
Abstract
Background
Rice is considered as a salt-sensitive plant, particularly at early vegetative stage, and its production is suffered from salinity due to expansion of salt affected land in areas under cultivation. Hence, significant increase of rice productivity on salinized lands is really necessary. Today genome-wide association study (GWAS) is a method of choice for fine mapping of QTLs involved in plant responses to abiotic stresses including salinity stress at early vegetative stage. In this study using > 33,000 SNP markers we identified rice genomic regions associated to early stage salinity tolerance. Eight salinity-related traits including shoot length (SL), root length (RL), root dry weight (RDW), root fresh weight (RFW), shoot fresh weight (SFW), shoot dry weight (SDW), relative water content (RWC) and TW, and 4 derived traits including SL-R, RL-R, RDW-R and RFW-R in a diverse panel of rice were evaluated under salinity (100 mM NaCl) and normal conditions in growth chamber. Genome-wide association study (GWAS) was applied based on MLM(+Q + K) model.
Results
Under stress conditions 151 trait-marker associations were identified that were scattered on 10 chromosomes of rice that arranged in 29 genomic regions. A genomic region on chromosome 1 (11.26 Mbp) was identified which co-located with a known QTL region SalTol1 for salinity tolerance at vegetative stage. A candidate gene (Os01g0304100) was identified in this region which encodes a cation chloride cotransporter. Furthermore, on this chromosome two other candidate genes, Os01g0624700 (24.95 Mbp) and Os01g0812000 (34.51 Mbp), were identified that encode a WRKY transcription factor (WRKY 12) and a transcriptional activator of gibberellin-dependent alpha-amylase expression (GAMyb), respectively. Also, a narrow interval on the same chromosome (40.79–42.98 Mbp) carries 12 candidate genes, some of them were not so far reported for salinity tolerance at seedling stage. Two of more interesting genes are Os01g0966000 and Os01g0963000, encoding a plasma membrane (PM) H+-ATPase and a peroxidase BP1 protein. A candidate gene was identified on chromosome 2 (Os02g0730300 at 30.4 Mbp) encoding a high affinity K+ transporter (HAK). On chromosome 6 a DnaJ-encoding gene and pseudouridine synthase gene were identified. Two novel genes on chromosome 8 including the ABI/VP1 transcription factor and retinoblastoma-related protein (RBR), and 3 novel genes on chromosome 11 including a Lox, F-box and Na+/H+ antiporter, were also identified.
Conclusion
Known or novel candidate genes in this research were identified that can be used for improvement of salinity tolerance in molecular breeding programmes of rice. Further study and identification of effective genes on salinity tolerance by the use of candidate gene-association analysis can help to precisely uncover the mechanisms of salinity tolerance at molecular level. A time dependent relationship between salt tolerance and expression level of candidate genes could be recognized.
Similar content being viewed by others
Background
Drought and salinity stresses seriously threat crop production worldwide (Munns 2011). The area of saline and salt affected land will be continuously increased mainly because of climate change (Islam et al. 2019). Rice (Oryza sativa L.) is one of the most important cereals as it is the source of nutrition for more than 50% of the world population (Xu et al. 2016). Rice is considered as a salt-sensitive plant and its production is suffered from salinity due to expansion of salt affected land in areas under cultivation of this crop (Islam et al. 2019). Hence, significant increase of rice productivity on salinized lands is really necessary (Al-Tamimi et al. 2016). Because of the sensitivity of rice plant at the seedling and reproductive phases, salinity stress is considered to be a major limitation for the production of rice (Maas and Hoffman 1977; Singh and Flowers 2010; Reddy et al. 2017). Thus, for improvement of salt tolerance in rice, it is better to target more sensitive growth stages to salt stress such as seedling stage (Walia et al. 2005). A lot of studies show that salinity tolerance is a complex trait which is controlled by quantitative trait loci (QTL) (Roy et al. 2011), and using QTL mapping in segregating populations many QTL regions were reported for different traits under salinity stress in rice. Prasad et al. (2000) using a DH population identified a QTL for seminal root length under salt stress on chromosome 6 which explained 18.9% of phenotypic variation. Using a RIL population and AFLP markers, Koyama et al. (2001) identified a QTL for dry mass under salt stress on chromosome 6 explaining 9.7% of phenotypic variation. They also identified 3 QTLs for Na+ uptake/concentration on chromosomes 1, 4 and 6, and identified 4 QTLs for K+ uptake/concentration on chromosomes 1, 4, 6 and 9. They also identified 2 QTLs for Na+:K+ ratio on chromosomes 1 and 4. Takehisa et al. (2004) identified 12 QTLs for shoot length under salt stress on chromosomes 1, 3 and 7 that explained 12% to 30% of phenotypic variation. Aman et al. (2007) identified 5 QTLs for salt injury on chromosomes 1, 3, 4 and 5 that explained 5.8% to 25.8% of phenotypic variation. Lee et al. (2007) identified 4 QTLs for salt tolerance in RIL population on chromosomes 1 and 3 that explained 9.1% to 27.8% of phenotypic variation. Using a BIL population (BC3F5), Kim et al. (2009) identified 1, 2 and 5 QTLs for relative seedling height, relative leaf area and relative dry weight on chromosomes 1, 6 and 7. Ul Haq et al. (2010) by using a RIL population identified 1, 2, 5, 5 and 5 QTLs for salt tolerance, shoot dry weight, shoot water content, Na+ concentration and Na+:K+ ratio on different chromosomes of rice. Ghomi et al. (2013) using a F2:4 population detected 41 QTLs for 12 physiological traits that were scattered on all rice chromosomes.
The aim of genetic mapping is to identify inherited markers related to loci that control the trait of interest, particularly complex quantitative traits. Generally, two strategies are used in the genetic mapping of traits in plants: (1) linkage mapping and (2) linkage disequilibrium (LD) mapping. Linkage mapping counts the recombination events between molecular markers and the unknown genes in a segregating population developed from crosses between 2 or more parents, whereas LD mapping or association mapping (AM) measures correlation between marker alleles and the given trait in a natural population (Rosyara and Joshi 2012). Association mapping identifies a single polymorphism within a locus or a within candidate gene that create the given phenotype. With this method one can search for genotype-phenotype correlations among unrelated individuals of a species. It has high resolution since the historical recombination events were accumulated in natural populations and collections of landraces, breeding materials and varieties. By exploiting broader genetic diversity, thus AM offers three main advantages over linkage mapping: higher mapping resolution, higher allele number and time saving in establishing a marker-trait association, and hence it is a better choice in genetic mapping and breeding programmes (Flint-Garcia et al. 2003). AM has been advocated as the method of choice for identifying loci involved in the inheritance of complex quantitative traits (Risch and Merikangas 1996; Slatkin 2008). In the last two decades, AM has been used in different plant species, and based on it a lot of QTLs have been mapped or cloned (Price 2006). However, a part of detected associations can also be the result of population structure which can create false marker-trait associations. To overcome this problem it is better to control for structure effect (Q coefficients) in the statistical model (Pritchard et al. 2000). AM is not only capable of identification and mapping of QTLs of interest, but also explains the reason for polymorphism within a gene which accounts for the difference between two phenotypes (Palaisa et al. 2003).
Association mapping has been applied to study the genetic control of quality traits, seed traits such as seed longevity (Li et al. 2014), dormancy, and seed vigor, and it was applied for quantitative traits, particularly for tolerance to biotic stresses such as sheath blight resistance, and tolerance to abiotic stresses such as cold tolerance and salinity and alkalinity tolerance (Kumar et al. 2015; Al-Tamimi et al. 2016). More recently, Frouin et al. (2018) in a GWAS research detected 27 QTLs for mild salinity-related traits that were mapped on 12 chromosomes of rice. Also, using GWAS, Naveed et al. (2018) reported 20 QTLs for 11 salinity tolerance-related traits at germination and seedling stages of rice. Because of higher sensitivity of rice plant to salinity stress at seedling stage, in this study we evaluated a panel of rice accessions from International Rice Research Institute (IRRI) at early vegetative stage under salt stress and normal conditions to finely identify genomic regions associated to salinity tolerance by dense map of SNP markers, to validate earlier candidate genes and to identify novel candidate genes affecting salinity tolerance.
Results
Salinity Response
The analysis of variance (ANOVA) showed that treatments (normal and salinity) had significant differences for all the studied traits except for the shoot length (SL). There was a significant difference between genotypes (G) and G × T interaction for all the studied traits (Supplementary Table S2).
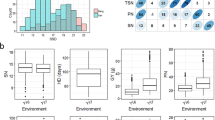
Mean comparisons showed that means of shoot length (SL) and turgid weight (TW) at salinity condition were not significantly differed from normal condition (Table 1). While the mean of most traits decreased significantly under salinity stress relative to normal condition; significant loss under salinity condition was observed for RL, SFW, RFW, RDW, SDW and RWC (Table 1). In addition, frequency distribution of traits under both conditions alleviated quantitative nature (Supplementary Fig. S1a&b) which is necessary for association mapping.
Correlation analysis showed that under both normal and salinity conditions there were significant correlations among most of the studied traits (Supplementary Tables S3&S4). In general, correlations among traits at both conditions alleviated similar trend and magnitude.
GWAS Results
Population Structure
After elimination of the monomorphic loci and loci with minor allele frequency (MAF) below 5%, from the total of 37,867 SNP markers, 33,864 markers remained in genotyping data set for analysis. The total length of the rice chromosomes is ~ 380 Mbp, hence, in average there was one SNP per 11.2 Kbp. The highest number of markers were on chromosome 1 (5272 SNPs), and the lowest number of markers were on chromosome 9 (2111 SNPs). As seen in supplementary Fig. S2a, based on Evanno et al. (2005) method the entire population consisted from 4 sub-populations which were depicted in a bar plot in supplementary Fig. S2b.
Identification of Associated Genomic Regions to the Studied Traits
The association between the SNP marker genotypes and the tolerance-related traits was assessed by the use of a mixed linear model (MLM) controlled for the population structure (Q matrix; Supplementary Table S5) and kinship coefficient of varieties (K). Significant marker-trait associations were identified by negative log10 (p value) and by r2 as part of the variations explained by SNP markers. The association analysis between 8 evaluated traits and SNP markers was conducted at normal and salinity stress conditions and the results have been presented in the form of Manhattan plots (Figs. 1 and 2).

Manhattan plots of p-values analyzed using mixed linear model (MLM) controlled for population structure and kinship of rice genotypes for different traits under normal condition

Manhattan plots of p-values analyzed using mixed linear model (MLM) controlled for population structure and kinship of rice genotypes for different traits under salinity condition
Under normal and stress conditions the MLM-Q-K analyses with –log10(p) > 4.0 identified 97 and 151 trait-marker associations, respectively (Supplementary Tables S6&S7). Chromosomes 3, 5 and 10 under normal condition, and chromosomes 3 and 7 under salinity conditions lake any marker-trait associations. In contrast, chromosome 12 under normal condition and chromosome 1 under salinity condition had maximum number of marker-trait associations.
For shoot length (SL) under normal condition, 5 genomic regions tagged to 65 SNP markers on chromosomes 1, 2, 6 and 12 showed a significant association (Fig. 1, Table 2). However, under salinity stress, the association analysis identified 2 genomic regions tagged to 2 SNPs on the chromosomes 1 and 12 (Fig. 2). These markers explained 9.26 to 10.14% of phenotype variation of the trait. The strongest QTL placed on chromosome 1 at position 32.48 Mbp (Table 2).
For root length (RL) under normal condition no SNP marker showed significant association. However, under salinity stress 6 genomic regions were identified with a significant association with RL (Fig. 2). Eight SNP markers on chromosomes 2, 6, 8 and 10 showed significant associations which explained 7.98 to 14.75% of phenotype variation of the trait. The strongest QTL placed on chromosome 6 at position 30.92 Mbp (Table 3).
In the case of root dry weight (RDW) under normal condition, 5 genomic regions tagged to 6 SNP markers on the chromosomes 1, 6 and 7 showed a significant association (Fig. 1). These markers explained 9.77 to 11.27% of phenotype variation of the trait (Table 4). However, under salinity stress condition, 7 genomic regions tagged to 39 SNP markers were identified which had a significant association with RDW (Fig. 2). These SNP markers were localized on chromosomes 1, 2, 4, 6 and 12 which explained 7.60 to 14.38% of phenotype variation of the trait. The strongest QTL was on chromosome 1 at position 41.35 Mbp (Table 4).
For root fresh weight (RFW) under normal condition, 7 genomic regions tagged to 14 SNPs showed a significant association (Fig. 1). The markers were localized on chromosomes 1, 4, 6, 8 and 12, and explained 6.62 to 11.63% of phenotype variation of the trait (Table 5). However, under the salinity stress condition the association analysis identified 15 genomic regions having a significant association with the root fresh weight (RFW) (Fig. 2). Sixty eight SNP markers on the chromosomes 1, 2, 5, 6, 8, 9, 10, 11 and 12 showed significant associations with the trait. These markers explained 5.58 to 14.62% of phenotype variation of the trait. The strongest QTL placed on chromosome 12 at position 18.46 Mbp (Table 5).
For shoot fresh weight (SFW) under normal condition, 5 genomic regions tagged to 5 SNP markers on chromosomes 2, 9 and 11 showed a significant association (Fig. 1). These markers explained 10.17 to 12.25% of phenotype variation of the trait (Table 6). However, under salt stress 4 genomic regions tagged to 6 SNP markers on chromosomes 1, 11 and 12 showed a significant association with SFW (Fig. 2). These markers explained 9.90 to 16.93% of phenotype variation of the trait. The strongest QTL placed on chromosome 1 at position 32.48 Mbp (Table 6).
For shoot dry weight (SDW) under normal condition, 4 genomic regions tagged to 4 SNP markers on chromosomes 1, 2, 9 and 11 showed a significant association (Fig. 1). These markers explained 10.60 to 13.66% of phenotype variation of this trait (Table 7). However, under salinity stress 2 genomic regions tagged to 2 SNP markers on chromosomes 1 and 11 were identified with a significant association with shoot dry weight (SDW) (Fig. 2). These markers explained 10.59 to 11.13% of phenotype variation of this trait. The strongest QTL placed on chromosome 1 at position 42.43 Mbp (Table 7).
For relative water content (RWC) under normal condition, no SNP marker was associated. These markers explained 8.36 to 10.08% of phenotype variation of this trait (Table 8). However, under salinity stress condition 3 genomic regions tagged to 5 SNP markers on chromosomes 1 and 11 were identified which were associated to RWC (Fig. 2). These markers explained 10.07 to 11.64% of phenotype variation of the trait. The strongest QTL placed on chromosome 1 at position 7.09 Mbp (Table 8).
For turgid weight (TW) under normal condition, 3 genomic regions tagged to 3 SNP markers on the chromosomes 2, 9 and 11 showed a significant association (Fig. 1). These markers explained 10.48 to 15.83% of phenotype variation of this trait (Table 9). However, under salt stress condition 4 genomic regions tagged to 7 SNP markers showed a significant association to TW (Fig. 2). The markers were located on chromosomes 1 and 11 which explained 9.82 to 21.17% of phenotype variation of the trait. The strongest QTL placed on chromosome 1 at position 32.48 Mbp (Table 9). All detected QTLs are illustrated on the rice chromosomal map (Fig. 3).

Distribution of GWAS-based detected QTLs on 10 chromosomes of rice. Distances on the map are in Mbp. The position of SalTol, a well-studied QTL at seedling stage, was shown on chromosome 1
For other derived traits including (SL-R, RL-R, RFW-R and RDW-R) 12 genomic regions tagged to 48 SNP markers on different chromosomes showed significant associations (supplementary Fig. S3). For SL-R two genomic regions on chromosomes 2 and 8 (at intervals of 30.42–30.55 and 14.06–14.99 Mbp, respectively) were identified that explained 9.37 to 16.04% of phenotypic variation of the trait. For RL-R three genomic regions on chromosomes 2, 7 and 9 were identified that explained 9.15 to 16.46% of phenotypic variation of the trait. For RFW-R four genomic regions on chromosomes 1, 2, 5 and 10 were identified that explained 9.84 to 18.64% of phenotypic variation of the trait. For RDW-R three genomic regions on chromosomes 1, 2 and 7 were identified that explained 10.06 to 18.05% of phenotypic variation of the trait (Table 10).
Co-Location of QTLs Under Salinity Condition
Investigation of co-location of the detected QTLs under salinity condition showed that 9 genomic regions harbored co-located QTLs (Fig. 3, Table 11). Most complex regions are two genomic regions on chromosome 1 each having 5 co-located QTLs; first one at a narrow interval of 32.33–32.54 Mbp comprised from qRDWs1.2, qRFWs1.3, qSLs1.1, qSFWs1.1, qTWs1.1, and second one at an adjacent region at 40.79–42.98 Mbp interval comprised from qRDWs1.3, qRFWs1.4, qSFWs1.2, qSDWs1.1, qTWs1.3. A genomic region on chromosome 11 also harbors 4 co-located QTLs at 24.96–25.85 Mbp interval including QTLs of qSDWs11.1, qSFWs11.1, qTWs11.1, qRWCs11.2. Furthermore, a genomic region of chromosome 6 at 29.76–31.21 Mbp interval carries 3 co-located QTLs including qRDWs6.1, qRFWs6.3, qRLs6.1. Eight other genomic regions on chromosomes 1, 2, 7, 10 and 12 carry a minimum number of co-located QTLs (e.g. two QTLs at each interval).
Identification of Candidate Genes for Salinity Tolerance
Comparison of the physical location of SNP markers that showed significant associations to studied traits in this study, with the physical location of rice genes deposited on the Rice Annotation Project database (https://rapdb.dna.affrc.go.jp/), resulted in the identification of 36 candidate genes that may be responsible to salinity tolerance (Table 12), such that among the significant markers related to one or more traits under the salinity stress, we found many SNP markers which physically placed within the gene sequences of RAP ID database. The results showed that these genes are distributed on different genomic regions of 5 chromosomes of rice. Twenty candidate genes were identified on chromosome 1 (Table 12; Fig. 4), 2 of which are uniquely related to root dry weight (RDW), 4 genes are related to root fresh weight (RFW), one gene is related to turgid weight (TW), one gene is related to relative water content (RWC) and 12 genes are related to both RDW and RFW. The latter 12 genes scattered between a coordinate of 42.25–42.98 Mbp which coincides to 4th co-located QTL region (defined in Table 11) carrying 5 QTLs of qRDWs1.3, qRFWs1.4, qSFWs1.2, qSDWs1.1, qTWs1.3.

Map location of salinity-tolerance candidate genes on 7 distinct regions of chromosome 1. Gene RAP locus IDs are in red
Furthermore, one candidate gene (Os05g0572900) on chromosome 5 was identified under salinity stress condition which showed a significant association to root fresh weight (RFW). Six candidate genes were identified on chromosome 6 under salinity stress condition (Table 12), one of which had a significant association to two traits including root fresh weight (RFW) and root length (RL), and 5 ones had unique associations to root fresh weight (RFW). Six candidate genes were identified on chromosome 8 under salinity stress, 2 of which had a significant association to root length (RL), and 4 other genes had a significant association to RFW. One and two candidate genes were identified on chromosome 11 with a significant association to RWC and RFW, respectively (Table 12).
Haplotype Analysis
Haplotype analysis revealed 2 distinct blocks on rice genome: first block on chromosome 1 spanning 42.25 to 42.65 Mbp which carry 7 candidate genes, and 2nd block on chromosome 8 spanning 26.72 to 26.93 Mbp which carry 3 candidate genes (supplementary Table S9). Two major haplotypes were identified within first block on chromosome 1; superior haplotype (with frequency of 90.2%) decreased both RFW and RDW, but inferior haplotype (with frequency of 9.1%) increased values of the 2 traits; so that average RFW and RDW of haplotype 1 was 125.1 and 17.3 mg/plant, respectively, while average of RFW and RDW of haplotype 2 was 180.0 and 24.4 mg/plant, respectively. In the case of 2nd block, 4 different haplotypes were identified with frequencies of 44.8, 22.7, 16.9 and 14.9%. Haplotype 4 had highest values for RFW and RDW (166.3 and 22.0 mg/plant) and haplotype 3 had lowest values of these traits (93.8 and 13.4 mg/plant). Superior haplotype (haplotype 1 with frequency of 44.8%) also produced relatively high values of RFW and RDW (142.9 and 19.6 mg/plant, respectively).
Expression Assay of Candidate Genes
Network analysis using riceFREND database revealed co-expression pattern of the candidate genes. For each candidate gene we detected 2 to 6 direct interactions in gene networks (supplementary Table S10). For example, in the case of Os01g0963000 (Peroxidase BP1 precursor) co-expressed with four genes including Os03g0225900 (Allene oxide synthase), Os06g0521500 (Haem peroxidase family protein), Os07g0542400 (Similar to Receptor protein kinase) and Os08g0508800 (Lipoxygenase, chloroplast precursor). Os01g0966000 (encoding a Plasma membrane H+-ATPase) co-expressed with Os03g0591000, Os08g0326100, Os11g0661000 and Os12g0613200.
For validation of the response to salinity of candidate genes, an expression assay was conducted under normal and salinity conditions using sensitive and tolerant accessions. The expression of PM H+-ATPase (Os01g0966000) at early times after stress was increased in both tolerant and sensitive cultivars but it decreased after 72 h in sensitive cultivar and increased in tolerant cultivar (Fig. 5). The expression of Peroxidase BP1 precursor (Os01g0963000) was increased up to 40 times at 72 h after stress in tolerant cultivar.

Changes in relative gene expression level of candidate genes at different times after salinity stress in different rice cultivars. a Peroxidase BP1, b PM H+-ATPase. T24, T48 and T72: 24, 48 and 72 h after salt stress. CV6 and CV178 (ARC6578 and Shoemed): sensitive cultivars. CV48 (Bombilla): tolerant cultivar
Discussion
Association study as an efficient method identifies the genomic regions controlling quantitative traits on the basis of linkage disequilibrium (LD) (Gupta et al. 2005). The amount of LD on the whole genome and chromosomes might significantly influence the mapping ability and selection efficacy by the help of the associated markers. In this study, the amount of LD on the basis of r2 values was variable from 0.254 within chromosome 11 to 0.395 within chromosome 3 with the mean value of 0.321 (data not shown). Altogether, 70.37% of the SNP markers showed significant LD (p < 0.01). In the association mapping method, the resolution and accuracy of the obtained map depend on the extent and amount of LD decline (Remington et al. 2001; Kim et al. 2007; Pritchard et al. 2000b; Bastien et al. 2014a, b). In another hand, LD measure in a mapping population presents an estimation of the number of required markers for the identification of QTLs (Remington et al. 2001; Kim et al. 2007; Pritchard et al. 2000; Bastien et al. 2014a, b). In the study of the core collection of rice by the use of SSR markers, it has been reported that 46.8% of the markers had significant LD (Zhang et al. 2011) which is lower than that of our study. In contrast, in the study of Jin et al. (2010) in a rice panel of 416 rice genotypes (including wild types, cultivar and breeding lines), 63% of the markers showed significant LD, which is comparable to LD obtained in our study. Zhang et al. (2011) stated that the low level of LD leads to unsuccessful scanning of the whole genome; and as a result, there would be a need for a higher number of markers. Considering the high and appropriate level of LD in our study (70.37%), it seems that the number of our SNP markers was enough to conduct association mapping.
As we showed, with a high density of SNP markers (1 SNP per 11 Kbp), the genome-wide association study (GWAS) method could facilitate fine mapping and identification of the genomic regions that control salinity tolerance-related traits at early vegetative stage. In our GWAS, many genomic regions were identified which were related to different traits under salt stress condition. Ninety seven and 151 marker-trait associations were identified by MLM-Q-K model under normal and salinity stress conditions, respectively. Kumar et al. (2015) also detected more association signals under salinity stress than under normal condition in their GWAS analysis using 4191 SNPs. The 151 associations signals under salinity stress were scattered on 10 chromosomes of rice (all chromosomes except for chromosomes 3 and 7), and altogether they were arranged in 29 genomic regions, 9 of which harbored 2 to 5 QTLs, and each of the remained 20 genomic regions comprised of only one QTL (Fig. 3). For eight studied traits viz. SL, RL, RDW, RFW, SFW, SDW, RWC and TW we identified 2, 6, 7, 15, 4, 2, 3 and 4 genomic regions under salinity condition.
The position of qRWCs1.1 on chromosome 1 (7.09 Mbp) is comparable to QTL #2138 for salt injury reported by Amman et al. (2007), and also it is comparable to q01_03 for SDW and RDW reported by Frouin et al. (2018) (supplementary Table S11). The position of qRFWs1.1 on the same chromosome (11.26 Mbp) is similar to that of QTLs #2181, #2182 and #2183 (for salt tolerance, shoot dry weight and leaf water content, respectively) reported by Ul Haq et al. (2010); also a QTL for leaf area (q01_04) was mapped in this region by Frouin et al. (2018). This region also co-locates with qSKC-1, known as SalTol1, a major QTL which controls salinity tolerance at vegetative stage of rice (Lin et al. 2004). Pandit et al. (2010) also mapped 3 co-located QTLs for salinity tolerance in the same genomic region (including qNaSH-1.1, qKSH-1.1 and qNa/KSH-1.1) using a RIL population from a cross between CSR27 and MI48. The positions of QTLs qRDWs1.1 and qRFWs1.2 on chromosome 1 (23.67 and 24.91–24.95 Mbp, respectively) are comparable to q01_5 and q01_6 for tiller number and specific leaf area reported by Frouin et al. (2018) and to qSNC1 for shoot Na+ concentration detected in a GWAS analysis by Naveed et al. (2018).
Among 8 genomic regions associated to different traits on chromosome 1, two genomic regions represented more importance for salinity tolerance; these are located in a coordinate of 32.33–34.51 Mbp interval showing association to SL, RDW, RFW, SFW and TW, and in a coordinate of 40.79–42.98 Mbp interval showing association to RDW, RFW, SFW, SDW and TW. Out of these, the location of QTL identified for turgid weight within this region (qTWs1.1 at 34.51 Mbp) is comparable to major QTL #2122 for shoot length reported by Takehisa et al. (2004). The locations of 2 QTLs of qRFWs1.4 and qRDWs1.3 on chromosome 1 (at positions of 40.79 and 41.12 Mbp, respectively) are comparable to QTLs #2123, #2124 and #2125 for shoot length under salinity reported by Takehisa et al. (2004). The QTL position also is similar to q01_09 for leaf area under salinity condition reported by Frouin et al. (2018). The location of QTL qRLs2.1 (27.27 Mbp) is comparable to 4 QTLs for tiller number detected by Takehisa et al. (2004) and to q02_10 for leaf area detected by Frouin et al. (2018). The QTL qRDWs4.1 on chromosome 4 (4.69 Mbp) co-locates to qGR4 for germination rate reported by Naveed et al. (2018), and the QTL qRFWs5.1 on chromosome 5 (28.54 Mbp) co-locates to QTL #2142 for salt injury reported by Amman et al. (2007).
The position of QTL qRFWs6.1 on chromosome 6 (4.58–4.61 Mbp) is similar to the position of q06_02 for leaf area reported by Frouin et al. (2018). The positions of 2 co-located QTLs qRLs6.1 and qRFWs6.3 on chromosome 6 (29.76–30.92 Mbp interval) are comparable to QTL #2098 for relative seminal root length reported by Prasad et al. (2000). Also, the location of QTLs qRLs8.1 on chromosome 8 (0.06–0.12 Mbp interval) co-locates to a08_04 for tiller number reported by Frouin et al. (2018), and the location of QTL qRLs8.2 on the same chromosome (2.33 Mbp) is comparable to QTL #2175 for leaf water content detected by Ul Haq et al. (2010). The positions of 2 QTLs qRLs8.3 and qRFWs8.1 on the same chromosome (at positions 21.51 and 26.73–26.94 Mbp) are similar to q08_05 and q08_06 for specific leaf area (SLA) reported by Frouin et al. (2018).
The location of qRFWs9.1 at 17.96 Mbp on chromosome 9 is comparable to QTL #2176 for leaf water content reported by Ul Haq et al. (2010) and to q09_05 for leaf area reported by Frouin et al. (2018), and the location of qRFWs11.1 at 21.68–21.98 Mbp interval on chromosome 11 is comparable to QTL #2178 for leaf water content reported by Ul Haq et al. (2010) and to q11_05 for leaf area reported by Frouin et al. (2018) and to qSKC11 for shoot Na+:K+ ratio detected by Naveed et al. (2018). The 2 QTLs including qRDWs12.1 and qRFWs12.1 on chromosome 12 (at 17.53 to 18.60 Mbp interval) locate to a similar region of QTLs #2168 for shoot Na+ concentration, #2173 for Na+:K+ ratio and #2174 for shoot dry weight (Ul Haq et al. 2010). Furthermore, the position of 2 co-located QTLs qSLs12.2 and qSFWs12.1 on this chromosome (21.08 Mbp) is comparable to q12_02 and q12_03 for root length reported by Frouin et al. (2018) and to qRL12 for root length reported by Naveed et al. (2018) (supplementary Table S11).
Based on comparison with locations of earlier reported QTLs, a complex genomic region on chromosome 1 at 32.33–32.54 interval, although it is adjacent to the QTL #2122 of Takehisa et al. (2004), it is adequately far from this QTL (~ 1.5 Mbp) to be distinguished as a different QTL, and hence it is a novel region containing 5 QTLs (qRDWs1.2, qRFWs1.3, qSLs1.1, qSFWs1.1, qTWs1.1) which was not, to our knowledge, reported before this study. Similarly, 5 other genomic regions detected in our GWAS signals are reported here for first that are associated to salinity-related traits: a genomic region at 35.26 Mbp on chromosome 2 (carrying qRDWs2.1 and qRFWs2.1), a genomic region at 20.04 Mbp on chromosome 6 (carrying qRFWs6.2), a genomic region at 6.12–7.32 Mbp interval on chromosome 10 (carrying qRLs10.1 and qRFWs10.1) and 2 genomic regions on chromosome 11 including 6.06 Mbp position (qRWCs11.1) and 24.96–25.85 Mbp interval (qRWCs11.2, qSFWs11.1, qSDWs11.1 and qTWs11.1).
In our study, the results for RDW and RFW were found more important than other traits, such that 7 and 15 QTL regions, respectively, tagged to 39 and 82 SNPs were identified on different chromosomes of rice. Out of these SNPs associated to RDW and RFW, 30 and 34 SNPs, respectively, located on chromosome 1 in a coordinate of 40.79–42.98 Mbp. This region carries 12 candidate genes (Table 11), some of them were not absolutely reported for salinity tolerance at seedling or any developmental stages in rice. More interesting genes are Os01g0966000 and Os01g0963000. First gene (Os01g0966000) encodes a plasma membrane (PM) H+-ATPase (EC 3.6.1.3). It was shown that PM H+-ATPase family members play a well-known role in tolerance to abiotic stresses such as cold (Iswari and Palta 1989; Martz et al. 2006), drought (Du et al. 2015) and acid rain (Liang et al. 2015). Many studies also documented the importance of PM H+-ATPase in response to salinity stress in Arabidopsis and cereal plants such as wheat and rice (Ayala et al. 1997; Vitart et al. 2001; Gévaudant et al. 2007; Morgan et al. 2014; Chen et al. 2015a, b; Falhof et al. 2016). Second gene (Os01g0963000) encodes a peroxidase BP1 protein. The main role of this peroxidase such as other type I peroxidases is the removal and scavenging of reactive oxygen species (ROS) including H2O2 (Asada 1992; Shigeoka et al. 2002). The important role of type I peroxidases was well documented in plant stress tolerance (Koussevitzky et al. 2008; Dietz 2016). In a more recently work by using proteome assay it was revealed that peroxidase is a major category of proteins differentially regulated under salt stress at seedling stage between non-tolerant and tolerant rice genotypes (Lakra et al. 2019). The role of other genes in this complex region of chromosome 1 (40.79–42.98 Mbp) remains to be elucidated.
In addition to 2 above-mentioned genes, we identified three other important genes on chromosome 1 in our GWAS signals, including Os01g0304100 (11.26 Mbp), Os01g0624700 (24.95 Mbp) and Os01g0812000 (34.51 Mbp). First gene resides in the vicinity of well-known SalTol QTL region and encodes a cation chloride cotransporter which was identified as a determinant of salt tolerance in previous works (Walia et al. 2005; reviewed in Waziri et al. 2016). Second gene (Os01g0624700) encodes a WRKY transcription factor (WRKY 12) which might play a role in salinity stress, but its precise mechanism of action remains to be determined. The involvement of WRKY family members in salinity tolerance were well documented (Hu et al. 2013; Li et al. 2013; Yan et al. 2014; Jiang et al. 2017). Third gene (Os01g0812000) encodes a transcriptional activator of gibberellin-dependent alpha-amylase expression (GAMyb) which is a unique gene of GAmyb class on chromosome 1 (see https://rapdb.dna.affrc.go.jp). Gibberlic acid (GA) has a known role in regulating α-amylase synthesis in aleurone cells via GAmyb which is necessary for expression of genes encoding α-amylase (Gubler et al. 1999). It was well documented that activation of α-amylases as major degradation factors of stored starches in the seed endosperm is needed for germination and post-germination growth (Lovegrove and Hooley 2000; Zou et al. 2008). Additional research has shown that GAmyb can transactivate other GA-regulated genes (Gubler et al. 1999; Cercós et al. 1999). Another GAmyb gene on chromosome 6 of rice at position 3.6 Mbp which was annotated as salt responsive DW40 3 (SRDW3), has a role in salt response (https://rapdb.dna.affrc.go.jp). This gene resides within two co-located QTL regions at normal condition (e. g qRDWn6.1 and qRFWn6.1) at a narrow coordinate on chromosome 6 (3.59–3.60 Mbp) (Tables 4 and 5). A DW40 type gene named BnSDW1 was characterized in Barssica napus which causes salinity tolerance (Lee et al. 2010). Taking together, it seems that the GAmyb identified on chromosome 1 in this study maybe play a role in salt response.
A candidate gene on chromosome 2 (Os02g0730300 at 30.4 Mbp) which is related to SL-R and RDW-R, encodes a high affinity potassium (K+) transporter (HAK). Potassium (K) affects all aspects of plant growth including resistance to pathogens and tolerance to abiotic stresses such as salinity, lodging, and drought (Ahmad et al. 2016). Limiting K loss supports osmotic adjustment, sustain cell expansion, ensures appropriate stomatal regulation and helps to sustain photosynthetic activity through photoassimilate translocation (Zörb et al. 2014), therefore, modulation of K transport is crucial under stress conditions. The putative function of the KT/HAK/KUP transporters has been predicted to play a key role in maintaining K homeostasis (Chen et al. 2015b; Li et al. 2017). Perception of osmotic stresses can trigger the transient K effluxes at the plasma membrane by impairing HAK5 activity (Brauer et al. 2016). OsHAK1 overexpression plants exhibited higher K acquisition efficiency, a stronger growth phenotype and increased grain yield, especially when grown in adverse environmental conditions (Chen et al. 2017). Regarding to these findings, we suppose that our candidate gene Os02g0730300 could play an inevitable role in response to salinity stress.
Another candidate gene on chromosome 6 encoding DnaJ domain protein C55 was identified in our GWAS signals. DnaJ as a heat shock protein has chaperon properties (Laufen et al. 1999; Miernyk 2001). A mitochondrial DnaJ protein (BIL2) in Arabidopsis thaliana confers growth and resistance against environmental stresses such as strong light and salinity stresses (Bekh-Ochir et al. 2013). More recently a few DnaJ genes were identified in rice that were up-regulated under salinity stress (Wang et al. 2015), all of which are different from our detected DnaJ (Os06g0535300). It seems that this gene is a novel DnaJ involved in salinity response. Another interesting candidate gene on chromosome 6 is Os06g0717400 which encodes a pseudouridine synthase domain containing protein. Pseudouridines do not occur naturally in mRNA, but mRNA pseudouridylation is induced at specific sites under some stress conditions such as heat shock in yeast (Schwartz et al. 2014) and it was shown that the rate of translational read-through was increased after pseudouridylation of stop codon (Karijolich and Yu 2011). This finding indicates that mRNA pseudouridylation can influence gene expression at post-transcriptional level (Lovejoy et al. 2014). Interestingly, it was observed that, in spite of being constitutive RNA modifications, a few pseudouridines in yeast can be induced in small nuclear RNAs (snRNAs) under some stress conditions such as heat shock (Wu et al. 2011; van der Feltz et al. 2018). These unusual post-transcriptional RNA modifications can alter regulation of splicing patterns under stress conditions (Lovejoy et al. 2014; Karijolich et al. 2015). Based on these findings, the candidate gene Os06g0717400 identified in our GWAS signals might play a role in salinity tolerance, but so far it was not reported the role of any pseudouridine synthase gene in stress tolerance in plants and hence, the mechanism of action of our candidate gene remains to be elucidated.
Six candidate genes were identified on chromosome 8, one of which was an ABI/VP1 transcription factor, ABI/VP1 TF (Os08g0101000). This gene was annotated as a gene involving in regulation of iron-deficiency response and tolerance in RAP-db (https://rapdb.dna.affrc.go.jp). A member of ABI/VP1 TF class in rice, ABI3/VP1 TF, is a IDE-binding TF and regulates expression of IDEF1 via binding to RY element of its promoter under iron deficiency condition (Kobayashi et al. 2007). Since under salinity stress the absorption of mineral nutrients (including iron) by plant roots is inhibited due to excessive accumulation of Na+ ions, the root cells activate IDE- type genes by mediating ABI/VP1 TF to absorb higher amounts of iron and to overcome iron deficiency. Although IDE-type genes (such as IDEF1 and IDEF2 in rice) are constitutively expressed (irrespective of Fe level) which indicates their role in sensing Fe deficiency signals, it was suggested that IDEF1 also plays an important role in activation of Fe deficiency-induced genes involved in Fe uptake under iron deficiency. Therefore, we suggest that ABI/VP1 TF gene identified in our research could play an indirect role in salinity tolerance via activating IDE-type genes which in turn transactivates the Fe deficiency-induced gene(s) to overcome iron deficiency by absorption higher amounts of Fe under salt stress. However, its essential role must be assessed experimentally via complementation tests such as genetic transformation and or RNAi. Another candidate gene on chromosome 8 is Os08g0538700 which encodes a retinoblastoma-related protein (RBR). To date there is no direct evidence on the role of RBR in salt stress tolerance in plants. In an early research (Åmellem et al. 1996) it was suggested that human RBR suppresses cell growth under hypoxic stress. This protein responds to mitogenic and anti-proliferative signals to coordinate cell-cycle control with the cellular environment (Daria et al. 2008). A more recently research revealed that a retinoblastoma-binding protein of WD40 family plays an important role in response to salinity stress in brine shrimp (Artemia sinica). In plants, however, it was reported that RBR has a role in stem cell maintenance and cell differentiation in Arabidopsis roots (Wildwater et al. 2005; Borghi et al. 2010). A study by Ausín et al. (2004) has revealed the role of a retinoblastoma-associated protein named FVE in regulation of flowering time in Arabidosis. Also, it was reported that RBR is involved in regulating flowering time and cold response in Arabidopsis (Jeon and Kim 2011). A comparative transcriptome analysis in foxtail millet (Setaria italica) showed that a putative retinoblastoma-related protein was up-regulated during dehydration stress (Lata et al. 2010). In regard to common nature of responses to different environmental stresses and evidences on RBR role in some above-mentioned stresses, perhaps the RBR gene detected in current study has a putative role in salinity tolerance, mechanism of which needs to be elucidated under salinity stress.
Three candidate genes on chromosome 11 were identified in our GWAS signals. One of them is Os11g0575600 which encodes a lipoxygense protein. The role of lipoxygenases in plant stress response was well documented (Babenko et al. 2017). Arabidopsis Lipoxygenase 1 (AtLox1) gene is involved in response to oxidative stress induced by cadmium exposure (Keunen et al. 2013). In pepper CaLox1 plays a role in response to different environmental stresses including high salinity stress (Lim et al. 2015). Rice Lox genes, specifically Lox2 are involved in response of rice to different environmental stresses including salinity stress (Rabbani et al. 2003; Walia et al. 2007; Roychoudhury et al. 2008; Liu et al. 2014; Islam et al. 2019). Another candidate gene on this chromosome (Os11g0578700) encodes an F-box protein. The F-box proteins are one of the largest class of functional proteins affecting diverse aspects of plant life. Involvement of F-box proteins in plant defense against abiotic stresses such as salinity stress was documented in previous researches (Jain et al. 2007; Zhang et al. 2008; Yan et al. 2011; Jia et al. 2017). More recently, Al-Tamimi et al. (2016) using a new association model identified an F-box encoding gene on chromosome 5 of rice for transpiration use efficiency (TUE) under saline condition. Accordingly, the candidate gene Os11g0575600 identified in our research could play a role in salinity response, but its precise mechanism of action remains to be elucidated. In addition, within a complex region of chromosome 11 carrying 4 co-located QTLs viz. qRDWs11.2, qSFWs11.1, qSDWs11.1 and qTWs11.1 (24.96–25.85 Mbp interval) there are two putative candidate genes including Os11g0644000 and Os11g0648000. Os11g0644000 encodes a receptor-like cytoplasmic kinase (RLCK 342). It was reported that OsSIK1, a receptor-like kinase enhances rice tolerance to drought and salt stress (Ouyang et al. 2010), and also it was reported that the receptor-like kinase OsSIT1 confers salt sensitivity in rice via regulating ethylene homeostasis (Li et al. 2014). Second gene (Os11g0648000) encodes a Na+/H+ antiporter annotated as OsNHX2. Four vacuolar Na+/H+ antiporters including OsNHX2 were reported in rice (Bassil et al. 2012). Na+/H+ antiporters enhance the compartmentalization of Na+ into the vacuoles, and hence improve salt tolerance (Kumar et al. 2013; Amin et al. 2016), an issue which was validated by overexpression of OsNHX1 in rice (Chen et al. 2007; Wang et al. 2012).
For validating the effect of identified candidate genes on salt tolerance, gene expression assays for 2 candidate genes (Peroxidase BP1 and PM H+-ATPase) were conducted in tolerant and sensitive rice cultivars. Expression of Peroxidase BP1 at early hours of salinity stress increased in both sensitive and tolerant cultivars, presumably to remove produced ROS, and then down-regulated, particularly in tolerant cultivar; in continue the expression of the gene was considerably increased at 72 h in both cultivars, but much more up-regulated in tolerant cultivar. In general, differential expression of peroxidases is one of factors involving in acquiring salinity tolerance in rice (Lakra et al. 2019). In the case of PM H+-ATPase also we observed similar expression pattern with minor differences. The gene was more up-regulated at 72 h after stress in tolerant cultivar, presumably to retain membrane function under salinity stress (Chen et al. 2015a, b; Falhof et al. 2016).
Conclusion
The results for RDW and RFW were found more important than other traits, and known or novel candidate genes in this research can be used for improvement of salinity tolerance in molecular breeding programmes. Further study and identification of effective genes on salinity tolerance by the use of candidate gene-association analysis can help to precisely uncover the mechanisms of salinity tolerance at molecular level. A time dependent relationship between salt tolerance and expression level of genes (e. g. PM H+-ATPase, and Peroxidase BP1) could be recognized.
Methods
Plant Materials and the Growth Conditions
One hundred fifty five varieties of rice (Table S1) were randomly selected from a larger panel of rice varieties (from the Chang Genetic Resources Center, International Rice Research Institute (IRRI), Philippines). The experiment was carried out in Shahid Beheshti University, Tehran, Iran. These accessions were cultured in early April (2017) in three replications under 2 environments including normal and salt stress conditions. Plant materials were evaluated in a factorial experiment (155 genotypes and two levels of salt stress induced by NaCl). To do this, seeds of each variety were surface-sterilized with 10% hypochlorite sodium, washed 3 times using distilled water and put in paper towels for germination in a germinator with 28 °C. After germination they were transferred to growth chamber with 25 °C, 60% relative humidity and 16/8 h light/dark regime.
Salinity Treatment and Phenotypic Evaluations
Phenotypic evaluations were conducted in a completely randomized design (CRD) with three replicates at controlled condition in a growth chamber. The control treatment (normal environment, N) included Yoshida solution, whereas the salinity stress treatment (100 mM NaCl) was induced by adding needed volume of 1 M NaCl to the Yoshida solution. The Yoshida solution included macro and micro-elements. During the growth of the seedlings, the solution pH was set to 5.5 and the medium salinity was set to the desired level every day. Also, every 3 days the solution was renewed.
In order to evaluate the given traits after normal and salt treatments (14 days after transfer to growth chamber), ten seedlings from each replicate were randomly selected and separated to root and shoot sections. The growth of seedling sections including root length (RL), shoot length (SL), root fresh weight (RFW), shoot fresh weight (SFW), was measured. Furthermore, fresh roots and shoots were oven-dried at 80 °C for 24 h. After that, root dry weight (RDW) and shoot dry weight (SDW) were also measured. Turgid weight (TW) was measured 24 h after floating the shoots in distilled water. The oven-dried seedling shoots were weighted to calculate relative water content (RWC). Also, some salinity-relative traits were calculated including SL-R, RL-R, RFW-R and RDW-R using appropriate formulae (for example SL-R = SLsalt/SLnormal). To determine differences among genotypes (G), salt treatment levels (T) and G × T interactions, the phenotypic data were analyzed by using general linear model (GLM) in SPSS v.19 software (Gray and Kinnear 2012). Differences between means were compared using LSD test at the 5% level of significance (p ≤ 0.5).
Genome-Wide Association Study (GWAS)
The genotypes of all rice varieties were determined at 37867 SNP loci which were downloaded from Gramene database (www.gramene.org). By the use of TASSEL software, these SNPs were filtered for monomorphic loci and loci with minor allele frequency (MAF) below 5%. In order to determine the number of the real subgroups (K) in the studied rice collection, the STRUCTURE software was used (Pritchard et al. 2000). The number of the initially assumed subpopulations (K) was considered to be between 1 and 10; and in order to increase the accuracy of the estimation of the parameters, 10 independent replicates were specified for each K subpopulation. Finally, the optimized number of the subpopulations (K) was estimated on the basis of the ΔK method according to Evanno et al. (2005). After determining the number of real subpopulations, the respective Q matrix was extracted to be used in the association mapping stage.
In most of the GWAS studies of plants, r2 is used for estimating the extent of linkage disequilibrium (LD) because it presents all of the information related to linkage between the marker and the QTL (Flint-Garcia et al. 2003, Remington et al. 2001; Courtois et al. 2013). r2 is estimated to be between 0.1 and 0.2 which is the minimum threshold for a significant association between the SNP marker and given traits. The association analysis was done by the use of SNP markers whose allele frequencies were higher than 0.05. The analysis was done through the MLM method by the use of TASSEL software (Bradbury et al. 2007). In the MLM method, in order to prevent the false marker-trait associations, in addition to Q matrix (coefficients of population structure), K matrix (the kinship coefficient of the varieties) was also used. Thus the used model was MLM-Q-K. In this method, the genotypic effects (SNP markers) and the coefficients of the population structure (Q) are taken as the fixed effects, and the varieties’ kinship coefficients (K) are taken as the random effects (Bradbury et al. 2007). MLM Model is as following:
Where P is the phenotype, M and Q are the genotypic fixed effects and population structure, respectively, and K is the effect of random kinship of the samples, and E is the residual effects. The association analysis was done separately for each condition, so that the mean value of replicates for each trait was used as the phenotypic data. After association analysis, the GWAS results were presented as Manhattan plots based on the negative log10 transformed observed p-values for each SNP-trait association. We used a threshold of 1e-04 to declare a SNP significant (Kaler et al. 2017).
Identification of the Candidate Genes and Haplotype Analysis
In order to identify the genes underlying the QTLs of salt tolerance, overlapping of the physical genomic regions of these QTLs (=their associated SNPs) and any gene deposited on the Rice Annotation Project database, RAP-db (https://rapdb.dna.affrc.go.jp), was assessed. The genes annotated as hypothetical, non-protein coding and transposable element were discarded. In addition, higher sensitivity was put on stress-related genes whose sequence coincided to associated SNPs. Then, from the gene lists, the candidate genes were selected based on the predicted function (biological processes) in relation with the trait of interest.
In haplotype analysis genomic blocks on each chromosome were scanned in adjacent regions using SNP data of candidate genes in our rice accession pannel. Block haplotypes with frequency lower than 5% were omitted.
Validation Assays
In the present study, the expression pattern of several new genes involved in salt tolerance of rice was investigated by real-time PCR in two sensitive (ARC6578 and Shoemed) and one tolerant (Bombilla) rice accessions that were selected based on previous work (Nayyeripasand et al. 2019). RNA samples were extracted from 20-day-old seedlings treated with NaCl (100 mM) at three times after salt stress (24, 48 and 72 h). Gene expression analysis was performed on 2 candidate genes (including Proxidase BP1 precursor and Plasma Membrane H+-ATPase) using real-time PCR. The Actin gene was used as the reference gene in this assay. Furthermore, a co-expression assay was conducted using riceFREND database (Sato et al. 2013) (https://ricefrend.dna.affrc.go.jp/) for all candidate genes.
Availability of Data and Materials
All relevant data have been provided as Tables, Figures with in the text and in the following supplementary data.
Abbreviations
- ANOVA:
-
Analysis of variance
- GWAS:
-
Genome-wide association study
- LD:
-
Linkage disequilibrium
- MLM:
-
Mixed linear model
- PM:
-
Plasma membrane
- QTL:
-
Quantitative trait loci
- RDW:
-
Root dry weight
- RFW:
-
Root fresh weight
- RL:
-
Root length
- RWC:
-
Relative water content
- SDW:
-
Shoot dry weight
- SFW:
-
Shoot fresh weight
- SL:
-
Shoot length
- SNP:
-
Single nucleotide polymorphism
- TW:
-
Turgid weight
References
Ahmad I, Mian A, Maathuis FJ (2016) Overexpression of the rice AKT1 potassium channel affects potassium nutrition and rice drought tolerance. J Exp Bot 67:2689–2698
Al-Tamimi N, Brien C, Oakey H, Berger B, Saade S, Ho YS, Schmöckel SM, Tester M, Negrao S (2016) Salinity tolerance loci revealed in rice using high-throughput non-invasive phenotyping. Nat Commun 7:13342
Åmellem Ø, Stokke T, Sandvik JA, Pettersen EO (1996) The retinoblastoma gene product is reversibly dephosphorylated and bound in the nucleus in S and G2 phases during hypoxic stress. Exp Cell Res 227(1):106–115
Amin US, Biswas S, Elias SM, Razzaque S, Haque T, Malo R, Seraj ZI (2016) Enhanced salt tolerance conferred by the complete 2.3 kb cDNA of the rice vacuolar Na+/H+ antiporter gene compared to 1.9 kb coding region with 5′-UTR in transgenic lines of rice. Front Plant Sci 7:14
Ammar MHM, Singh RK, Singh AK, Mohapatra T, Sharma TR, Singh NK (2007) Mapping QTLs for salinity tolerance at seedling stage in rice (Oryza sativa L.). In: African Crop Science Conference Proceedings, vol 8, pp 617–620
Asada K (1992) Ascorbate peroxidase–a hydrogen peroxide-scavenging enzyme in plants. Physiol Plant 85(2):235–241
Ausín I, Alonso-Blanco C, Jarillo JA, Ruiz-García L, Martínez-Zapater JM (2004) Regulation of flowering time by FVE, a retinoblastoma-associated protein. Nat Genet 36(2):162
Ayala F, Ashraf M, O'Leary JW (1997) Plasma membrane H+-ATPase activity in salt-tolerant and salt-sensitive lines of spring wheat (Triticum aestivum L.). Acta Bot Neerlandica 46(3):315–324
Babenko LM, Shcherbatiuk MM, Skaterna TD, Kosakivska IV (2017) Lipoxygenases and their metabolites in formation of plant stress tolerance. Ukr Biochem J 89(1):5–21
Bassil E, Coku A, Blumwald E (2012) Cellular ion homeostasis: emerging roles of intracellular NHX Na+/H+ antiporters in plant growth and development. J Exp Bot 63:5727–5740
Bastien M, Poirier P, Lemieux I, Després JP (2014a) Overview of epidemiology and contribution of obesity to cardiovascular disease. Prog Cardiovasc Dis 56(4):369–381
Bastien M, Sonah H, Belzile F (2014b) Genome wide association mapping of Sclerotinia sclerotiorum resistance in soybean with a genotyping-by-sequencing approach. Plant Genome 7(1):1–3
Bekh-Ochir D, Shimada S, Yamagami A, Kanda S, Ogawa K, Nakazawa M, Nakano T (2013) A novel mitochondrial DnaJ/Hsp40 family protein BIL2 promotes plant growth and resistance against environmental stress in brassinosteroid signaling. Planta 237(6):1509–1525
Borghi L, Gutzat R, Fütterer J, Laizet YH, Hennig L, Gruissem W (2010) Arabidopsis RETINOBLASTOMA-RELATED is required for stem cell maintenance, cell differentiation, and lateral organ production. Plant Cell 22(6):1792–1811
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23(19):2633–2635
Brauer EK, Ahsan N, Dale R, Kato N, Coluccio AE, Piñeros MA, Kochian LV, Thelen JJ, Popescu SC (2016) The Raf-like kinase ILK1 and the high affinity K+ transporter HAK5 are required for innate immunity and abiotic stress response. Plant Physiol 171:1470–1484
Cercós M, Gómez-Cadenas A, Ho THD (1999) Hormonal regulation of a cysteine proteinase gene, EBP1, in barley aleurone layers: Cis and trans-acting elements involved in the coordinated gene expression regulated by gibberellins and abscisic acid. Plant J 19:107–118
Chen G, Hu Q, Luo LE, Yang T, Zhang S, Hu Y, Yu L, Xu G (2015a) Rice potassium transporter OsHAK1 is essential for maintaining potassium-mediated growth and functions in salt tolerance over low and high potassium concentration ranges. Plant Cell Environ 38:2747–2765
Chen G, Liu C, Gao Z, Zhang Y, Jiang H, Zhu L, Ren D, Yu L, Xu G, Qian Q (2017) OsHAK1, a high-affinity potassium transporter, positively regulates responses to drought stress in rice. Front Plant Sci 8:1885
Chen J, Wang WH, Wu FH, He EM, Liu X, Shangguan ZP, Zheng HL (2015b) Hydrogen sulfide enhances salt tolerance through nitric oxide-mediated maintenance of ion homeostasis in barley seedling roots. Sci Rep 5:12516
Chen M, Chen Q, Niu X, Zhang R, Lin H, Xu C, Wang X, Wang G, Chen J (2007) Expression of OsNHX1 gene in maize confers salt tolerance and promotes plant growth in the field. Plant Soil Environ 53(11):490–498
Courtois B, Audebert A, Dardou A, Roques S, Ghneim-Herrera T, Droc G, Frouin J, Rouan L, Gozé E, Kilian A, Ahmadi N (2013) Genome-wide association mapping of root traits in a japonica rice panel. PLoS One 8(11):e78037
Daria D, Filippi MD, Knudsen ES, Faccio R, Li Z, Kalfa T, Geiger H (2008) The retinoblastoma tumor suppressor is a critical intrinsic regulator for hematopoietic stem and progenitor cells under stress. Blood 111(4):1894–1902
Dietz KJ (2016) Thiol-based peroxidases and ascorbate peroxidases: why plants rely on multiple peroxidase systems in the photosynthesizing chloroplast? Mol Cell 39(1):20
Du H, Zhou X, Yang Q, Liu H, Kurtenbach R (2015) Changes in H+-ATPase activity and conjugated polyamine contents in plasma membrane purified from developing wheat embryos under short-time drought stress. Plant Growth Reg 75(1):1–10
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14(8):2611–2620
Falhof J, Pedersen JT, Fuglsang AT, Palmgren M (2016) Plasma membrane H+-ATPase regulation in the center of plant physiology. Mol Plant 9(3):323–337
Flint-Garcia SA, Thornsberry JM, Buckler ES IV (2003) Structure of linkage disequilibrium in plants. Ann Rev Plant Biol 54(1):357–374
Frouin J, Languillaume A, Mas J, Mieulet D, Boisnard A, Labeyrie A, Turquay P (2018) Tolerance to mild salinity stress in japonica rice: a genome-wide association mapping study highlights calcium signaling and metabolism genes. PLoS One 13(1):e0190964
Gévaudant F, Duby G, von Stedingk E, Zhao R, Morsomme P, Boutry M (2007) Expression of a constitutively activated plasma membrane H+-ATPase alters plant development and increases salt tolerance. Plant Physiol 144(4):1763–1776
Ghomi K, Rabiei B, Sabouri H, Sabouri A (2013) Mapping QTLs for traits related to salinity tolerance at seedling stage of rice (Oryza sativa L.): an agrigenomics study of an Iranian rice population. OMICS 17:242–251
Gray C, Kinnear PR (2012) IBM SPSS statistics 19 made simple. Psychology Press
Gubler F, Raventos D, Keys M, Watts R, Mundy J, Jacobsen JV (1999) Target genes and regulatory domains of the GAMYB transcriptional activator in cereal aleurone. Plant J 17:1–9
Gupta PK, Rustgi S, Kulwal PL (2005) Linkage disequilibrium and association studies in higher plants: present status and future prospects. Plant Mol Biol 57(4):461–485
Haq TU, Gorham J, Akhtar J, Akhtar N, Steele KA (2010) Dynamic quantitative trait loci for salt stress components on chromosome 1 of rice. Funct Plant Biol 37(7):634–645
Hu Y, Chen L, Wang H, Zhang L, Wang F, Yu D (2013) A rabidopsis transcription factor WRKY 8 functions antagonistically with its interacting partner VQ 9 to modulate salinity stress tolerance. Plant J 74(5):730–745
Islam F, Wang J, Farooq MA, Yang C, Jan M, Mwamba TM, Zhou W (2019) Rice responses and tolerance to salt stress: deciphering the physiological and molecular mechanisms of salinity adaptation. In: Advances in rice research for abiotic stress tolerance. Woodhead Publishing, pp 791–819
Iswari S, Palta JP (1989) Plasma membrane ATPase activity following reversible and irreversible freezing injury. Plant Physiol 90:1088–1095
Jain M, Nijhawan A, Arora R, Agarwal P, Ray S, Sharma P, Khurana JP (2007) F-box proteins in rice. Genome-wide analysis, classification, temporal and spatial gene expression during panicle and seed development, and regulation by light and abiotic stress. Plant Physiol 143(4):1467–1483
Jeon J, Kim J (2011) FVE, an Arabidopsis homologue of the retinoblastoma-associated protein that regulates flowering time and cold response, binds to chromatin as a large multiprotein complex. Mol Cell 32(3):227–234
Jia Q, Xiao ZX, Wong FL, Sun S, Liang KJ, Lam HM (2017) Genome-wide analyses of the soybean F-box gene family in response to salt stress. Inter J Mol Sci 18(4):818
Jiang J, Ma S, Ye N, Jiang M, Cao J, Zhang J (2017) WRKY transcription factors in plant responses to stresses. J Integr Plant Biol 59(2):86–101
Jin H, White SR, Shida T, Schulz S, Aguiar M, Gygi SP, Nachury MV (2010) The conserved Bardet-Biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell 141(7):1208–1219
Kaler AS, Ray JD, Schapaugh WT, King CA, Purcell LC (2017) Genome-wide association mapping of canopy wilting in diverse soybean genotypes. Theor Appl Genet 130(10):2203–2217
Karijolich J, Yi C, Yu YT (2015) Transcriptome-wide dynamics of RNA pseudouridylation. Nat Rev Mol Cell Biol 16(10):581
Karijolich J, Yu YT (2011) Converting nonsense codons into sense codons by targeted pseudouridylation. Nature 474:395–398
Keunen E, Remans T, Opdenakker K, Jozefczak M, Gielen H, Guisez Y, Cuypers A (2013) A mutant of the Arabidopsis thaliana LIPOXYGENASE1 gene shows altered signalling and oxidative stress related responses after cadmium exposure. Plant Physiol Biochem 63:272–280
Kim DM, Ju HG, Kwon TR, Oh CS, Ahn SN (2009) Mapping QTLs for salt tolerance in an introgression line population between japonica cultivars in rice. J Crop Sci Biotechnol 12(3):121
Kim JK, Bamba T, Harada K, Fukusaki E, Kobayashi A (2007) Time-course metabolic pro fi ling in Arabidopsis thaliana cell cultures after salt stress treatment. J Exp Bot 58:415–424
Kobayashi T, Ogo Y, Itai RN, Nakanishi H, Takahashi M, Mori S, Nishizawa NK (2007) The transcription factor IDEF1 regulates the response to and tolerance of iron deficiency in plants. Proc Natl Acad Sci U S A 104:19150–19155
Koussevitzky S, Suzuki N, Huntington S, Armijo L, Sha W, Cortes D, Shulaev V, Mittler R (2008) Ascorbate peroxidase 1 plays a key role in the response of Arabidopsis thaliana to stress combination. J Biol Chem 283(49):34197–34203
Koyama ML, Levesley A, Koebner RM, Flowers TJ, Yeo AR (2001) Quantitative trait loci for component physiological traits determining salt tolerance in rice. Plant Physiol 125(1):406–422
Kumar K, Kumar M, Kim SR, Ryu H, Cho YG (2013) Insights into genomics of salt stress response in rice. Rice 6(1):27
Kumar V, Singh A, Mithra SA, Krishnamurthy SL, Parida SK, Jain S, Khurana JP (2015) Genome-wide association mapping of salinity tolerance in rice (Oryza sativa). DNA Res 22(2):133–145
Lakra N, Kaur C, Singla-Pareek SL, Pareek A (2019) Mapping the ‘early salinity response’triggered proteome adaptation in contrasting rice genotypes using iTRAQ approach. Rice 12(1):3
Lata C, Sahu PP, Prasad M (2010) Comparative transcriptome analysis of differentially expressed genes in foxtail millet (Setaria italica L.) during dehydration stress. Biochem Biophys Res Commun 393(4):720–727
Laufen T, Mayer MP, Beisel C, Klostermeier D, Moor A, Reinstein J, Bakau B (1999) Mechanism of regulation of hsp70 chaperones by DnaJ cochaperones. Proc Natl Acad Sci U S A 96:5452–5457
Lee S, Lee J, Paek KH, Kwon SY, Cho HS, Kim SJ, Park JM (2010) A novel WD40 protein, BnSWD1, is involved in salt stress in Brassica napus. Plant Biotechnol Rep 4(2):165–172
Lee SY, Ahn JH, Cha YS, Yun DW, Lee MC, Ko JC, Eun MY (2007) Mapping QTLs related to salinity tolerance of rice at the young seedling stage. Plant Breeding 126(1):43–46
Li CH, Wang G, Zhao JL, Zhang LQ, Ai LF, Han YF, Sun DY, Zhang SW, Sun Y (2014) The receptor-like kinase SIT1 mediates salt sensitivity by activating MAPK3/6 and regulating ethylene homeostasis in rice. Plant Cell 26(6):2538–2553
Li H, Gao Y, Xu H, Dai Y, Deng D, Chen J (2013) ZmWRKY33, a WRKY maize transcription factor conferring enhanced salt stress tolerances in Arabidopsis. Plant Growth Reg 70(3):207–216
Li W, Xu G, Alli A, Yu L (2017) Plant HAK/KUP/KT K+ transporters: function and regulation. Semin Cell Dev Biol 74:133–141
Liang C, Ge Y, Su L, Bu J (2015) Response of plasma membrane H+-ATPase in rice (Oryza sativa) seedlings to simulated acid rain. Environ Sci Pollut Res 22(1):535–545
Lim CW, Han SW, Hwang IS, Kim DS, Hwang BK, Lee SC (2015) The pepper lipoxygenase CaLOX1 plays a role in osmotic, drought and high salinity stress response. Plant Cell Physiol 56(5):930–942
Lin HX, Zhu MZ, Yano M, Gao JP, Liang ZW, Su WA, Hu XH, Ren ZH, Chao DY (2004) QTLs for Na+ and K+ uptake of the shoots and roots controlling rice salt tolerance. Theor Appl Genet 108(2):253–260
Liu CW, Chang TS, Hsu YK, Wang AZ, Yen HC, Wu YP, Wang CS, Lai CC (2014) Comparative proteomic analysis of early salt stress responsive proteins in roots and leaves of rice. Proteomics 14(15):1759–1775
Lovegrove A, Hooley R (2000) Gibberellin and abscisic acid signalling in aleurone. Trends Plant Sci 5:102–110
Lovejoy AF, Riordan DP, Brown PO (2014) Transcriptome-wide mapping of pseudouridines: pseudouridine synthases modify specific mRNAs in S. cerevisiae. PLoS One 9(10):e110799
Maas EV, Hoffman GJ (1977) Crop salt tolerance- current assessment. J Irrig Drain Div 103:115–134
Martz F, Sutinen ML, Kiviniemi S, Palta JP (2006) Changes in freezing tolerance, plasma membrane H+-ATPase activity and fatty acid composition in Pinus resinosa needles during cold acclimation and de-acclimation. Tree Physiol 26(6):783–790
Miernyk JA (2001) The J-domain proteins of Arabidopsis thaliana: an unexpectedly large and diverse family of chaperones. Cell Stress Chaperones 6(3):209
Morgan SH, Maity PJ, Geilfus CM, Lindberg S, Mühling KH (2014) Leaf ion homeostasis and plasma membrane H+-ATPase activity in Vicia faba change after extra calcium and potassium supply under salinity. Plant Physiol Biochem 82:244–253
Munns R (2011) Plant adaptations to salt and water stress: differences and commonalities. Adv Bot Res 57:1–32 Academic Press
Naveed SA, Zhang F, Zhang J, Zheng TQ, Meng LJ, Pang YL, Xu JL, Li ZK (2018) Identification of QTN and candidate genes for salinity tolerance at the germination and seedling stages in rice by genome-wide association analyses. Sci Rep 8(1):1
Nayyeripasand L, Garosi GA, Ahmadikhah A (2019) Selection for salinity tolerance in an international rice collection at vegetative stage. Aust J Crop Sci 13(6):837–846
Ouyang SQ, Liu YF, Liu P, Lei G, He SJ, Ma B, Zhang WK, Zhang JS, Chen SY (2010) Receptor-like kinase OsSIK1 improves drought and salt stress tolerance in rice (Oryza sativa) plants. Plant J 62(2):316–329
Palaisa KA, Morgante M, Williams M, Rafalski A (2003) Contrasting effects of selection on sequence diversity and linkage disequilibrium at two phytoene synthase loci. Plant Cell 15:1795–1806
Pandit A, Rai V, Bal S, Sinha S, Kumar V, Chauhan M, Gautam RK, Singh R, Sharma PC, Singh AK, Gaikwad K (2010) Combining QTL mapping and transcriptome profiling of bulked RILs for identification of functional polymorphism for salt tolerance genes in rice (Oryza sativa L.). Mol Gen Genomics 284(2):121–136
Prasad SR, Bagali PG, Hittalmani S, Shashidhar HE (2000) Molecular mapping of quantitative trait loci associated with seedling tolerance to salt stress in rice (Oryza sativa L.). Curr Sci 25:162–164
Price AH (2006) Believe it or not, QTLs are accurate! Trends Plant Sci 11:213–216
Pritchard JK, Stephens M, Rosenberg NA, Donnelly P (2000) Association mapping in structured populations. Am J Hum Genet 67:170–181
Rabbani MA, Maruyama K, Abe H, Khan MA, Katsura K, Ito Y, Yoshiwara K, Seki M, Shinozaki K, Yamaguchi-Shinozaki K (2003) Monitoring expression profiles of rice genes under cold, drought, and high-salinity stresses and abscisic acid application using cDNA microarray and RNA gel-blot analyses. Plant Physiol 133(4):1755–1767
Reddy INBL, Kim BK, Yoon IS, Kim KH, Kwon TR (2017) Salt tolerance in rice: focus on mechanisms and approaches. Rice Sci 24(3):123–144
Remington DL, Thornsberry JM, Matsuoka Y, Wilson LM, Whitt SR, Doebley J, Buckler ES (2001) Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc Natl Acad Sci 98(20):11479–11484
Risch N, Merikangas K (1996) The future of genetic studies of complex human diseases. Science 273(5281):1516–1517
Rosyara UR, Joshi BK (2012) Association mapping for improvement of quantitative traits in plant breeding populations. Nepal J Biotechnol 2(1):72–89
Roy SJ, Tucker EJ, Tester M (2011) Genetic analysis of abiotic stress tolerance in crops. Curr Opin Plant Biol 14:232–239
Roychoudhury A, Basu S, Sarkar SN, Sengupta DN (2008) Comparative physiological and molecular responses of a common aromatic indica rice cultivar to high salinity with non-aromatic indica rice cultivars. Plant Cell Rep 27(8):1395
Sato Y, Namiki N, Takehisa H, Kamatsuki K, Minami H, Ikawa H, Ohyanagi H, Sugimoto K, Itoh J, Antonio B, Nagamura Y (2013) RiceFREND: a platform for retrieving coexpressed gene networks in rice. Nucl Acids Res 41:D1214–D1221
Schwartz S, Bernstein DA, Mumbach MR, Jovanovic M, Herbst RH, León-Ricardo BX, Fink G (2014) Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell 159(1):148–162
Shigeoka S, Ishikawa T, Tamoi M, Miyagawa Y, Takeda T, Yabuta Y, Yoshimura K (2002) Regulation and function of ascorbate peroxidase isoenzymes. J Exp Bot 53(372):1305–1319
Singh R, Flowers T (2010) Physiology and molecular biology of the effects of salinity on rice, pp 899–939
Slatkin M (2008) Linkage disequilibrium-understanding the evolutionary past and mapping the medical future. Nat Rev Genet 9(6):477
Takehisa H, Shimodate T, Fukuta Y, Ueda T, Yano M, Yamaya T, Sato T (2004) Identification of quantitative trait loci for plant growth of rice in paddy field flooded with salt water. Field Crop Res 89(1):85–95
van der Feltz C, DeHaven AC, Hoskins AA (2018) Stress-induced pseudouridylation alters the structural equilibrium of yeast U2 snRNA stem II. J Mol Biol 430(4):524–536
Vitart V, Baxter I, Doerner P, Harper JF (2001) Evidence for a role in growth and salt resistance of a plasma membrane H+-ATPase in the root endodermis. Plant J 27(3):191–201
Walia H, Wilson C, Condamine P, Liu X, Ismail AM, Zeng L, Close TJ (2005) Comparative transcriptional profiling of two contrasting rice genotypes under salinity stress during the vegetative growth stage. Plant Physiol 139:822–835
Walia H, Wilson C, Zeng L, Ismail AM, Condamine P, Close TJ (2007) Genome-wide transcriptional analysis of salinity stressed japonica and indica rice genotypes during panicle initiation stage. Plant Mol Biol 63(5):609–623
Wang H, Zhang M, Guo R, Shi D, Liu B, Lin X, Yang C (2012) Effects of salt stress on ion balance and nitrogen metabolism of old and young leaves in rice (Oryza sativa L.). BMC Plant Biol 12:194
Wang WS, Zhao XQ, Li M, Huang LY, Xu JL, Zhang F, Cui YR, Fu BY, Li ZK (2015) Complex molecular mechanisms underlying seedling salt tolerance in rice revealed by comparative transcriptome and metabolomic profiling. J Exp Bot 67(1):405–419
Waziri A, Kumar P, Purty RS (2016) Saltol QTL and their role in salinity tolerance in rice. Austin J Biotechnol Bioeng 3(3):1067
Wildwater M, Campilho A, Perez-Perez JM, Heidstra R, Blilou I, Korthout H, Chatterjee J, Mariconti L, Gruissem W, Scheres B (2005) The RETINOBLASTOMA-RELATED gene regulates stem cell maintenance in Arabidopsis roots. Cell 123(7):1337–1349
Wu G, Xiao M, Yang C, Yu YT (2011) U2 snRNA is inducibly pseudouridylated at novel sites by Pus7p and snR81 RNP. EMBO J 30:79–89
Xu R, Yang Y, Qin R, Li H, Qiu C, Li L, Wei P, Yang J (2016) Rapid improvement of grain weight via highly efficient CRISPR/Cas9-mediated multiplex genome editing in rice. J Genet Genomics 43:529–532
Yan H, Jia H, Chen X, Hao L, An H, Guo X (2014) The cotton WRKY transcription factor GhWRKY17 functions in drought and salt stress in transgenic Nicotiana benthamiana through ABA signaling and the modulation of reactive oxygen species production. Plant Cell Physiol 55(12):2060–2076
Yan YS, Chen XY, Yang K, Sun ZX, Fu YP, Zhang YM, Fang RX (2011) Overexpression of an F-box protein gene reduces abiotic stress tolerance and promotes root growth in rice. Mol Plant 4(1):190–197
Zhang X, Morrissey C, Sun S, Ketchandji M, Nelson PS, True LD, Plymate SR (2011) Androgen receptor variants occur frequently in castration resistant prostate cancer metastases. PLoS One 6(11):e27970
Zhang YE, Xu W, Li Z, Deng XW, Wu W, Xue Y (2008) F-box protein DOR functions as a novel inhibitory factor for abscisic acid-induced stomatal closure under drought stress in Arabidopsis. Plant Physiol 148(4):2121–2133
Zörb C, Senbayram M, Peiter E (2014) Potassium in agriculture-status and perspectives. J Plant Physiol 171:656–669
Zou X, Neuman D, Shen QJ (2008) Interactions of two transcriptional repressors and two transcriptional activators in modulating gibberellin signaling in aleurone cells. Plant Physiol 148(1):176–186
Acknowledgments
This research was conducted in Biotechnology laboratory, Department of Plant Sciences and Biotechnology, Shahid Beheshti University, Iran. We thank Iran National Biotechnology Council (INBC) for partial financial support of the work. In addition we express grateful thanks to International Rice Research Institute (IRRI) in Philippines for providing the seeds of rice cultivars.
Funding
Iran National Biotechnology Council (INBC) partially supported the research.
Author information
Authors and Affiliations
Contributions
Leila Nayyeripasand performed experiments and prepared the draft of manuscript. Ghasem Ali Garoosi co-supervised the research and carried out phenotypic data analysis. Asadollah Ahmadikhah co-supervised the research, provided all samples used in the project and carried out the GWAS analysis. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics Approval and Consent to Participate
Not applicable as no animal experiments were involved in the entire project.
Consent for Publication
All the authors have given their consent to publish the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Rice varieties used in this study. Table S2. Analysis of variance for the studied traits in germplasm set of rice under normal and salinity stress conditions. Table S3. Pearson correlation coefficients between different traits under normal condition. Table S4. Pearson correlation coefficients between different traits under salinity condition. Table S5. Co-ancestory coefficients (Q) in 4 sub-populations for germplasm set used in the study. Table S6. Results of association mapping using MLM-Q-K model for detection of SNPs with significant association to the studied traits under normal condition. Table S7. Results of association mapping using MLM-Q-K model for detection of SNPs with significant association to the studied traits under salinity condition. Table S8. Approximate expression levels of candidate genes (in view of transcript per million, TPM) inferred from EST sources in Unigene database. Table S9. Haplotype analysis of candidate genes on rice chromosomes. Table S10. Co-expression analysis of candidate genes using riceFREND database. Table S11. Comparison of the locations of the detected genomic regions in this study (QTLs) and the known reported QTLs.
Additional file 2: Figure S1.
a. A histogram showing the frequency distribution of 8 traits phenotyped for germplasm set used in the study under control condition. b. A histogram showing the frequency distribution of 8 traits phenotyped for germplasm set used in the study under salinity condition. Figure S2 Results of clustering the entire population. (A) Determining the optimized number of K clusters based on Evanno et al. (2005) method. (B) Bar plot of the clustering result showing the population structure derived from the STRUCTURE software. Figure S3. Manhattan plots of p-values analyzed using mixed linear model (MLM) controlled for population structure and kinship of rice genotypes for derived traits under salinity condition.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nayyeripasand, L., Garoosi, G.A. & Ahmadikhah, A. Genome-Wide Association Study (GWAS) to Identify Salt-Tolerance QTLs Carrying Novel Candidate Genes in Rice During Early Vegetative Stage. Rice 14, 9 (2021). https://doi.org/10.1186/s12284-020-00433-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12284-020-00433-0