compound 34, 3-chloroaniline was treated with phenyl chloroformate, and then reacted with the intermediate 3-(2,4-difluorobenzyl)tetrahydropyrimidin-4(1H)-one from the same procedure to afford 35. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.44–7.37 (m, 1H), 7.37–7.28 (m, 2H), 7.24–7.12 (m, 2H), 7.06–6.96 (m, 1H), 6.88–6.73 (m, 2H), 4.87 (s, 2H), 4.60 (s, 2H), 3.69 (t, J = 6.6 Hz, 2H), 2.63 (t, J = 6.6 Hz, 2H); Calculated for C18H16ClF2N3O2, 379.1; observed (M + H)+ 380.6.
3-(2,4-difluorobenzyl)-N-(3-(difluoromethyl)-4-fluorophenyl)-4-oxotetrahydropyrimidine-1(2H)-carboxamide (36)
According to the procedure for preparation of compound 34, 3-(difluoromethyl)-4-fluoroaniline was treated with phenyl chloroformate, and then reacted with the intermediate 3-(2,4-difluorobenzyl)tetrahydropyrimidin-4(1H)-one from the same procedure to afford 36. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.55–7.45 (m, 2H), 7.36–7.25 (m, 1H), 7.07–6.98 (m, 1H), 6.86–6.73 (m, 2H), 7.02–6.60 (m, 1H, CHF2, broad), 4.87 (s, 2H), 4.60 (s, 2H), 3.69 (t, J = 6.6 Hz, 2H), 2.62 (t, J = 6.6 Hz, 2H); Calculated for C19H16F5N3O2, 413.1; observed (M + H)+ 414.6.
3-(2,4-difluorobenzyl)-N-(2,3-dimethylphenyl)-4-oxotetrahydropyrimidine-1(2H)-carboxamide (37)
According to the procedure for preparation of compound 34, 3-(difluoromethyl)-4-fluoroaniline was treated with phenyl chloroformate, and then reacted with the intermediate 3-(2,4-difluorobenzyl)tetrahydropyrimidin-4(1H)-one from the procedure of compound 27 to afford 37. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.36–7.28 (m, 1H), 7.12–6.94 (m, 3H), 6.85–6.70 (m, 2H), 4.85 (s, 2H), 4.59 (s, 2H), 3.68 (t, J = 6.6 Hz, 2H), 2.74 (t, J = 6.6 Hz, 2H), 2.25 (s, 3H), 2.04 (s, 3H); Calculated for C20H21F2N3O2, 373.2; observed (M + H)+ 374.6.
3-(2,4-difluorobenzyl)-N-(3,4-difluorophenyl)-4-oxotetrahydropyrimidine-1(2H)-carboxamide (38)
According to the procedure for preparation of compound 34, 3,4-difluoroaniline was treated with phenyl chloroformate, and then reacted with the intermediate 3-(2,4-difluorobenzyl)tetrahydropyrimidin-4(1H)-one from the procedure of compound 27 to afford 38. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.57–7.45 (m, 1H), 7.37–7.25 (m, 2H), 7.20–7.10 (m, 2H), 7.06–6.93 (m, 1H), 4.82 (s, 2H), 4.51 (s, 2H), 3.75–3.65 (m, 2H), 2.55–2.30 (m, 2H); Calculated for C18H15F4N3O2, 381.1; observed (M + H)+ 382.5.
N-(2,4-dichlorophenyl)-3-(2,4-difluorobenzyl)-4-oxotetrahydropyrimidine-1(2H)-carboxamide (39)
According to the procedure for preparation of compound 34, 2,4-dichloroaniline was treated with phenyl chloroformate, and then reacted with the intermediate 3-(2,4-difluorobenzyl)tetrahydropyrimidin-4(1H)-one from the procedure of compound 27 to afford 39. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.90–7.84 (m, 1H), 7.34–7.29 (m, 1H), 7.20–7.14 (m, 2H), 6.85–6.69 (m, 3H), 4.87 (s, 2H), 4.58 (s, 2H), 3.70 (t, J = 6.6 Hz, 2H), 2.66 (t, J = 6.6 Hz, 2H); Calculated for C18H15Cl2F2N3O2, 413.0; observed (M + H)+ 414.5.
3-(2,4-difluorobenzyl)-4-oxo-N-(3,4,5-trifluorophenyl)tetrahydropyrimidine-1(2H)-carboxamide (40)
According to the procedure for preparation of compound 34, 3,4,5-trifluoroaniline was treated with phenyl chloroformate, and then reacted with the intermediate 3-(2,4-difluorobenzyl)tetrahydropyrimidin-4(1H)-one from the procedure of compound 27 to afford 40. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.40–7.20 (m, 1H), 7.15–7.03 (m, 2H), 6.86–6.72 (m, 2H), 4.84 (s, 2H), 4.59 (s, 2H), 3.71–3.61 (m, 2H), 2.65–2.55 (m, 2H); Calculated for C18H14F5N3O2, 399.1; observed (M + H)+ 400.5.
N-(2-chloro-4,5-difluorophenyl)-3-(2,4-difluorobenzyl)-4-oxotetrahydropyrimidine-1(2H)-carboxamide (41)
According to the procedure for preparation of compound 34, 2-chloro-4,5-difluoroaniline was treated with phenyl chloroformate, and then reacted with the intermediate 3-(2,4-difluorobenzyl)tetrahydropyrimidin-4(1H)-one from the procedure of compound 27 to afford 41. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.89–7.78 (m, 1H), 7.34–7.20 (m, 1H), 6.95–6.84 (m, 1H), 6.84–6.70 (m, 2H), 4.85 (s, 2H), 4.57 (s, 2H), 3.72–3.60 (m, 2H), 2.66–2.57 (m, 2H); Calculated for C18H14ClF4N3O2, 415.1; observed (M + H)+ 416.5.
N-(3,5-dichloro-4-fluorophenyl)-3-(2,4-difluorobenzyl)-4-oxotetrahydropyrimidine-1(2H)-carboxamide (42)
According to the procedure for preparation of compound 34, 3,5-dichloro-4-fluoroaniline was treated with phenyl chloroformate, and then reacted with the intermediate 3-(2,4-difluorobenzyl)tetrahydropyrimidin-4(1H)-one from the procedure of compound 27 to afford 42. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.41–7.33 (m, 2H), 7.33–7.26 (m, 1H), 6.86–6.72 (m, 2H), 4.88–4.80 (m, 2H), 4.59 (s, 2H), 3.70–3.60 (m, 2H), 2.64–2.54 (m, 2H); Calculated for C18H14Cl2F3N3O2, 431.0; observed (M + H)+ 432.5.
3-(2,4-difluorobenzyl)-N-(2-fluorobenzyl)-4-oxotetrahydropyrimidine-1(2H)-carboxamide (43)
According to the procedure for preparation of compound 34, 2-fluoroaniline was treated with phenyl chloroformate, and then reacted with the intermediate 3-(2,4-difluorobenzyl)tetrahydropyrimidin-4(1H)-one from the procedure of compound 27 to afford 43. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.31–7.27 (m, 2H), 7.24–7.17 (m, 1H), 7.10–6.94 (m, 2H), 6.82–6.69 (m, 2H), 4.80 (s, 2H), 4.56 (s, 2H), 4.39 (s, 2H), 3.53 (t, J = 6.6 HZ, 2H), 2.55 (t, J = 6.6 Hz, 2H); Calculated for C19H18F3N3O2, 377.1; observed (M + H)+ 378.5.
N-(3-chloro-4-fluorophenyl)-3-(2,4-difluorobenzyl)-6-methyl-4-oxotetrahydropyrimidine-1(2H)-carboxamide (44)
According to the procedure for preparation of compound 21, Boc-beta-HoAla-OH (50 mg, 0.246 mmol) was treated with 2,4-difluorobenzylamine (32 μL, 0.271 mmol), DCC (56 mg, 0.271 mmol), and HOBt·H2O (41 mg, 0.271 mmol), and then continued the same procedure to afford 44 (23.3 mg, 23%). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.46–7.40 (m, 1H), 7.35–7.27 (m, 1H), 7.20–7.10 (m, 1H), 7.06–6.96 (m, 1H), 6.86–6.70 (m, 2H), 5.20–5.12 (m, 1H), 4.74–4.64 (m, 1H), 4.56–4.46 (m, 2H), 4.26–4.14 (m, 1H), 2.80–2.70 (m, 1H), 2.40–2.30 (m, 1H), 1.30–1.24 (m, 3H); Calculated for C19H17ClF3N3O2, 411.1; observed (M + H)+ 412.4.
N-(3-chloro-4-fluorophenyl)-3-(2,4-difluorobenzyl)-4-oxo-6-(2,4,5-trifluorobenzyl)tetrahydropyrimidine-1(2H)-carboxamide (45)
According to the procedure for preparation of compound 21, (R)-3-((tert-butoxycarbonyl)amino)-4-(2,4,5-trifluorophenyl)butanoic acid (60 mg, 0.180 mmol) was treated with 2,4-difluorobenzylamine (24 μL, 0.198 mmol), DCC (41 mg, 0.198 mmol), and HOBt·H2O (30 mg, 0.198 mmol), and then continued the same procedure to afford 45. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.44–7.8 (m, 2H), 7.12–6.90 (m, 4H), 6.90–6.72 (m, 2H), 5.14–5.06 (m, 1H), 4.76–4.64 (m, 1H), 4.64–4.52 (m, 2H), 4.34–4.22 (m, 1H), 3.09–2.98 (m, 1H), 2.78–2.65 (m, 2H), 2.50–2.38 (m, 1H); Calculated for C25H18ClF6N3O2, 541.1; observed (M + H)+ 542.6.
N-(3-chloro-4-fluorophenyl)-3-(2,4-difluorobenzyl)-6-((4,4-difluoropiperidin-1-yl)methyl)-4-oxotetrahydropyrimidine-1(2H)-carboxamide (46)
2,4-difluorobenzylamine (0.12 mL, 1.003 mmol), DCC (207 mg, 1.003 mmol), and HOBt·H2O (154 mg, 1.003 mmol) was added to 3-((tert-butoxycarbonyl)amino)-4-hydroxybutanoic acid (200 mg, 0.912 mmol) in 5 mL DCM, and stirred overnight. After filtering, the filtrate was purified by CombiFlash to afford tert-butyl (4-((2,4-difluorobenzyl)amino)-1-hydroxy-4-oxobutan-2-yl)carbamate. Oxalyl chloride (0.16 mL, 1.824 mmol) and DMSO (0.19 mL, 2.73 mmol) was dissolved in 8 mL DCM at −78 °C in a dry ice/acetone bath. After 20 min, tert-butyl (4-((2,4-difluorobenzyl)amino)-1-hydroxy-4-oxobutan-2-yl)carbamate in 7 mL DCM was added dropwise. After 2 h, triethylamine (0.51 mL, 3.65 mmol) was added dropwise, and the reaction was stirred overnight, allowing it to gradually go to room temperature. The completed reaction was quenched with saturated aqueous NH4Cl, and then extracted with DCM twice to afford tert-butyl (4-((2,4-difluorobenzyl)amino)-1,4-dioxobutan-2-yl)carbamate. 4,4-difluoropiperidine hydrochloride (131 mg, 0.912 mmol) in 2 mL DCE was stirred with a few drops of triethylamine to release it from the HCl salt, and then NaBH(OAc)3 (290 mg, 1.368 mmol) was added. After stirring for several minutes, tert-butyl (4-((2,4-difluorobenzyl)amino)-1,4-dioxobutan-2-yl)carbamate in 2 mL DCE was added dropwise. After letting the reaction stir overnight, it was quenched with saturated aqueous NaHCO3 and stirred vigorously. The phases were separated, and the aqueous phase was extracted with DCM twice. All the organic phases were combined and purified by CombiFlash. It was then dissolved in 1:1 MeOH (2 mL) to 4 M HCl in dioxane (2 mL) for 1 h, and then dried on high vacuum. The residue was refluxed in 4 mL EtOH at 70 °C overnight with 10 N aqueous NaOH (73 μL, 0.730 mmol) and paraformaldehyde (18 mg, 0.593 mmol). The reaction was diluted with EtOAc and washed with brine. The organic phase was concentrated down and dissolved in acetonitrile. DIPEA and 4-DMAP was added, along with phenyl (3-chloro-4-fluorophenyl)carbamate. After refluxing at 110 °C overnight, the reaction was diluted with EtOAc and washed with 2 N HCl thrice, saturated NaHCO3 once, and brine once. It was purified by CombiFlash to afford 46. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.43–7.38 (m, 1H) 7.37–7.32 (m, 1H), 7.08–6.98 (m, 2H), 6.86–6.70 (m, 2H), 5.24–5.16 (m, 1H), 4.90–4.78 (m, 1H), 4.50–4.34 (m, 2H), 4.32–4.15 (m, 1H), 3.05–2.74 (m, 6H), 2.72–2.58 (m, 2H), 2.40–2.28 (m, 1H), 2.16–1.96 (m, 4H); Calculated for C24H24ClF5N4O2, 530.2; observed (M + H)+ 531.6.
N-(3-chloro-4-fluorophenyl)-3-(2,4-difluorobenzyl)-6-((3,3-difluoropyrrolidin-1-yl)methyl)-4-oxotetrahydropyrimidine-1(2H)-carboxamide (47)
According to the procedure for preparation of compound 46, 3,3-difluoropyrrolidine hydrochloride was used instead of 4,4-difluoropiperidine hydrochloride to afford 47 (8.5 mg, 2%). 1H NMR (300 MHz, CD3OD): δ (ppm) 7.50–7.44 (m, 1H), 7.42–7.30 (m, 1H), 7.18–7.10 (m, 2H), 6.93–6.82 (m, 2H), 5.18–5.12 (m, 1H), 4.84–4.68 (m, 3H), 4.62–4.53 (m, 1H), 3.30–2.90 (m, 6H), 2.84–2.74 (m, 1H), 2.66–2.37 (m, 4H); Calculated for C23H22ClF5N4O2, 516.1; observed (M + H)+ 517.6.
N-(3-chloro-4-fluorophenyl)-3-(2,4-difluorobenzyl)-5,5-dimethyl-4-oxotetrahydropyrimidine-1(2H)-carboxamide (48)
According to the procedure for preparation of compound 21, 3-((tert-butoxycarbonyl)amino)-2,2-dimethylpropanoic acid (37 mg, 0.168 mmol) was treated with 2,4-difluorobenzylamine (20 μL, 0.168 mmol), DCC (35 mg, 0.168 mmol), and HOBt·H2O (23 mg, 0.168 mmol), and then continued the same procedure to afford 48 (22.5 mg, 31%). 1H NMR (300 MHz, CD3OD): δ (ppm) 7.56–7.50 (m, 1H), 7.40–7.22 (m, 2H), 7.18–7.08 (m, 1H), 7.00–6.89 (m, 2H), 4.93 (s, 2H), 4.60 (s, 2H), 3.67 (s, 2H), 1.22 (s, 6H); Calculated for C20H19ClF3N3O2, 425.1; observed (M + H)+ 426.5.
N-(3-chloro-4-fluorophenyl)-3-(2,4-difluorobenzyl)-4-oxooctahydro-1H-cyclopenta[d]pyrimidine-1-carboxamide (49)
According to the procedure for preparation of compound 21, 2-((tert-butoxycarbonyl)amino)cyclopentanecarboxylic acid (39 mg, 0.168 mmol) was treated with 2,4-difluorobenzylamine (20 μL, 0.168 mmol), DCC (35 mg, 0.168 mmol), and HOBt·H2O (23 mg, 0.168 mmol), and then continued the same procedure to afford 49 (10.5 mg, 14%). 1H NMR (300 MHz, CD3OD): δ (ppm) 7.54–7.46 (m, 1H), 7.42–7.30 (m, 1H), 7.28–7.18 (m, 1H), 7.18–7.08 (m, 1H), 6.96–6.84 (m, 2H), 5.21–5.12 (m, 1H), 4.77–4.56 (m, 4H), 3.06–2.94 (m, 1H), 2.34–2.04 (m, 2H), 1.92–1.76 (m, 2H), 1.76–1.46 (m, 2H); Calculated for C21H19ClF3N3O2, 437.1; observed (M + H)+ 438.6.
N-(3-chloro-4-fluorophenyl)-3-(2,4-difluorobenzyl)-4-oxo-5-phenyltetrahydropyrimidine-1(2H)-carboxamide (50)
According to the procedure for preparation of compound 21, 3-((tert-butoxycarbonyl)amino)-2-phenylpropanoic acid (45 mg, 0.168 mmol) was treated with 2,4-difluorobenzylamine (20 μL, 0.168 mmol), DCC (35 mg, 0.168 mmol), and HOBt·H2O (23 mg, 0.168 mmol), and then continued the same procedure to afford 50 (23.6 mg, 29%). 1H NMR (300 MHz, CD3OD): δ (ppm) 7.52–7.42 (m, 1H), 7.40–7.20 (m, 6H), 7.12–7.06 (m, 2H), 7.06–6.90 (m, 2H), 5.22–5.14 (m, 1H), 5.00–4.90 (m, 1H), 4.82–4.64 (m, 2H), 4.14–4.04 (m, 1H), 4.04–3.95 (m, 1H), 3.95–3.82 (m, 1H); Calculated for C24H19ClF3N3O2, 473.1; observed (M + H)+ 474.6.
5-benzyl-N-(3-chloro-4-fluorophenyl)-3-(2,4-difluorobenzyl)-4-oxotetrahydropyrimidine-1(2H)-carboxamide (51)
According to the procedure for preparation of compound 21, 2-benzyl-3-((tert-butoxycarbonyl)amino)propanoic acid (60 mg, 0.215 mmol) was treated with 2,4-difluorobenzylamine (31 mg, 0.215 mmol), EDC·HCl (41 mg, 0.215 mmol), HOBt·H2O (33 mg, 0.215 mmol), and excess triethylamine, and then continued the same procedure to afford 51. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.41–7.36 (m, 1H), 7.36–7.23 (m, 4H), 7.23–7.17 (m, 2H), 7.15–7.07 (m, 1H), 7.04–6.96 (m, 1H), 6.88–6.74 (m, 2H), 5.03–4.95 (m, 1H), 4.76–4.70 (m, 1H), 4.70–4.55 (m, 2H), 3.64–3.55 (m, 1H), 3.45–3.36 (m, 1H), 3.00–2.85 (m, 1H), 2.74–2.62 (m, 1H); Calculated for C25H21ClF3N3O2, 487.1; observed (M + H)+ 488.6.
N-(3-chloro-4-fluorophenyl)-5-(4-cyclopropyl-1H-1,2,3-triazol-1-yl)-3-(2,4-difluorobenzyl)-4-oxotetrahydropyrimidine-1(2H)-carboxamide (52)
2,4-difluorobenzylamine (34 mg, 0.239 mmol), EDC·HCl (46 mg, 0.239 mmol), HOBt·H2O (37 mg, 0.239 mmol), and excess triethylamine was added to 2-azido-3-((tert-butoxycarbonyl)amino)propanoic acid 56 (50 mg, 0.217 mmol) in 2 mL DCM. The reaction was stirred overnight. After concentrating down, it was dissolved in 1:1 MeOH to 4 M HCl in dioxane for several hours and then dried overnight on high vacuum. Then, it was refluxed in acetonitrile at 50 °C overnight in a sealed flask with paraformaldehyde (8 mg, 0.251 mmol) and cyanuric chloride (4 mg, 0.0217 mmol). The reaction was diluted with EtOAc and washed with brine. The organic phase was concentrated down and dissolved in 3 mL DCM to react with phenyl (3-chloro-4-fluorophenyl)carbamate. After stirring overnight, the reaction was diluted with EtOAc and washed with 2 N HCl, saturated NaHCO3, and brine. The azido-intermediate, 5-azido-N-(3-chloro-4-fluorophenyl)-3-(2,4-difluorobenzyl)-4-oxotetrahydropyrimidine-1(2H)-carboxamide 60 (10.0 mg, 11%) was obtained after HPLC separation. Calculated for C18H14ClF3N6O2, 438.79; observed (M + H)+ 439.5.
Ethynylcyclopropane (1 mg, 0.0137 mmol), CuI (1 mg, 0.00571 mmol), and triethylamine was added to the azido-intermediate 60 (5 mg, 0.0114 mmol) prepared above in 1 mL THF under argon. After stirring overnight, the reaction was concentrated down and purified by HPLC to afford 52. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.54 (s, 1H), 7.50–7.44 (m, 1H), 7.38–7.28 (m, 1H), 7.20–7.12 (m, 1H), 7.06–6.98 (m, 1H), 6.88–6.76 (m, 2H), 5.16-5.06 (m, 2H), 4.83–4.48 (m, 4H), 4.30–4.12 (m, 1H), 1.98–1.85 (m, 1H), 0.98–0.90 (m, 2H), 0.80–0.72 (m, 2H); Calculated for C23H20ClF3N6O2, 504.1; observed (M + H)+ 505.6.
N-(3-chloro-4-fluorophenyl)-5-(2,4-difluorobenzamido)-3-(2,4-difluorobenzyl)-4-oxotetrahydropyrimidine-1(2H)-carboxamide (53)
Triphenylphosphine (32 mg, 0.123 mmol) was added to the azido-intermediate 60 prepared in the procedure for 52 (18 mg, 0.0411 mmol) in 2 mL THF. Water was added until the solution became turbid (about 1.8 mL). THF was added dropwise until the solution became clear again. After stirring overnight, the reaction was diluted in EtOAc and washed with brine. The organic phase was purified by HPLC to afford the reduced amine intermediate 61, 5-amino-N-(3-chloro-4-fluorophenyl)-3-(2,4-difluorobenzyl)-4-oxotetrahydropyrimidine-1(2H)-carboxamide. 2,4-difluorobenzoyl chloride (2 μL, 0.0121 mmol) was added to 61 (5 mg, 0.0121 mmol) in 1:1 EtOAc (1 ml) to saturated aqueous NaHCO3 (1 mL). After stirring overnight, the organic phase was purified by HPLC to afford 53. 1H NMR (300 MHz, CDCl3): δ (ppm) 8.08–7.97 (m, 1H), 7.40–7.30 (m, 1H), 7.25–7.19 (m, 1H), 7.07–6.76 (m, 5H), 5.03–4.90 (m, 2H), 4.85–4.77 (m, 1H), 4.74–4.64 (m, 1H), 4.56–4.44 (m, 2H), 3.30–3.20 (m, 1H); Calculated for C25H18ClF5N4O3, 552.1; observed (M + H)+ 553.7.
N-(3-chloro-4-fluorophenyl)-3-(2,4-difluorobenzyl)-5-(4-methylphenylsulfonamido)-4-oxotetrahydropyrimidine-1(2H)-carboxamide (54)
According to the procedure for preparation of compound 53, the reduced amino intermediate 61 (5 mg, 0.0121 mmol) was treated with 4-toluenesulfonyl chloride (3 mg, 0.0121 mmol) to afford 54. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.96–7.90 (m, 1H), 7.56–7.44 (m, 2H), 7.37–7.26 (m, 2H), 7.24–7.18 (m, 2H), 7.08–7.00 (m, 1H), 6.86–6.74 (m, 2H), 5.02–4.94 (m, 1H), 4.71–4.76 (m, 1H), 4.58–4.53 (m, 2H), 4.18–4.10 (m, 1H), 3.70–3.62 (m, 1H), 3.55–3.43 (m, 1H), 2.71 (s, 3H); Calculated for C25H22ClF3N4O4S, 566.1; observed (M + H)+ 567.7.
5-acrylamido-N-(3-chloro-4-fluorophenyl)-3-(2,4-difluorobenzyl)-4-oxotetrahydropyrimidine-1(2H)-carboxamide (55)
According to the procedure for preparation of compound 53, the reduced amino intermediate 61 (5 mg, 0.0121 mmol) was treated with acryloyl chloride (1 μL, 0.0121 mmol) to afford 55. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.55–7.50 (m, 1H), 7.36–7.18 (m, 2H), 7.06–6.98 (m, 1H), 6.88–6.76 (m, 2H), 6.37–6.28 (m, 1H), 6.24–6.13 (m, 1H), 5.76–5.70 (m, 1H), 4.92 (s, 2H), 4.79–4.70 (m, 1H), 4.60–4.49 (m, 2H), 4.41–4.32 (m, 1H), 3.22–3.13 (m, 1H); Calculated for C21H18ClF3N4O3, 466.1; observed (M + H)+ 467.6.
Biological evaluation
Materials
AML12HBV10, AML12HBV_DE11, and HepDES19 cells are immortalized mouse hepatocyte (AML12)- and human hepatoma cell (HepG2)-derived stable cell lines supporting the replication of a stably-transfected envelope protein-deficient HBV genome in a tetracycline-inducible manner [38, 39]. These cell lines were maintained in DMEM/F12 medium (Corning) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 µg/ml streptomycin, 1 µg/ml tetracycline and 200 µg/ml G-418. When cultured in medium without tetracycline, HBV pgRNA transcription will be activated and viral DNA replication occurs subsequently. ENAN-34017 was synthesized in house [34]. Bay 41-4109 is a gift from Dr. Lai Wei at Peking University, Beijing China. Entecavir is a gift from Dr. William S. Mason at Fox Chase Cancer Center, Philadelphia.
Antiviral and cytotoxicity assays in AML12HBV10 cells
The cells were seeded into 96-well plates at a density of 2 × 104 cells per well and cultured in the absence of tetracycline. One day after seeding, cells were mock treated or treated with a serial twofold dilution of compound, ranging from 10 to 0.08 µM, for 48 h and lysed by addition of 100 µl per well of lysis buffer containing 10 mM Tris-HCl (pH 7.5), 1 mM EDTA, 100 mM NaCl, and 1% NP-40. Half of the lysate was added to DNA denaturing solution containing 1.5M NaCl and 1M NaOH. After 5 min of incubation at room temperature, 100 µl of neutralization solution containing 1.5M NaCl, 1M Tris-HCl (pH 7.4) was added. Using a 96-well dot-blot manifold (Bio-Rad), the lysates were applied to a Hybond-N+ membrane (Amersham). HBV DNA in the cell lysates was detected by hybridization with alpha-32P-UTP-labeled (800 Ci/mmol, PerkinElmer) riboprobe specific for HBV minus strand DNA. After overnight incubation, membrane was washed twice, 1 h each, with buffer containing 0.1X SSC and 0.1% SDS at 65 °C, and exposed to a phosphoimager screen (GE Healthcare). Quantification done by QuantityOne software was used to determine the concentration that reduces the amount of HBV DNA by 50% (EC50). To determine the cytotoxicity, the cells were treated with a serial 2-fold dilution of compound, ranging from 50 to 1.56 µM, for 48 h under the same culture condition for the antiviral assay. The cell viability was inspected under microscopy and quantified by a MTT assay (Sigma) and expressed as the concentration of compound that reduced the viability of the cells by 50% (CC50).
Antiviral and cytotoxicity assays in HepDES19 cells
For antiviral activity assay, HepDES19 cells were seeded into 24-well plates and cultured in the absence of tetracycline for 2 days. The cells were then mock treated or treated with a serial twofold dilution of compound for an additional 4 days. Cytoplasmic HBV core DNA were extracted and quantified by a qPCR assay as previously described. The antiviral activity (EC50) was determined from biologically triplicated experiments by regression method of GraphPad Prism. To determine the cytotoxicity, HepDES19 cells seeded in 96-well plates were treated with a serial threefold dilution of compound, ranging from 30 to 0.12 µM, for 4 days under the same culture condition for the antiviral assay. The cell viability was inspected under microscopy and CC50 value was determined by a MTT assay.
Particle gels assay
AML12HBV_DE11 cells were seeded into 24-well plates and cultured in the absence of tetracycline for 6 h and then mock treated or treated with compounds at desired concentrations for an additional 30 h. The cells were lysed by a lysis buffer containing 10 mM Tris-HCl (pH 8.0), 1 mM EDTA, 100 mM NaCl, and 0.5% NP-40. Cell debris was removed by centrifugation at 12,000 × g for 10 min and the lysates were subjected to electrophoresis through native 1.8% agarose gels. HBV capsids were transferred onto a Hybond N+ membrane (Amersham). After fixing the membrane in 2.5% paraformaldehyde and then in 1:1 methanol:PBS, membrane was blocked with 5% milk in TBST for 2 h at room temperature. Capsids were detected with an antibody against HBV Cp (Santa Cruz, Cat. No. sc-52406). Capsid-associated HBV DNA was detected by hybridization with an α-32P-UTP (800 Ci/mmol, PerkinElmer) labeled full-length riboprobe specific for HBV minus strand.
Western blot assay
AML12HBV_DE11 cells were lysed by 1× LDS loading buffer (Invitrogen, catalog No. NP0007). Cell lysate was boiled at 100 °C for 20 min and resolved in a NuPAGE 12% Bis-Tris protein gel (Invitrogen, catalog No. NP0342PK2), using MOPs running buffer (Genscript, catalog No. M00138) and then transferred onto a polyvinylidene difluoride (PVDF) membrane (Thermo Fisher, catalog No. IB24001). The membrane was probed with a rabbit polyclonal antibody against C-terminal 14 amino acid peptide of HBV Cp and he bound antibody was revealed by IRDye secondary antibodies and imaged in the LI-COR Odyssey system (LI-COR).
References
Polaris Observatory Collaborators. Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: a modelling study. Lancet Gastroenterol Hepatol. 2018;3:383–403.
Liang TJ, Block TM, McMahon BJ, Ghany MG, Urban S, Guo JT, et al. Present and future therapies of hepatitis B: from discovery to cure. Hepatology. 2015;62:1893–908.
Chang J, Guo F, Zhao X, Guo JT. Therapeutic strategies for a functional cure of chronic hepatitis B virus infection. Acta Pharm Sin B. 2014;4:248–57.
Block TM, Gish R, Guo H, Mehta A, Cuconati A, Thomas London W, et al. Chronic hepatitis B: what should be the goal for new therapies? Antivir Res. 2013;98:27–34.
Hu J, Cheng J, Tang L, Hu Z, Luo Y, Li Y, et al. Virological basis for the cure of chronic hepatitis B. ACS Infect Dis. 2019;5:659–74.
Viswanathan U, Mani N, Hu Z, Ban H, Du Y, Hu J, et al. Targeting the multifunctional HBV core protein as a potential cure for chronic hepatitis B. Antivir Res. 2020;182:104917.
Nijampatnam B, Liotta DC. Recent advances in the development of HBV capsid assembly modulators. Curr Opin Chem Biol. 2019;50:73–9.
Zlotnick A, Venkatakrishnan B, Tan Z, Lewellyn E, Turner W, Francis S. Core protein: a pleiotropic keystone in the HBV lifecycle. Antivir Res. 2015;121:82–93.
Campagna MR, Liu F, Mao R, Mills C, Cai D, Guo F, et al. Sulfamoylbenzamide derivatives inhibit the assembly of hepatitis B virus nucleocapsids. J Virol. 2013;87:6931–42.
Zhou Z, Hu T, Zhou X, Wildum S, Garcia-Alcalde F, Xu Z, et al. Heteroaryldihydropyrimidine (HAP) and sulfamoylbenzamide (SBA) inhibit hepatitis B virus replication by different molecular mechanisms. Sci Rep. 2017;13:42374.
Vandyck K, Rombouts G, Stoops B, Tahri A, Vos A, Verschueren W, et al. Synthesis and evaluation of N-phenyl-3-sulfamoyl-benzamide derivatives as capsid assembly modulators inhibiting hepatitis B virus (HBV). J Med Chem. 2018;61:6247–60.
Sari O, Boucle S, Cox BD, Ozturk T, Russell OO, Bassit L, et al. Synthesis of sulfamoylbenzamide derivatives as HBV capsid assembly effector. Eur J Med Chem. 2017;29:407–21.
Lam AM, Espiritu C, Vogel R, Ren S, Lau V, Kelly M, et al. Preclinical characterization of NVR 3-778, a first-in-class capsid assembly modulator against hepatitis B virus. Antimicrob Agents Chemother. 2018;63:e01734–18.
Mani N, Cole AG, Phelps JR, Ardzinski A, Cobarrubias KD, Cuconati A, et al. Preclinical profile of AB-423, an inhibitor of hepatitis B virus pregenomic RNA encapsidation. Antimicrob Agents Chemother. 2018;62:e00082–18.
Hartman GD. Azepane derivatives and methods of treating hepatitis b infections. 2015. WO/2015/073774.
Lu D, Liu F, Xing W, Tong X, Wang L, Wang Y, et al. Optimization and synthesis of pyridazinone derivatives as novel inhibitors of hepatitis B virus by inducing genome-free capsid formation. ACS Infect Dis. 2017;3:199–205.
Pei Y, Wang C, Ben H, Wang L, Ma Y, Ma Q, et al. Discovery of new hepatitis B virus capsid assembly modulators by an optimal high-throughput cell-based assay. ACS Infect Dis. 2019;5:778–87.
Qiu YL, Li W, Cao H, Jin M, Gao X, Peng X, et al. Hepatitis B antiviral agents. 2016. WO2016183266A1.
Pan T, Ding Y, Wu L, Liang L, He X, Li Q, et al. Design and synthesis of aminothiazole based Hepatitis B Virus (HBV) capsid inhibitors. Eur J Med Chem. 2019;15:480–501.
Hu T, Han X, Kou B, Shen H, Yan S, Zhang Z. Pyrazine compounds for the treatment of infectious diseases. 2016. WO2016113273A1.
Vandyck K, HACHÉ GYP, Kesteleyn BRR, Raboisson PJMB. Carboxamide derivatives and the use thereof as medicaments for the treatment of hepatitis B. 2015. WO2015059212A1.
Tang J, Huber AD, Pineda DL, Boschert KN, Wolf JJ, Kankanala J, et al. 5-Aminothiophene-2,4-dicarboxamide analogues as hepatitis B virus capsid assembly effectors. Eur J Med Chem. 2019;15:179–92.
Na HG, Imran A, Kim K, Han HS, Lee YJ, Kim MJ, et al. Discovery of a new sulfonamide hepatitis B capsid assembly modulator. ACS Med Chem Lett. 2020;11:166–71.
Vandyck K, HACHÉ GYP, Last SJ, Mc Gowan DC, Rombouts G, Verschueren WG, et al. Sulphamoylpyrrolamide derivatives and the use thereof as medicaments for the treatment of hepatitis B. 2014. WO2014184350A1.
Kapoor TM, Miller RM. Leveraging chemotype-specific resistance for drug target identification and chemical biology. Trends Pharmacol Sci. 2017;38:1100–9.
Takahashi K, Sumino N, Yamamoto S, Sugitani M, Uegaki A, Nakatani S, et al. Nitrogenous heterocyclic compound and medical use thereof. 2004. WO2004101529A1.
Zheng Z, Ensinger CL, Adams SP. Cell adhesion inhibitors. 1998. WO1998004247A1.
Pikul S, Mcdow-Dunham KL, De B, Taiwo YO, Almstead NG, Bradley RS, et al. Heterocyclic metalloprotease inhibitors. 1988. WO1998008823A1.
Tuccinardi T, Nuti E, Ortore G, Rossello A, Avramova SI, Martinelli A. Development of a receptor-based 3D-QSAR study for the analysis of MMP2, MMP3, and MMP9 inhibitors. Bioorg Med Chem. 2008;16:7749–58. https://doi.org/10.1016/j.bmc.2008.07.004.
Zhu Z, McKittrick, B, Sun, ZY, Ye YC, Voigt, JH, Strickland C, et al. Preparation of imidazolidin-2-imines and their analogs as aspartyl protease inhibitors for treating various diseases. 2008. WO2008103351A2.
Schiedler DA, Vellucci JK, Beaudry CM. Formation of carbon-carbon bonds using aminal radicals. Org Lett. 2012;14:6092–5.
Sharma M, Pandey S, Chauhan K, Sharma D, Kumar B, Chauhan PM. Cyanuric chloride catalyzed mild protocol for synthesis of biologically active dihydro/spiro quinazolinones and quinazolinone-glycoconjugates. J Org Chem. 2012;77:929–37.
Wu S, Zhao Q, Zhang P, Kulp J, Hu L, Hwang N, et al. Discovery and mechanistic study of benzamide derivatives that modulate hepatitis B virus capsid assembly. J Virol. 2017;91:e00519–7.
Guo F, Zhao Q, Sheraz M, Cheng J, Qi Y, Su Q, et al. HBV core protein allosteric modulators differentially alter cccDNA biosynthesis from de novo infection and intracellular amplification pathways. PLoS Pathog. 2017;13:e1006658.
Wu S, Luo Y, Viswanathan U, Kulp J, Cheng J, Hu Z, et al. CpAMs induce assembly of HBV capsids with altered electrophoresis mobility: Implications for mechanism of inhibiting pgRNA packaging. Antivir Res. 2018;159:1–12.
Zhao Q, Hu Z, Cheng J, Wu S, Luo Y, Chang J. et al. Hepatitis B virus core protein dephosphorylation occurs during pregenomic RNA encapsidation. J Virol. 2018;92:e02139–17.
Hu Z, Ban H, Zheng H, Liu M, Chang J, Guo JT. Protein phosphatase 1 catalyzes HBV core protein dephosphorylation and is co-packaged with viral pregenomic RNA into nucleocapsids. PLoS Pathog. 2020;16:e1008669.
Guo H, Jiang D, Zhou T, Cuconati A, Block TM, Guo JT. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation. J Virol. 2007;81:12472–84.
Xu C, Guo H, Pan XB, Mao R, Yu W, Xu X, et al. Interferons accelerate decay of replication-competent nucleocapsids of hepatitis B virus. J Virol. 2010;84:9332–40.
Acknowledgements
This work was supported by grants from the National Institutes of Health, USA (AI113267) and appreciation of The Commonwealth of Pennsylvania through the Hepatitis B Foundation.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Hwang, N., Ban, H., Chen, J. et al. Synthesis of 4-oxotetrahydropyrimidine-1(2H)-carboxamides derivatives as capsid assembly modulators of hepatitis B virus. Med Chem Res 30, 459–472 (2021). https://doi.org/10.1007/s00044-020-02677-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-020-02677-3