Astrocyte-Derived TGFβ1 Facilitates Blood–Brain Barrier Function via Non-Canonical Hedgehog Signaling in Brain Microvascular Endothelial Cells

 ,
,
Abstract
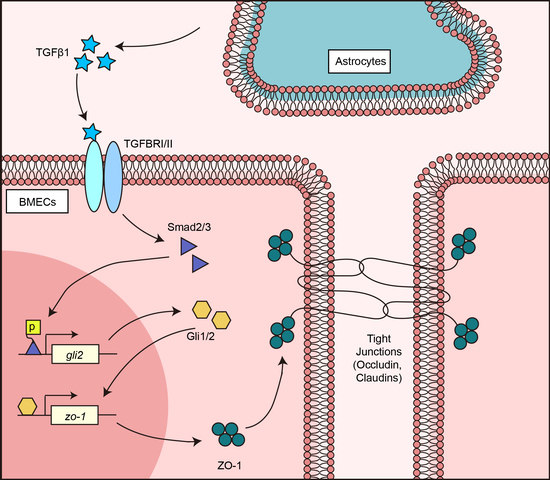
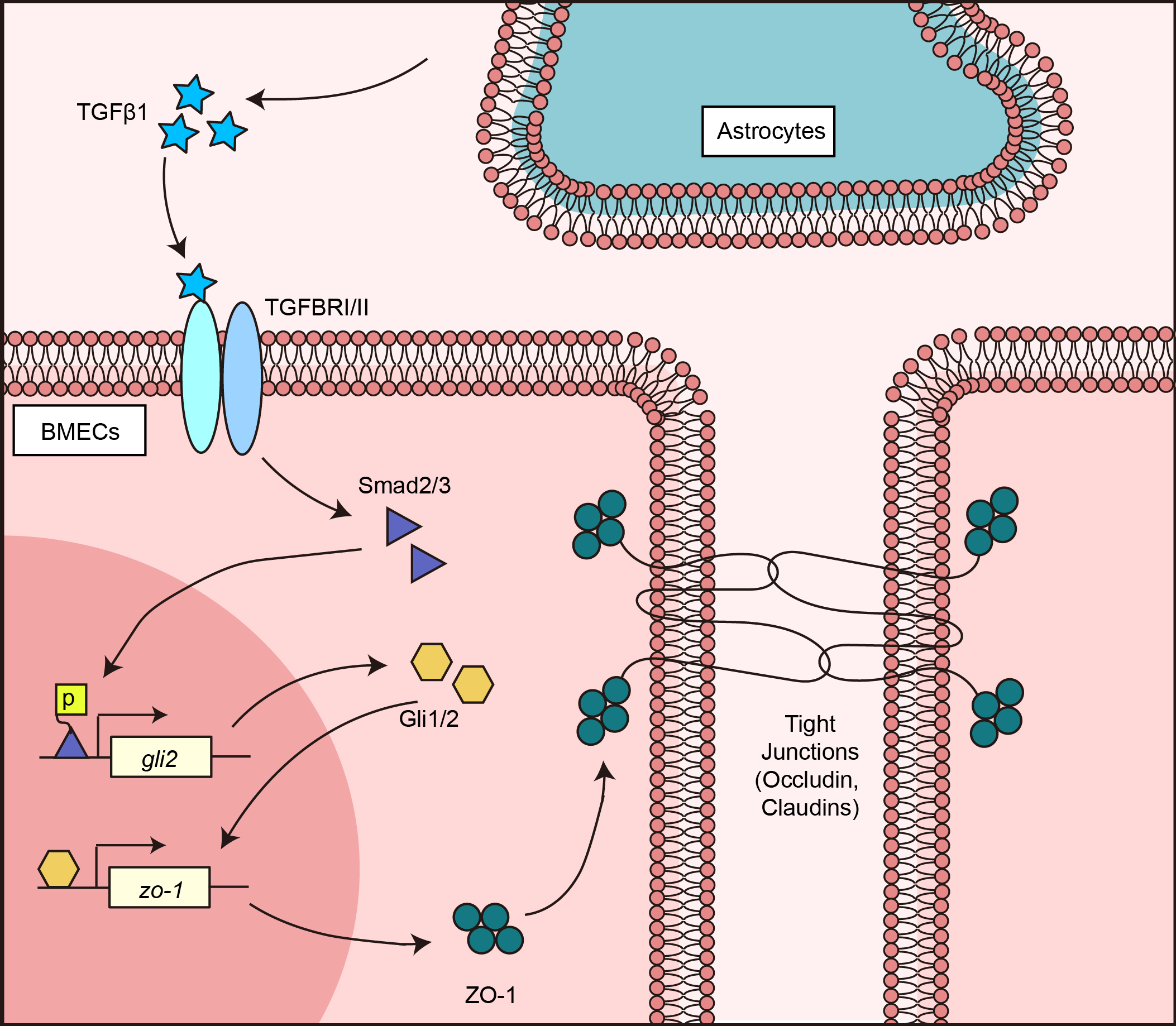
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Reagents and Antibodies
2.3. Mouse Assays
2.4. hBMECs and U251 Co-Cultivation
2.5. Western Blot
2.6. RT-PCR and qPCR
2.7. Enzyme-Linked Immunosorbent Assay (ELISA)
2.8. Immunofluorescence (IF)
2.9. Electric Cell-Substrate Impedance Sensing
2.10. CRISPR/Cas9 Genomic Editing
2.11. Transfection
2.12. Dual-Luciferase Reporter Assay
2.13. Chromatin Immunoprecipitation (ChIP)
2.14. Statistical Analysis
3. Results
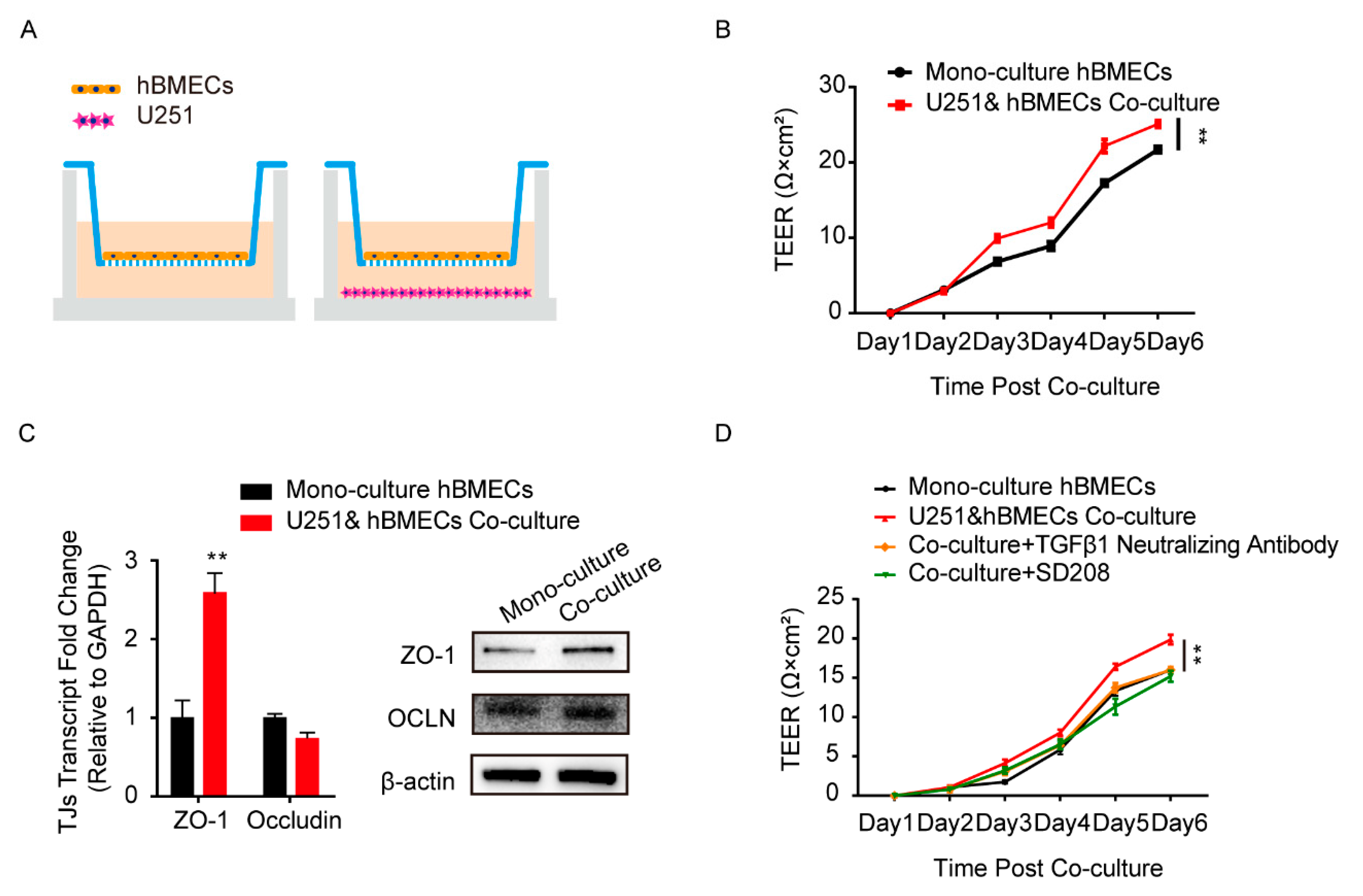
3.1. Astrocyte-Derived TGFβ1 Enhanced the Barrier Function of BMECs
3.2. TGFβ1 Promoted TJ Protein ZO-1 Expression via Smad2/3
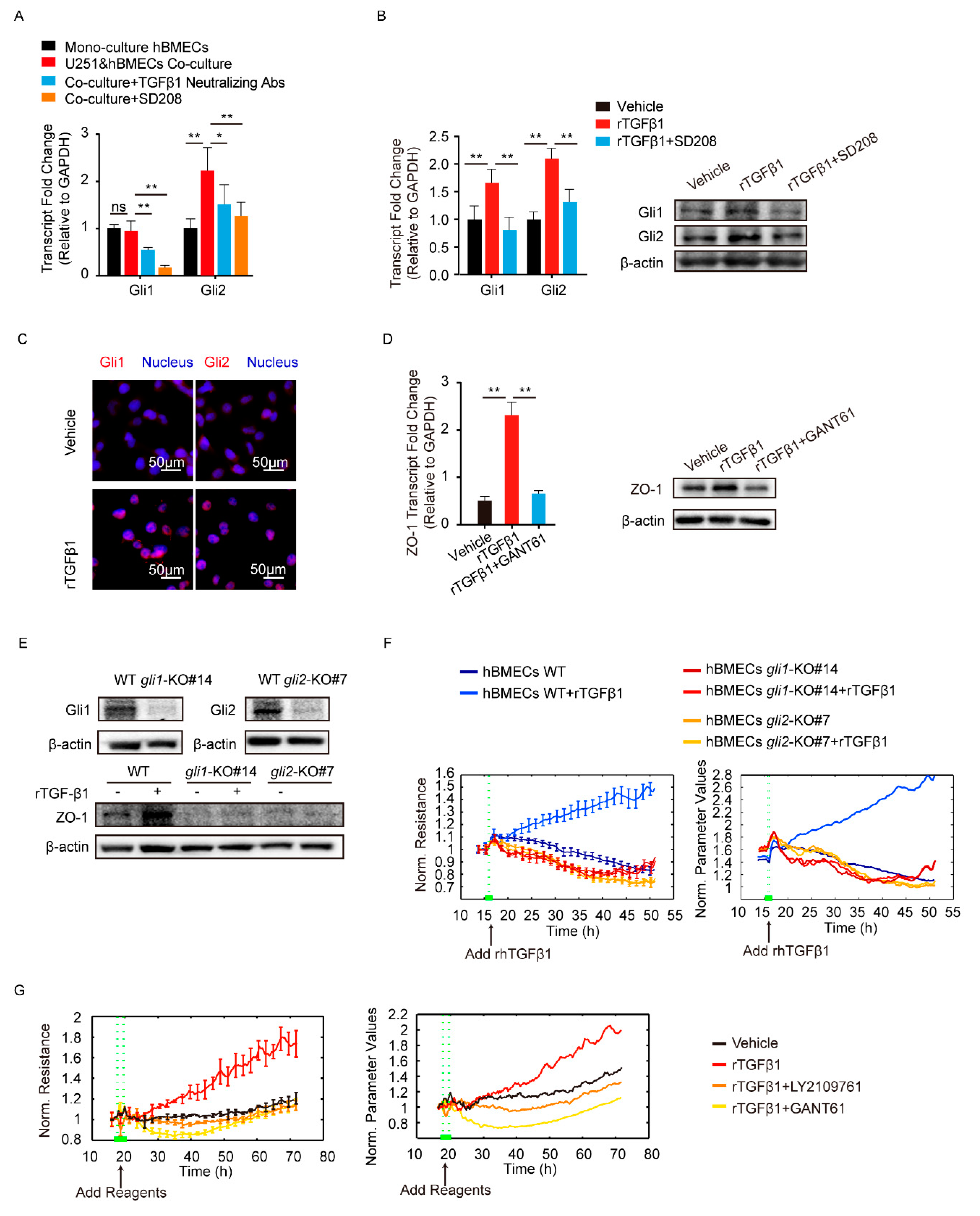
3.3. TGFβ1 Activated Non-Canonical Hedgehog Signaling to Enhance ZO-1 Expression
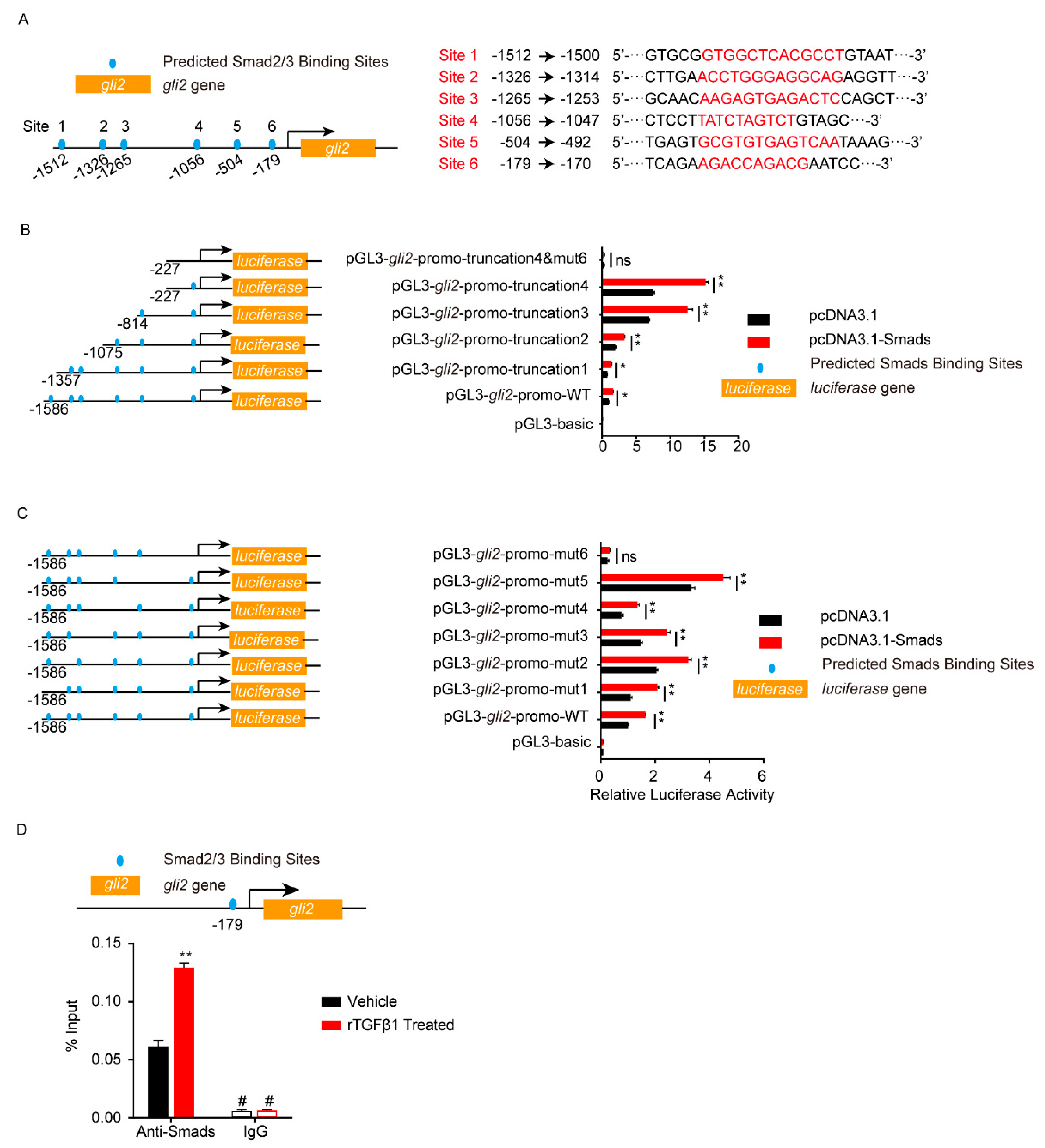
3.4. TGFβ1 Triggered the SMADS/Gli2/ZO-1 Regulatory Cascades
4. Discussion
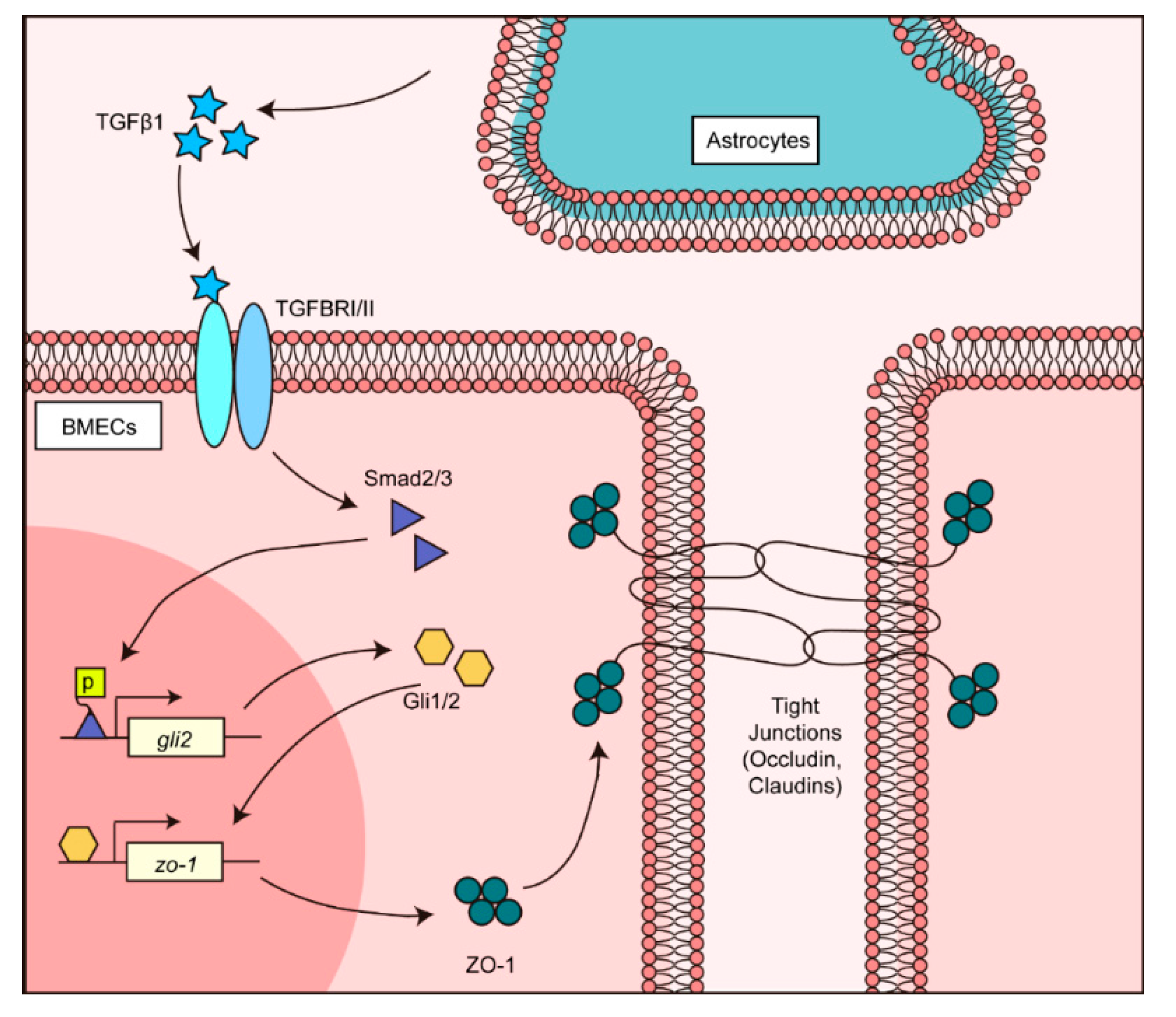
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Keaney, J.; Campbell, M. The dynamic blood-brain barrier. FEBS J. 2015, 282, 4067–4079. [Google Scholar] [CrossRef] [PubMed]
- Correale, J.; Villa, A. Cellular elements of the blood-brain barrier. Neurochem. Res. 2009, 34, 2067–2077. [Google Scholar] [CrossRef] [PubMed]
- Saunders, N.R.; Ek, C.J.; Habgood, M.D.; Dziegielewska, K.M. Barriers in the brain: A renaissance? Trends Neurosci. 2008, 31, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Edwards, V.L.; Wang, L.C.; Dawson, V.; Stein, D.C.; Song, W. Neisseria gonorrhoeae breaches the apical junction of polarized epithelial cells for transmigration by activating EGFR. Cell. Microbiol. 2013, 15, 1042–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dejana, E.; Tournier-Lasserve, E.; Weinstein, B.M. The control of vascular integrity by endothelial cell junctions: Molecular basis and pathological implications. Dev. Cell 2009, 16, 209–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballabh, P.; Braun, A.; Nedergaard, M. The blood-brain barrier: An overview: Structure, regulation, and clinical implications. Neurobiol. Dis. 2004, 16, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Tietz, S.; Engelhardt, B. Brain barriers: Crosstalk between complex tight junctions and adherens junctions. J. Cell Biol. 2015, 209, 493–506. [Google Scholar] [CrossRef] [Green Version]
- Haseloff, R.F.; Dithmer, S.; Winkler, L.; Wolburg, H.; Blasig, I.E. Transmembrane proteins of the tight junctions at the blood-brain barrier: Structural and functional aspects. Semin. Cell Dev. Biol. 2015, 38, 16–25. [Google Scholar] [CrossRef]
- Almutairi, M.M.; Gong, C.; Xu, Y.G.; Chang, Y.; Shi, H. Factors controlling permeability of the blood-brain barrier. Cell. Mol. Life Sci. 2016, 73, 57–77. [Google Scholar] [CrossRef]
- Igarashi, Y.; Utsumi, H.; Chiba, H.; Yamada-Sasamori, Y.; Tobioka, H.; Kamimura, Y.; Furuuchi, K.; Kokai, Y.; Nakagawa, T.; Mori, M.; et al. Glial cell line-derived neurotrophic factor induces barrier function of endothelial cells forming the blood-brain barrier. Biochem. Biophys. Res. Commun. 1999, 261, 108–112. [Google Scholar] [CrossRef]
- Haj-Yasein, N.N.; Vindedal, G.F.; Eilert-Olsen, M.; Gundersen, G.A.; Skare, O.; Laake, P.; Klungland, A.; Thoren, A.E.; Burkhardt, J.M.; Ottersen, O.P.; et al. Glial-conditional deletion of aquaporin-4 (Aqp4) reduces blood-brain water uptake and confers barrier function on perivascular astrocyte endfeet. Proc. Natl. Acad. Sci. USA 2011, 108, 17815–17820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wosik, K.; Cayrol, R.; Dodelet-Devillers, A.; Berthelet, F.; Bernard, M.; Moumdjian, R.; Bouthillier, A.; Reudelhuber, T.L.; Prat, A. Angiotensin II controls occludin function and is required for blood brain barrier maintenance: Relevance to multiple sclerosis. J. Neurosci. 2007, 27, 9032–9042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hata, A.; Chen, Y.G. TGF-β Signaling from Receptors to Smads. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Heldin, C.H. The regulation of TGF-β signal transduction. Development 2009, 136, 3699–3714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massague, J. TGF-β signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Liu, J.; Derynck, R. Post-translational regulation of TGF-β receptor and Smad signaling. FEBS Lett. 2012, 586, 1871–1884. [Google Scholar] [CrossRef] [Green Version]
- Weiss, A.; Attisano, L. The TGF-β superfamily signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 47–63. [Google Scholar] [CrossRef]
- Gomes, F.C.; Sousa Vde, O.; Romao, L. Emerging roles for TGF-β1 in nervous system development. Int. J. Dev. Neurosci. 2005, 23, 413–424. [Google Scholar] [CrossRef]
- Diniz, L.P.; Matias, I.; Siqueira, M.; Stipursky, J.; Gomes, F.C.A. Astrocytes and the TGF-β1 Pathway in the Healthy and Diseased Brain: A Double-Edged Sword. Mol. Neurobiol. 2018. [Google Scholar] [CrossRef]
- Aimaiti, Y.; Yusufukadier, M.; Li, W.; Tuerhongjiang, T.; Shadike, A.; Meiheriayi, A.; Abudusalamu, A.; Wang, H.; Tuerganaili, A.; Shao, Y.; et al. TGF-β1 signaling activates hepatic stellate cells through Notch pathway. Cytotechnology 2019, 71, 881–891. [Google Scholar] [CrossRef]
- Wang, Q.; Zhou, C.; Zhang, D.; Zou, J.; Liu, W.; Cai, L.; Cui, Y.; Lai, W.; Xie, J. The involvement of the ERK-MAPK pathway in TGF-β1-mediated connexin43-gap junction formation in chondrocytes. Connect. Tissue Res. 2019, 60, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Luo, K. Signaling Cross Talk between TGF-β/Smad and Other Signaling Pathways. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osterlund, T.; Kogerman, P. Hedgehog signalling: How to get from Smo to Ci and Gli. Trends Cell Biol. 2006, 16, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Dessaud, E.; McMahon, A.P.; Briscoe, J. Pattern formation in the vertebrate neural tube: A sonic hedgehog morphogen-regulated transcriptional network. Development 2008, 135, 2489–2503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haan, N.; Goodman, T.; Najdi-Samiei, A.; Stratford, C.M.; Rice, R.; El Agha, E.; Bellusci, S.; Hajihosseini, M.K. Fgf10-expressing tanycytes add new neurons to the appetite/energy-balance regulating centers of the postnatal and adult hypothalamus. J. Neurosci. 2013, 33, 6170–6180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, J.I.; Dodelet-Devillers, A.; Kebir, H.; Ifergan, I.; Fabre, P.J.; Terouz, S.; Sabbagh, M.; Wosik, K.; Bourbonniere, L.; Bernard, M.; et al. The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science 2011, 334, 1727–1731. [Google Scholar] [CrossRef] [Green Version]
- Javelaud, D.; Pierrat, M.J.; Mauviel, A. Crosstalk between TGF-β and hedgehog signaling in cancer. FEBS Lett. 2012, 586, 2016–2025. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Yao, Y.; Tsirka, S.E.; Cao, Y. Cell-culture models of the blood-brain barrier. Stroke 2014, 45, 2514–2526. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Liu, W.; Miao, L.; Yang, X.; Fu, J.; Dou, B.; Cai, A.; Zong, X.; Tan, C.; Chen, H.; et al. Induction of VEGFA and Snail-1 by meningitic Escherichia coli mediates disruption of the blood-brain barrier. Oncotarget 2016, 7, 63839–63855. [Google Scholar] [CrossRef] [Green Version]
- Szulcek, R.; Bogaard, H.J.; van Nieuw Amerongen, G.P. Electric cell-substrate impedance sensing for the quantification of endothelial proliferation, barrier function, and motility. J. Vis. Exp. 2014. [Google Scholar] [CrossRef] [Green Version]
- Abbott, N.J.; Ronnback, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nature reviews. Neuroscience 2006, 7, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Yuan, Y.; Wang, D.; Su, Z. Heterogeneous astrocytes: Active players in CNS. Brain Res. Bull. 2016, 125, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Suarez, E.; Caldwell, A.L.; Allen, N.J. Role of astrocyte-synapse interactions in CNS disorders. J. Physiol. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao-Cheng, J.H.; Nagy, Z.; Brightman, M.W. Tight junctions of brain endothelium in vitro are enhanced by astroglia. J. Neurosci. 1987, 7, 3293–3299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zacharek, A.; Chen, J.; Cui, X.; Li, A.; Li, Y.; Roberts, C.; Feng, Y.; Gao, Q.; Chopp, M. Angiopoietin1/Tie2 and VEGF/Flk1 induced by MSC treatment amplifies angiogenesis and vascular stabilization after stroke. J. Cereb. Blood Flow Metab. 2007, 27, 1684–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, N.D.; Correale, J.; Schreiber, S.S.; Fisher, M. Transforming growth factor-β mediates astrocyte-specific regulation of brain endothelial anticoagulant factors. Stroke 1999, 30, 1671–1678. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Grumet, M. BMP and LIF signaling coordinately regulate lineage restriction of radial glia in the developing forebrain. Glia 2007, 55, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Yosef, N.; Ubogu, E.E. GDNF restores human blood-nerve barrier function via RET tyrosine kinase-mediated cytoskeletal reorganization. Microvasc. Res. 2012, 83, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Chang, H.M.; Yi, Y.; Yao, Y.; Leung, P.C.K. TGF-β1 Increases GDNF Production by Upregulating the Expression of GDNF and Furin in Human Granulosa-Lutein Cells. Cells 2020, 9, 185. [Google Scholar] [CrossRef] [Green Version]
- Diemel, L.T.; Jackson, S.J.; Cuzner, M.L. Role for TGF-β1, FGF-2 and PDGF-AA in a myelination of CNS aggregate cultures enriched with macrophages. J. Neurosci. Res. 2003, 74, 858–867. [Google Scholar] [CrossRef]
- Racke, M.K.; Dhib-Jalbut, S.; Cannella, B.; Albert, P.S.; Raine, C.S.; McFarlin, D.E. Prevention and treatment of chronic relapsing experimental allergic encephalomyelitis by transforming growth factor-β1. J. Immunol. 1991, 146, 3012–3017. [Google Scholar] [PubMed]
- Johns, L.D.; Flanders, K.C.; Ranges, G.E.; Sriram, S. Successful treatment of experimental allergic encephalomyelitis with transforming growth factor-β1. J. Immunol. 1991, 147, 1792–1796. [Google Scholar] [PubMed]
- Zoller, T.; Schneider, A.; Kleimeyer, C.; Masuda, T.; Potru, P.S.; Pfeifer, D.; Blank, T.; Prinz, M.; Spittau, B. Silencing of TGF-β signalling in microglia results in impaired homeostasis. Nat. Commun. 2018, 9, 4011. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Yang, G.Y.; Ahlemeyer, B.; Pang, L.; Che, X.M.; Culmsee, C.; Klumpp, S.; Krieglstein, J. Transforming growth factor-β1 increases bad phosphorylation and protects neurons against damage. J. Neurosci. 2002, 22, 3898–3909. [Google Scholar] [CrossRef] [PubMed]
- Westerhausen, D.R., Jr.; Hopkins, W.E.; Billadello, J.J. Multiple transforming growth factor-β-inducible elements regulate expression of the plasminogen activator inhibitor type-1 gene in Hep G2 cells. J. Biol. Chem. 1991, 266, 1092–1100. [Google Scholar]
- Buisson, A.; Nicole, O.; Docagne, F.; Sartelet, H.; Mackenzie, E.T.; Vivien, D. Up-regulation of a serine protease inhibitor in astrocytes mediates the neuroprotective activity of transforming growth factor-β1. FASEB J. 1998, 12, 1683–1691. [Google Scholar] [CrossRef] [Green Version]
- Finco, I.; LaPensee, C.R.; Krill, K.T.; Hammer, G.D. Hedgehog signaling and steroidogenesis. Annu. Rev. Physiol. 2015, 77, 105–129. [Google Scholar] [CrossRef] [Green Version]
- Byrd, N.; Grabel, L. Hedgehog signaling in murine vasculogenesis and angiogenesis. Trends Cardiovasc. Med. 2004, 14, 308–313. [Google Scholar] [CrossRef]
- Kim, J.; Wang, S.; Lee, C.; Sung, S.; Shin, Y.; Song, K.S.; Cha, H.J.; Ock, M.; Jung, Y. Blood-Stage Plasmodium Berghei ANKA Infection Promotes Hepatic Fibrosis by Enhancing Hedgehog Signaling in Mice. Cell. Physiol. Biochem. 2018, 50, 1414–1428. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, J.; Li, L.; Huo, D.; Zhi, S.; Yang, R.; Yang, B.; Xu, B.; Zhang, T.; Dai, M.; Tan, C.; et al. Astrocyte-Derived TGFβ1 Facilitates Blood–Brain Barrier Function via Non-Canonical Hedgehog Signaling in Brain Microvascular Endothelial Cells. Brain Sci. 2021, 11, 77. https://doi.org/10.3390/brainsci11010077
Fu J, Li L, Huo D, Zhi S, Yang R, Yang B, Xu B, Zhang T, Dai M, Tan C, et al. Astrocyte-Derived TGFβ1 Facilitates Blood–Brain Barrier Function via Non-Canonical Hedgehog Signaling in Brain Microvascular Endothelial Cells. Brain Sciences. 2021; 11(1):77. https://doi.org/10.3390/brainsci11010077
Chicago/Turabian StyleFu, Jiyang, Liang Li, Dong Huo, Shuli Zhi, Ruicheng Yang, Bo Yang, Bojie Xu, Tao Zhang, Menghong Dai, Chen Tan, and et al. 2021. "Astrocyte-Derived TGFβ1 Facilitates Blood–Brain Barrier Function via Non-Canonical Hedgehog Signaling in Brain Microvascular Endothelial Cells" Brain Sciences 11, no. 1: 77. https://doi.org/10.3390/brainsci11010077