
Abstract
Neuronal apoptosis has an important role in early brain injury (EBI) following subarachnoid hemorrhage (SAH). TRAF3 was reported as a promising therapeutic target for stroke management, which covered several neuronal apoptosis signaling cascades. Hence, the present study is aimed to determine whether downregulation of TRAF3 could be neuroprotective in SAH-induced EBI. An in vivo SAH model in mice was established by endovascular perforation. Meanwhile, primary cultured cortical neurons of mice treated with oxygen hemoglobin were applied to mimic SAH in vitro. Our results demonstrated that TRAF3 protein expression increased and expressed in neurons both in vivo and in vitro SAH models. TRAF3 siRNA reversed neuronal loss and improved neurological deficits in SAH mice, and reduced cell death in SAH primary neurons. Mechanistically, we found that TRAF3 directly binds to TAK1 and potentiates phosphorylation and activation of TAK1, which further enhances the activation of NF-κB and MAPKs pathways to induce neuronal apoptosis. Importantly, TRAF3 expression was elevated following SAH in human brain tissue and was mainly expressed in neurons. Taken together, our study demonstrates that TRAF3 is an upstream regulator of MAPKs and NF-κB pathways in SAH-induced EBI via its interaction with and activation of TAK1. Furthermore, the TRAF3 may serve as a novel therapeutic target in SAH-induced EBI.
Similar content being viewed by others

Introduction
Subarachnoid hemorrhage (SAH) is a major cause of morbidity and mortality in neurosurgery practice1,2. Early brain injury (EBI) is regarded as the main factor for poor prognosis after SAH3,4. Neuronal apoptosis has been demonstrated to have a vital role in EBI5,6. Although inhibition of a single pathway could reduce neuronal apoptosis and improve the prognosis in experimental SAH, few of these research results have been translated into clinical practice7,8. Accordingly, targeting multiple neuronal apoptosis pathways simultaneously is currently considered to be a novel treatment strategy for EBI.
Mitogen-activated protein kinases (MAPKs) involving c-Jun-N-terminal kinases (JNK), P38, and nuclear factor-kappa B (NF-κB) signaling pathways have been demonstrated to have an important role in the development of EBI after SAH9,10,11. Specifically, they have been shown to aggravate brain damage by initiating apoptotic pathways11. Therefore, therapies that can regulate both MAPKs and NF-κB pathways are superior to single-target agents.
The tumor necrosis factor receptor-associated factor (TRAF) family has been shown to mediate signal transduction of the IL-1 superfamily, TLRs, and TNF receptors12. TRAF3, one of the seven members of the TRAF family, deemed as a general immune-related signal transduction regulator, has a variety of biological functions. In some neurological diseases, TRAF3 serves its multiple purposes via interacting with diverse regulatory proteins, kinases, and receptors13. Previous research has revealed that neuronal expression of TRAF3 could be induced by ischemic injury, and TRAF3 activated the NF-κB and JNK pathways through regulation of TAK114. Our previous research has demonstrated that TAK1 was a key upstream mediator of neuronal apoptosis in EBI following SAH15. Therefore, we hypothesize that TRAF3 might also have an important role in regulating the complex pathological mechanisms of SAH-induced EBI.
Materials and methods
Animals
Healthy adult male C57BL/6J mice (22–25 g) and pregnant C57BL/6J mice at 13 days of gestation were purchased from the Nanjing Biomedical Research Institute of Nanjing University. All the experiments were carried out under the guidance of the Guide for the Care and Use of Laboratory Animals which was published by NIH and was approved by the Committee for Animal Experimentation at the Drum Tower Hospital.
SAH model
The endovascular perforation SAH model was used as previously described16. A sharpened prolene filament (Ethicon, Inc., Taipei, Taiwan) was inserted into the intracranial ICA via the cervical ICA and then advanced 3 mm further to puncture the bifurcation-level of ICA while the mice were under isoflurane anesthesia (2% in oxygen gas, 400 ml/min). The animals in the sham group underwent similar operations, with the omission of ICA puncture.
Exclusion criteria
The neurological score was blindly evaluated by an independent observer using a Modified Garcia scale score 24 h post-SAH17. Mice with a Modified Garcia scale score ≤6 or ≥15 were excluded17.
Experimental protocol
All mice were randomly assigned to the following experiments as described (Fig. 1).

The experimental designs in vivo (A) and in vitro (B). WB western blotting, IF immunofluorescence staining, MGS Modified Garcia Scale, siRNA control siRNA, and TRAF3 siRNA; Co-IP co-immunoprecipitation; adenovirus AdTRAF3, AdTRAF3-M, and AdGFP.
Experimental design 1
Animals were randomly assigned to six groups: sham (n = 12), 6 h post-SAH (n = 6), 12 h post-SAH (n = 6), 24 h post-SAH (n = 12), 48 h post-SAH (n = 6), and 72 h post-SAH (n = 6). At the scheduled time points, tissue was harvested for evaluation of TRAF3 expression (n = 6 each). Cellular distribution of TRAF3 was determined by double-staining immunofluorescence in 24 h post-SAH and sham groups (n = 6 each).
Experimental design 2
Mice were randomly allocated into six groups: (1) sham group (n = 6), in which mice underwent surgery without ICA puncture; (2) SAH group (n = 6), in which mice underwent ICA puncture; (3) SAH + control siRNA group (n = 6), in which mice underwent ICA puncture and intraventricular injection of control siRNA; (4) SAH + 125 pmol of TRAF3 siRNA group (n = 6), in which mice underwent ICA puncture and intraventricular injection of 125 pmol of TRAF3 siRNA; (5) SAH + 250 pmol of TRAF3 siRNA group (n = 6), in which mice underwent ICA puncture and intraventricular injection of 250 pmol of TRAF3 siRNA; (6) SAH + 500 pmol of TRAF3 siRNA group (n = 6), in which mice underwent ICA puncture and intraventricular injection of 500 pmol of TRAF3 siRNA. Brain tissue was harvested for evaluation of TRAF3 expression 24 h post-SAH via western blotting.
Experimental design 3
Mice were randomly allocated into four groups: (1) sham group (n = 22), in which mice underwent surgery without ICA puncture; (2) SAH group (n = 22), in which mice underwent ICA puncture; (3) SAH + control siRNA group (n = 22), in which mice underwent ICA puncture and intraventricular injection of control siRNA; (4) SAH + TRAF3 siRNA (n = 22), in which mice underwent ICA puncture and intraventricular injection of 250 pmol of TRAF3 siRNA. Out of each group, 12 mice were sacrificed to carry out western blotting and TUNEL staining to determine the effect of TRAF3 in SAH-induced EBI 24 h post-SAH. The remaining mice were assessed using the rotarod test and Modified Garcia scale score on days 3 and 7 following SAH and then sacrificed prior to Nissl staining.
Primary neuronal cell culture
Primary neuronal cell cultures were prepared from the cortex of fetal C57BL/6 J mice at 13 days of gestation as a modification of a previously described method18. Cells were seeded in poly-d-lysine-coated plates (Sigma, Darmstadt, Germany) with neurobasal medium containing 0.5 mM GlutaMax-I (Gibco Company, USA) and 2% B27 supplement (Gibco Company, USA). The medium was replaced 4 h after seeding. Subsequently, half of the medium was replaced every 3 days following seeding. For the in vitro SAH model, cells were incubated with hemoglobin (Millipore Sigma, Darmstadt, Germany) for 24 h at a concentration of 25 μM. Primary neuronal cell cultures were randomly assigned to different groups.
Patient recruitment and brain tissue collection
All patients in the study were admitted to the Department of Neurosurgery of the Nanjing Drum Tower Hospital between January 2017 and July 2019. Clinical variables are shown in Tables 1 and 2. A total of 4 patients with deep brain tumors that needed surgical resection were selected as control. The samples were obtained from the pathway during surgical removal of the deep tumors. In addition, 4 SAH patients with encephalocele who required surgery were recruited. Written informed consent was obtained from all patients or their family members. The study was conducted in accordance with the Declaration of Helsinki and was approved by the Ethics Committee of Drum Tower Hospital. The team blinded to the clinical parameters of the patients performed the immunofluorescence analysis.
SiRNA transfection
Transfection of siRNA with i-Fect siRNA Delivery Kits (NI35150, Neuromics) was carried out to achieve TRAF3 downregulation. The TRAF3 siRNA (SC-36712, Santa Cruz Biotech) and control siRNA (SC-37007, Santa Cruz Biotech) were mixed with i-Fect siRNA Delivery Kits according to the manufacturer’s instructions. In the in vivo experiments, the siRNA was injected into the right lateral ventricles using a Hamilton microsyringe. In the in vitro experiments, the siRNA was mixed with neurobasal medium. The siRNA was transfected 72 h before the SAH inducing procedure19.
Adenoviral vector construction and neuronal infection
We constructed adenoviruses carrying sequences encoding mouse TRAF3 (AdTRAF3), mutated TRAF3 with a mutant 267–376 aa domain (AdTRAF3-M)14. AdGFP was used as control. Primary neurons were infected with the adenovirus at a multiplicity of infection (MOI) of 100 for 48 h prior to SAH.
Western blotting
Brain tissue and primary neural cells were harvested and then homogenized and lysed with RIPA buffer (ThermoFisher Scientific) and phosphatase inhibitor (ThermoFisher Scientific) as previously described. Protein extracts were resolved by 7.5%~12.5% SDA-PAGE (EpiZyme Scientific) and transferred to the Immobilon-FL membranes (Millipore Sigma, Darmstadt, Germany). Then, membranes were incubated with the following primary antibodies at 4°C overnight: anti-TRAF3 (1:500, G-6, Santa Cruz Biotech), anti-P38 (1:1000, 8690, Cell Signaling Technology), anti-P44/42 (1:1000, 4695, Cell Signaling Technology), anti-JNK (1:1000, 9252, Cell Signaling Technology), anti-p-P38 (1:1000, 4511, Cell Signaling Technology), anti-p-P44/42 (1:1000, 4370, Cell Signaling Technology), anti-p-JNK (1:1000, 4668, Cell Signaling Technology), anti-P65 (1:1000, 8242, Cell Signaling Technology), anti-p-P65 (1:1000, 3033, Cell Signaling Technology), anti-IκBα (1:1000, 4812, Cell Signaling Technology), anti-Bax (1:1000, 5023, Cell Signaling Technology), anti-Bcl-xL (1:1000, 2764, Cell Signaling Technology) anti-caspase-3 (1:1000, 9662, Cell Signaling Technology), anti-cleaved caspase-3 (1:1000, 9661, Cell Signaling Technology), anti-TAK1 (1:500, SC-7916, Santa Cruz Biotech), anti-p-TAK1 (1:1000, AF3019, Affinity Biosciences), and anti-GAPDH (1:1000, 5147, Cell Signaling Technology). Then, membranes were incubated with corresponding secondary antibodies (Bioworld). Membranes were visualized using an enhanced chemiluminescence kit (Pekin Elmer, Waltham, MA, USA). The relative band intensities were quantified using ImageJ (National Institutes of Health, Bethesda, MD, USA) software and normalized against GAPDH20,21.
Immunofluorescence
Mice were perfused with PBS and 4% paraformaldehyde after anesthetization. Human brain tissue was postfixed in paraformaldehyde immediately after collection for immunofluorescence staining. Sections of 10 μm were collected from brains after being postfixed in paraformaldehyde for 24 h. Primary neurons seeded in 24-well plates were postfixed in paraformaldehyde for 30 min for immunofluorescence analysis. Cells were stained for TRAF3 (1:100, AB36988, Abcam), NeuN (1:200, ABN78, Millipore), Iba1 (1:200, AB5076, Abcam), GFAP (1:200, AB7260, Abcam) overnight at 4 °C. Primary antibodies were visualized using corresponding secondary antibodies (1:200, Millipore). Immunofluorescence images were captured using the microscope (ZEISS, HB050, Germany)22.
Modified Garcia scale
The Modified Garcia scale contains 6 tests covering spontaneous activity and movement of the four limbs, forepaw outstretching, climbing, body proprioception, and response to whisker stimulation. The 18-point scoring system was applied to assess the functional defects at 1, 3, and 7 days post-SAH by an observer who was blind to groups23.
Rotarod test
The rotarod test was conducted to measure the motor coordination of mice 3 and 7 days post-SAH by an observer who was blind to groups as previously described24. Animals were placed on a non-skid rotating rod measuring 3 cm in diameter. The latency (RR time) to fall was measured at 30 rpm for up to 5 min after 6 training sessions given during 3 days24.
TUNEL staining
For terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining, a TUNEL detection kit (EpiZyme Scientific) was used according to the manufacturer’s instructions. After incubation with a primary antibody against NeuN (1:200, ABN78, Millipore) at 4 °C overnight, the coverslips were incubated with the TUNEL reaction mixture for 45 min and then counterstained using DAPI (Sigma). The positive cells were identified, counted, and analyzed with a fluorescence microscope by two investigators blinded to the grouping.
Nissl staining
Nissl staining was performed as described in our previous study25. Brain sections were stained with cresyl violet and then hydrated in 1% toluidine blue for 10 min. After washing with double-distilled water and mounting with Permount Mounting Medium, the mean number of intact and normal neurons in the views was counted. Six sections from each animal and 6 random high-power views (at ×400 magnification) were chosen for quantification.
Lactate dehydrogenase (LDH) assay
LDH release detected with the LDH Cytotoxicity Assay Kit (88953, ThermoFisher Scientific) was used to quantify SAH-induced neuronal injury at 24 h post-SAH quantitatively according to the manufacturer’s instructions. Neurons seeded in 96-well plates were incubated with kit reagents. Then, the absorbance at 490 and 680 nm were measured, and LDH units per sample were calculated and expressed as a ratio of LDH released/LDH total (LDH released + LDH extracted post lysis).
Immunoprecipitation
SAH neurons were harvested and lysed with RIPA buffer (ThermoFisher Scientific). The extracts were precleared and then incubated with 1 μg of TRAF3 antibody (G-6 Santa Cruz Biotech), TAK1 antibody (SC-7916, Santa Cruz Biotech), normal mouse IgG (sc-2025, Santa Cruz Biotech) and 10 μl of protein A/G-agarose (Millipore Sigma, Darmstadt, Germany) beads on a rocking platform at 4 °C overnight. The immunocomplex was collected, washed, and blotted using the TAK1 antibody (1:500, SC-7916, Santa Cruz Biotech) and TRAF3 antibody (1:500, G-6 Santa Cruz Biotech)14.
Statistical analysis
Statistical analysis was performed using Prism (GraphPad Software, La Jolla, CA, USA). All data were first tested for normality via Kolmogorov–Smirnov test. One-way ANOVA analysis was used to analyze differences among multiple groups, followed by Tukey’s post hoc test. The TRAF3 expression of immunofluorescence image was statistically analyzed using the Student’s t-test. Values of p < 0.05 were considered as statistically significant.
Results
Mortality and exclusion
The overall mortality of SAH mice was 17.5% (28/160). No mice died in the sham group. According to the modified Garcia scores on postoperative day 1, a total of 9 mice were excluded from this study (Table 3).
Expression of TRAF3 in the cortex after SAH
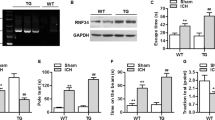
We observed an upregulation of TRAF3 protein expression in temporal cortex samples of SAH mice. The western blotting analysis showed a statistically significant increase of TRAF3 protein expression at 12 h, followed by a gradual decrease after 24 h, and a return to normal levels at 72 h (Fig. 2A, B). This result was confirmed by double immunofluorescence staining. Moreover, our results showed that TRAF3 was mainly expressed in neurons, with sparse expression in microglia and no expression in astrocytes (Fig. 2E–H).

A Western blotting analysis revealed that TRAF3 expression levels increased post-SAH. B Quantitative analysis of TRAF3/GAPDH. N = 6 mice per group. *p < 0.05 versus the sham group, #P < 0.05 versus the SAH 48 h group. C, D Western blotting analysis revealed that the expression of TRAF3 in SAH + 125 pmol of TRAF3 siRNA group showed no significant change compared with SAH + control siRNA group. However, 250 and 500 pmol of TRAF3 siRNA significantly reduced the TRAF3 expression. GAPDH served as a loading control. N = 6 mice per group. *p < 0.05 versus the SAH group. E Representative double immunofluorescence images showed that TRAF3 expression was elevated 24 h following SAH and was mainly expressed in neurons, with sparse expression in microglia and no expression in astrocytes. Scale bar: 50 μm. The TRAF3 positive index of neurons (F), microglia (G), and astrocytes (H). N = 6 mice per group. *p < 0.05 versus the sham group.
TRAF3 siRNA reduced TRAF3 expression in SAH
To evaluate the role of TRAF3 in EBI after SAH, TRAF3 siRNA was used to reduce the expression of TRAF3 in SAH mice. The levels of TRAF3 were increased in the SAH and SAH + control siRNA groups in comparison to the sham group. Moreover, the expression of TRAF3 in the SAH + 125 pmol of the TRAF3 siRNA treatment group showed no significant difference compared to the SAH + control siRNA group. However, both 250 pmol and 500 pmol of TRAF3 siRNA did significantly downregulate TRAF3 expression in SAH. There was no significant difference between the SAH + 125 pmol of TRAF3 siRNA group and SAH + 250 pmol of TRAF3 siRNA group (Fig. 2C, D). These data led us to use 250 pmol of TRAF3 siRNA in the subsequent experiments.
Downregulation of TRAF3 attenuated neuronal apoptosis in EBI
Since neuronal apoptosis is a major pathological process in EBI, western blotting was used to examine protein expression of indicators of early-stage apoptosis, including Bcl-xL, Bax, caspase-3, and cleaved caspase-3 (Fig. 3A–E). The protein levels of Bcl-xL and caspase-3 were significantly decreased, and the protein levels of Bax and cleaved caspase-3 were significantly increased in the SAH and SAH + control siRNA groups compared with the sham group. However, treatment with TRAF3 siRNA significantly reduced Bax and cleaved caspase-3 expression, meanwhile, increased Bcl-xL and caspase-3 expression. Additionally, TUNEL staining was used to assess temporal neuronal apoptosis at 24 h following SAH. As shown in Fig. 3F, G, the number of TUNEL-positive cells in the temporal lobe was increased in the SAH and SAH + control siRNA groups. However, downregulation of TRAF3 decreased the number of TUNEL-positive cells in SAH mice. To further analyze whether downregulation of TRAF3 could have an effect on the longer-term improvement of neurons, Nissl staining was used to assess neuron loss at 7 d after SAH. The results showed that the percentage of necrotic neurons in SAH mice treated with TRAF3 siRNA was greatly reduced at 7 d following SAH compared to SAH and SAH + control siRNA groups (Fig. 3H, I).

A Representative western blot images of Bcl-xL, Bax, Caspase-3, and Cleaved caspase-3. Quantitative analysis showed that downregulation of TRAF3 increased the expression of Bcl-xL (B) and Caspase-3 (D), while decreased the expression of Bax (C) and Cleaved caspase-3 (E) compared with SAH and SAH + control siRNA groups. N = 6 per mice group. *p < 0.05 versus the sham group, #p < 0.05 versus the SAH group. F Double staining of TUNEL (red) and NeuN (green); nuclei were counterstained with DAPI (blue). Scale bar: 100 μm. G The TUNEL-positive index of neuron in the temporal lobe was increased in the SAH and SAH + control siRNA groups, TRAF3 siRNA treatment significantly decreased the number of TUNEL-positive cells in SAH mice at 24 h post-SAH. N = 6 mice per group. *p < 0.05 versus the sham group, #p < 0.05 versus the SAH group. H, I Nissl staining results showed that TRAF3 siRNA treatment significantly decreased the percentage of necrotic neurons at 7 days post-SAH. Scale bar: 20 μm. N = 6 mice per group. *p < 0.05 versus the sham group, #p < 0.05 versus the SAH group. J Downregulation of TRAF3 could ameliorate motor coordination scores on days 3 and 7 post-SAH. K The RR time on days 3 and 7 post-SAH in the SAH and SAH + control siRNA groups were lower compared to the sham group. The RR time was increased by TRAF3 siRNA treatment. N = 10 mice per group. Data represent mean ± SEM. *p < 0.05 versus the sham group, #p < 0.05 versus the SAH group.
Downregulation of TRAF3 improved neurological behavior in SAH mice
The Modified Garcia score was used to evaluate the neurological function at postoperative days 3 and 7. The results showed that the Modified Garcia scores of the SAH and SAH + control siRNA groups were lower compared to the sham group. Moreover, the Modified Garcia score was significantly increased in the TRAF3 siRNA treatment group (Fig. 3J). The effects of TRAF3 downregulation on motor coordination were assessed using the rotarod test. As shown in Fig. 3K, SAH mice demonstrated significant impairment of motor coordination compared to the sham group. In contrast, downregulation of TRAF3 ameliorated motor coordination on postoperative days 3 and 7.
Downregulation of TRAF3 inhibited SAH-induced activation of a MAPKs signaling pathway in vivo
To further explore the molecular mechanism of neuroprotection by downregulation of TRAF3, western blotting was used to evaluate protein levels of MAPK family members and their phosphorylation, including P38, P44/42, JNK, p-P38, p-P44/42, and p-JNK. A significant increase in the expression levels of p-P38, p-P44/42, and p-JNK was observed in the SAH group in comparison with the sham group. In addition, TRAF3 siRNA treatment significantly reduced the expressions of p-p38, p-p44/42, and p-JNK (Fig. 4A–D).

A Western blotting of MAPKs at 24 h after SAH. Quantitative analysis results revealed that SAH increased the expression of p-P38 (B), p-P44/42 (C), and p-JNK (D). TRAF3 siRNA treatment significantly reduced the expression of p-P38, p-P44/42, and p-JNK. E Western blotting of the NF-κB signal pathway at 24 h after SAH. The protein expression levels of NF-κB nuclear subunits p-P65 (F) and P65 (G) were increased in SAH and SAH + control siRNA groups compared with the sham group and downregulated in the SAH + TRAF3 siRNA group. The expression of IκBα (H) was increased in the sham group compared to the SAH and SAH + control siRNA groups; it was substantially reduced and reversed in the SAH + TRAF3 siRNA group. N = 6 mice per group. *p < 0.05 versus the sham group, #p < 0.05 versus the SAH group.
Downregulation of TRAF3 inhibited SAH-induced activation of NF-κB signaling pathway in vivo
Previous studies have demonstrated that the NF-κB pathway has an essential role in the SAH-induced EBI pathogenesis8,10. To determine whether the NF-κB signaling pathway is involved in downregulation of TRAF3 following SAH, the expression of IκBα, an inhibitor of NF-κB, was investigated using western blotting. The protein level of IκBα was high in the sham group, whereas it was reduced in the SAH and SAH + control siRNA groups. However, the IκBα protein level of the SAH + TRAF3 siRNA group was higher than SAH and SAH + control siRNA groups. Moreover, the expression levels of P65 and p-P65 were increased in SAH and SAH + control siRNA groups compared with the sham group. However, P65 and p-P65 levels were also decreased in the SAH + TRAF3 siRNA group (Fig. 4E–H).
Expression of TRAF3 in primary neuronal cells after SAH
We used hemoglobin to mimic the condition of SAH in primary neurons to determine whether TRAF3 would have an effect on SAH-induced neuronal apoptosis in vitro. Western blot analysis showed that the level of TRAF3 was upregulated at 12 h after SAH and reached a peak at 24 h following SAH (Fig. 5A, B). Additionally, this result was confirmed by double immunofluorescence staining (Fig. 5C, D).

Western blotting analysis represented that the level of TRAF3 increased significantly at 24 h following SAH (A, B). N = 6 wells per group. *p < 0.05 versus the control group, #p < 0.05 versus the SAH 48 h group. C, D Overlapped images with TRAF3 and NeuN demonstrated that TRAF3 expression was elevated after SAH. Scale bar: 50 μm. N = 6 wells per group. *p < 0.05 versus control group. E, F Western blotting analysis showed that the level of TRAF3 could be downregulated via TRAF3 siRNA after SAH. N = 6 wells per group. *p < 0.05 versus the control group, #p < 0.05 versus the SAH group. G Representative western blot images and quantitative analyses showed that downregulation of TRAF3 increased the expression of Bcl-xL (H), Caspase-3 (J) and decreased the expression of Bax (I) and C Hayakawa leaved caspase-3 (K) compared to SAH and SAH + control siRNA groups. N = 6 wells per group. *p < 0.05 versus the control group, #p < 0.05 versus the SAH group. L LDH release rates demonstrated that TRAF3 siRNA treatment decreased LDH release rates compared to SAH and SAH + control siRNA groups. N = 10 wells per group. Data represent mean ± SEM. *p < 0.05 versus the control group, #p < 0.05 versus the SAH group. M Light microscopy of neurons revealed the decrease of neuronal synapses and cell shrinkage was rescued via downregulating TRAF3. N = 3 wells per group. Scale bar: 20 μm.
TRAF3 siRNA downregulated TRAF3 expression and attenuated neuronal apoptosis in SAH primary neural cells
Primary neurons were then transfected with TRAF3 siRNAs at 72 h before SAH. As shown in Fig. 5E, F, TRAF3 protein level in SAH + TRAF3 siRNA group was significantly decreased compared with the SAH group. Compared with SAH and SAH + control siRNA groups, the downregulation of TRAF3 increased the expression of Bcl-xL and caspase-3 and decreased the expression of Bax and cleaved caspase-3 (Fig. 5G–K). The lactate dehydrogenase (LDH) assay was used to assess cytotoxicity levels in primary neurons. The study showed increased LDH release rates in both SAH and SAH + control siRNA groups compared to the control group. In contrast, TRAF3 siRNA treatment decreased LDH release rates (Fig. 5L). Light microscopy of neurons showed reduced neuronal synapses and cell shrinkage in the SAH and SAH + control siRNA groups compared to the control group. When compared to SAH and SAH + control siRNA groups neuronal damage was reduced in the SAH + TRAF3 siRNA group (Fig. 5M). These results indicated that the downregulation of TRAF3 did not only improve the neuron damage in vivo SAH model but also produce similar effects in vitro.
Downregulation of TRAF3 inhibited SAH-induced activation of MAPKs and the NF-κB signaling pathways in vitro
Similarly, the expression of MAPKs and NF-κB pathway was examined using western blotting. The results showed that the downregulation of TRAF3 decreased the expression of SAH-induced p-P38, p-P44/42, and p-JNK in vitro. Besides, SAH reduced the level of IκBα and increased the level of P65 and p-P65 in vitro. Moreover, these changes in protein expression were reversed by downregulating TRAF3 (Fig. 6A–H).

A Western blotting of MAPKs at 24 h after SAH. The P38, P44/42, and JNK expression levels were unaltered. the expression of p-P38, p-P44/42, and p-JNK increased in the SAH group compared with the control group. Quantitative analyses showed that the downregulation of TRAF3 reduced the activation of p-P38 (B), p-P44/42 (C), and p-JNK (D). E Western blotting of the NF-κB signal pathway in 24 h after SAH. The protein expression levels of NF-κB nuclear subunits p-P65 (F) and P65 (G) were increased in SAH and SAH + control siRNA groups and downregulated in the SAH + TRAF3 siRNA group. Levels IκBα was decreased after SAH, TRAF3 siRNA treatment increased the protein level of IκBα (H). N = 6 wells per group. *p < 0.05 versus the control group, #p < 0.05 versus the SAH group.
TRAF3–TAK1 interaction and phosphorylation of TAK1 is required for TRAF3-dependent neuronal apoptosis in SAH
TRAF3 has been reported as a critical upstream regulator of MAPKs and NF-κB pathways via regulation of TAK114. Therefore, we assumed that TRAF3 facilitated SAH-induced neuronal death via direct phosphorylation of TAK1. As respected, the western blotting results revealed that downregulation of TRAF3 significantly inhibited the phosphorylation of TAK1 after SAH, both in vivo and in vitro (Fig. 7A–D).

Western blot analysis revealed that TRAF3 siRNA reduced the phosphorylation of TAK1 after SAH both in vivo (A, B) and in vitro (C, D). N = 6 mice or wells per group. *p < 0.05 versus the control group, #p < 0.05 versus the SAH group. E Co-immunoprecipitation showed that the expression of TRAF3 could interact with TAK1 in SAH primary neural cells. F, G Western blotting analysis showed that AdTRAF3-M failed to increase the expression of p-TAK1 in primary neural cells while AdTRAF3 succeeded. H–N Western blotting analysis showed that the activation of MAPKs and NF-κB signaling was potentiated by AdTRAF3, but abrogated by AdTRAF3-M. N = 6 wells per group. *p < 0.05 versus the control group.
We next investigated whether a direct interaction existed between TRAF3 and TAK1. Co-immunoprecipitation was observed between TRAF3 and TAK1 in SAH primary neural cells (Fig. 7E). Since the 267–376 amino acid (aa) domain of TRAF3 is responsible for the capacity to interact with TAK114,26,27, we further constructed AdTRAF3-M to transfect primary neural cells. Compared with the control adenoviral vectors infection group, the AdTRAF3 infection significantly increased the p-TAK1 expression, whereas the AdTRAF3-M infection did not enhance the p-TAK1 expression (Fig. 7F, G). Furthermore, the primary neuronal cells were infected with AdTRAF3 and AdTRAF3-M. As expected, the activation of MAPKs and NF-κB signaling was potentiated by AdTRAF3, but abrogated by AdTRAF3-M (Fig. 7H–N). Collectively, these results suggest that TRAF3 regulates neuronal apoptosis in SAH probably depends on the TRAF3–TAK1 interaction and activation of TAK1.
TRAF3 expression in the cortex increased in brain tissue of patients with SAH
To initially validate the feasibility of TRAF3 as an early therapeutic target for EBI in patients with clinical SAH, we evaluated TRAF3 expression levels in brain tissue excised from patients who had experienced SAH using double immunofluorescence staining. Accordingly, our results demonstrated that TRAF3 expression levels were elevated following SAH and it was mainly expressed in neurons (Fig. 8A, B).

A Immunofluorescence staining results confirmed that TRAF3 (red) expression was elevated after SAH and was mainly expressed in neurons (green). Scale bar: 50 μm. B The TRAF3 positive index of neurons. N = 4 people per group. *p < 0.05 versus control group. C Schematic illustrating the possible mechanisms of antiapoptotic role of TRAF3 in EBI after SAH.
Discussion
As has been shown in a large number of studies, complex molecular mechanisms and multiple pathophysiological events are involved in EBI induced by SAH6. Consequently, using a single target that could regulate multiple signaling pathways in SAH-induced EBI may serve as a promising strategy. In the present study, we have demonstrated the following (Fig. 8C): (1) the expression of TRAF3 was upregulated after SAH both in mouse cortex and in primary cortical neurons; (2) after silencing TRAF3 gene by siRNA, the activation of TAK1/MAPK and NF-κB signaling pathway were inhibited; (3) the downregulation of TRAF3 was sufficient to reduce neuronal apoptosis both in vivo and in vitro following SAH; (4) the downregulation of TRAF3 alleviated the neurological deficits of SAH mice as demonstrated by the reduction in the Modified Garcia scores; (5) the TRAF3 expression was upregulated in neurons in patients who had experienced SAH. Considering the above findings, TRAF3 may serve as an ideal therapeutic target in SAH-induced EBI.
TRAF3 participates in modulating diverse cellular functions via its zinc finger motifs in the N-terminal interacting with adaptors, receptors, kinases, and regulatory proteins28,29,30. It has been reported that neuronal TRAF3 deficiency inhibited neuronal apoptosis after brain ischemic stroke and spinal cord injury14,31,32. Moreover, neuronal apoptosis involving multiple pathophysiological events in SAH-induced EBI is the main predictor of poor prognosis in SAH4,5. In the current study, we report that the expression of TRAF3 was significantly increased following SAH. In addition, TRAF3 was mainly expressed in neurons, with sparse expression in microglia and no expression in astrocytes. Importantly, TRAF3 siRNA treatment inhibits neuronal apoptosis by downregulating Bax and cleaved caspase-3 expression, while upregulating Bcl-xL and caspase-3 expression. Furthermore, we demonstrated that downregulating TRAF3 could reduce neuronal loss and ameliorate neurological deficits after SAH. Therefore, TRAF3 has a vital role in EBI. In the current study, we further observed that TRAF3 was also upregulated in microglia after SAH. When activated, microglia will secrete proinflammatory cytokines and reactive oxygen species, which disrupt the blood–brain barrier and induce secondary neuronal injury after SAH33. Thus, it remains to be studied whether TRAF3 has a functional role in EBI through modulating microglial activation.
TRAF3 has been demonstrated to promote neuronal apoptosis via the NF-κB pathway34,35. NF-κB regulates cellular pathophysiological processes, such as the activity of apoptotic mediators and inflammatory cytokines in SAH-induced EBI15, therefore, it has been considered to be a potential therapeutic target in EBI prevention36. In the current study, our research demonstrates that the downregulation of TRAF3 inhibits the activation of IκBα–P65 axis. Based on these observations, we concluded that the neuroprotective effect of TRAF3 could be partially mediated through the regulation of the NF-κB pathway.
The MAPKs, including ERK, p38 MAPK, and JNK, have a central role in stress-induced cell death as well as in apoptosis. TRAF3 also regulates apoptosis using MAPK signaling pathways14,37,38. In ischemic stroke, TRAF3 overexpression resulted in JNK phosphorylation, leading to neuronal apoptosis14. In hepatic ischemic injury, the expression of p-JNK was elevated by TRAF3 upregulation and suppressed by TRAF3 deletion26. Further, JNK and P38 have been confirmed to have an important role in the development of EBI after SAH11,15. In the current study, we found that the downregulation of TRAF3 led to decreased JNK and P38 phosphorylation, which further regulated downstream effectors.
Our previous study demonstrated that TAK1, a member of the MAP3Ks family, acts as a key regulator of NF-κB and the MAP kinases JNK and P38 and modulates post-SAH apoptosis15. Targeting TAK1 may have great potential to alleviate the EBI following SAH. Despite the important role of TAK1 in EBI, the mechanism by which TAK1 kinase is activated remains poorly understood. Previous studies showed that TRAF3 could negatively regulate TAK1/MAPK immune signaling39. In contrast, TRAF3 can stimulate TAK1 phosphorylation in ischemic stroke and hepatic ischemic injury14,26,27. The differences in these findings could be attributed to variations in disease models, animals, cell types, and stimuli involved. In the current study, we demonstrated that the downregulation of TRAF3 decreased TAK1 phosphorylation in SAH-induced EBI, suggesting that it has a pivotal role in the regulation of TAK1 activity.
Further, we investigated how TRAF3 regulated the phosphorylation of TAK1 in SAH. The co-immunoprecipitation result indicates that TRAF3 directly interacts with TAK1 in SAH. Previous studies have shown that the interaction between the 267–376 amino acid (aa) domain of TRAF3 and the 481–579 aa domain of TAK1 contributes to the phosphorylation and activation of TAK114,27. In our study, AdTRAF3-M (adenoviruses carrying sequences encoding mouse TRAF3 with a mutant 267–376 aa domain) failed to increase the expression of p-TAK1 in primary neural cells while AdTRAF3 succeeded. Besides, AdTRAF3 but not AdTRAF3-M could potentiate the activation of MAPKs and NF-κB signaling pathways. Therefore, we deduce that the 267–376 amino acid (aa) domain of TRAF3 is essential for its capacity to interact with TAK1 in SAH.
Despite numerous basic discoveries in SAH animal research, few have turned out to be effective in clinical trials on humans. Moreover, oftentimes, animal research findings do not translate into human medicine due to incomplete replication of human pathophysiology40,41,42. Here, we have demonstrated that TRAF3 expression was elevated after clinical SAH and was mainly expressed in neurons, suggesting that downregulation of TRAF3 may have potential clinical application in the treatment of EBI. Considering that only patients with very severe SAH requiring surgery could provide brain tissue in our research, further studies are warranted to determine whether TRAF3 expression is increased in less severe SAH patients.
In conclusion, our research has revealed that downregulation of TRAF3 inhibited the activation of MAPKs and NF-κB pathways via inhibiting the phosphorylation of TAK1, which effectively reduced neuronal apoptosis and neurological deficits in SAH. Thus, TRAF3 is a novel mediator of neuronal apoptosis in EBI following SAH. More importantly, we demonstrated that TRAF3 expression was elevated in human neurons following SAH. Therefore, TRAF3 may serve as a potential therapeutic target for improving prognosis in SAH patients.
References
Schuette, A. J. & Barrow, D. L. Epidemiology and long-term mortality in subarachnoid hemorrhage. World Neurosurg. 80, 264–265 (2013).
Connolly, E. S. Jr. et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 43, 1711–1737 (2012).
Fujii, M. et al. Early brain injury, an evolving frontier in subarachnoid hemorrhage research. Transl. Stroke Res. 4, 432–446 (2013).
Sabri, M., Lass, E. & Macdonald, R. L. Early brain injury: a common mechanism in subarachnoid hemorrhage and global cerebral ischemia. Stroke Res. Treat. 2013, 394036 (2013).
Sehba, F. A., Hou, J., Pluta, R. M. & Zhang, J. H. The importance of early brain injury after subarachnoid hemorrhage. Prog. Neurobiol. 97, 14–37 (2012).
Kusaka, G., Ishikawa, M., Nanda, A., Granger, D. N. & Zhang, J. H. Signaling pathways for early brain injury after subarachnoid hemorrhage. J. Cereb. Blood Flow. Metab. 24, 916–925 (2004).
Zhang, X. S. et al. Sirtuin 1 activation protects against early brain injury after experimental subarachnoid hemorrhage in rats. Cell Death Dis. 7, e2416 (2016).
Sun, Q. et al. Glycyrrhizic acid confers neuroprotection after subarachnoid hemorrhage via inhibition of high mobility group box-1 protein: a hypothesis for novel therapy of subarachnoid hemorrhage. Med. Hypotheses 81, 681–685 (2013).
You, W. C. et al. Activation of nuclear factor-kappaB in the brain after experimental subarachnoid hemorrhage and its potential role in delayed brain injury. PLoS ONE 8, e60290 (2013).
Wang, Z. et al. Progesterone administration modulates cortical TLR4/NF-kappaB signaling pathway after subarachnoid hemorrhage in male rats. Mediat. Inflamm. 2011, 848309 (2011).
Cahill, J., Calvert, J. W. & Zhang, J. H. Mechanisms of early brain injury after subarachnoid hemorrhage. J. Cereb. Blood Flow. Metab. 26, 1341–1353 (2006).
Cullell, N. et al. Role of TRAF3 in neurological and cardiovascular diseases: an overview of recent studies. Biomol. Concepts 8, 197–202 (2017).
Hacker, H., Tseng, P. H. & Karin, M. Expanding TRAF function: TRAF3 as a tri-faced immune regulator. Nat. Rev. Immunol. 11, 457–468 (2011).
Gong, J. et al. Neuron-specific tumor necrosis factor receptor-associated factor 3 is a central regulator of neuronal death in acute ischemic stroke. Hypertension 66, 604–616 (2015).
Zhang, D. et al. TGFbeta-activated kinase 1 (TAK1) inhibition by 5Z-7-oxozeaenol attenuates early brain injury after experimental subarachnoid hemorrhage. J. Biol. Chem. 290, 19900–19909 (2015).
Egashira, Y., Shishido, H., Hua, Y., Keep, R. F. & Xi, G. New grading system based on magnetic resonance imaging in a mouse model of subarachnoid hemorrhage. Stroke 46, 582–584 (2015).
Tao, T. et al. DHEA attenuates microglial activation via induction of JMJD3 in EXPERIMENTAL SUBARACHNOID HAEmorrhage. J. Neuroinflammation 16, 243 (2019).
Feiler, S., Friedrich, B., Scholler, K., Thal, S. C. & Plesnila, N. Standardized induction of subarachnoid hemorrhage in mice by intracranial pressure monitoring. J. Neurosci. Methods 190, 164–170 (2010).
Lu, Y. et al. Peroxiredoxin 2 activates microglia by interacting with Toll-like receptor 4 after subarachnoid hemorrhage. J. Neuroinflammation 15, 87 (2018).
Wu, L. Y. et al. Roles of Pannexin-1 channels in inflammatory response through the TLRs/NF-kappa B signaling pathway following experimental subarachnoid hemorrhage in rats. Front. Mol. Neurosci. 10, 175 (2017).
Boyer, J. G. et al. ERK1/2 signaling induces skeletal muscle slow fiber-type switching and reduces muscular dystrophy disease severity. JCI Insight 5, https://doi.org/10.1172/jci.insight.127356 (2019).
Zhang, D. et al. Inhibition of transforming growth factor beta-activated kinase 1 confers neuroprotection after traumatic brain injury in rats. Neuroscience 238, 209–217 (2013).
Yagi, K. et al. Therapeutically targeting tumor necrosis factor-alpha/sphingosine-1-phosphate signaling corrects myogenic reactivity in subarachnoid hemorrhage. Stroke 46, 2260–2270 (2015).
Hayakawa, K. et al. Delayed treatment with minocycline ameliorates neurologic impairment through activated microglia expressing a high-mobility group box1-inhibiting mechanism. Stroke 39, 951–958 (2008).
Zhang, X. S. et al. Amelioration of oxidative stress and protection against early brain injury by astaxanthin after experimental subarachnoid hemorrhage. J. Neurosurg. 121, 42–54 (2014).
Hu, J. et al. Targeting TRAF3 signaling protects against hepatic ischemia/reperfusions injury. J. Hepatol. 64, 146–159 (2016).
Wang, P. X. et al. Hepatocyte TRAF3 promotes liver steatosis and systemic insulin resistance through targeting TAK1-dependent signalling. Nat. Commun. 7, 10592 (2016).
Wang, Y., Zhang, P., Liu, Y. & Cheng, G. TRAF-mediated regulation of immune and inflammatory responses. Sci. China Life Sci. 53, 159–168 (2010).
Inoue, J. et al. Tumor necrosis factor receptor-associated factor (TRAF) family: adapter proteins that mediate cytokine signaling. Exp. Cell Res. 254, 14–24 (2000).
Jiang, X. et al. Tumor necrosis factor receptor-associated factor 3 is a positive regulator of pathological cardiac hypertrophy. Hypertension 66, 356–367 (2015).
Yao, S., Tang, B., Li, G., Fan, R. & Cao, F. miR-455 inhibits neuronal cell death by targeting TRAF3 in cerebral ischemic stroke. Neuropsychiatr. Dis. Treat. 12, 3083–3092 (2016).
Wu, Y. et al. Spatiotemporal pattern of TRAF3 expression after rat spinal cord injury. J. Mol. Histol. 45, 541–553 (2014).
Zhang, Z. Y. et al. Activation of mGluR5 attenuates microglial activation and neuronal apoptosis in early brain injury after experimental subarachnoid hemorrhage in rats. Neurochem. Res. 40, 1121–1132 (2015).
Chen, S. et al. Controversies and evolving new mechanisms in subarachnoid hemorrhage. Prog. Neurobiol. 115, 64–91 (2014).
Zirlik, A. et al. TRAF-1, -2, -3, -5, and -6 are induced in atherosclerotic plaques and differentially mediate proinflammatory functions of CD40L in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 27, 1101–1107 (2007).
Hasegawa, Y., Suzuki, H., Sozen, T., Altay, O. & Zhang, J. H. Apoptotic mechanisms for neuronal cells in early brain injury after subarachnoid hemorrhage. Acta Neurochir. Suppl. 110, 43–48 (2011).
Hildebrand, J. M. et al. Roles of tumor necrosis factor receptor associated factor 3 (TRAF3) and TRAF5 in immune cell functions. Immunol. Rev. 244, 55–74 (2011).
Yi, Z., Lin, W. W., Stunz, L. L. & Bishop, G. A. Roles for TNF-receptor associated factor 3 (TRAF3) in lymphocyte functions. Cytokine Growth Factor Rev. 25, 147–156 (2014).
Tseng, P. H. et al. Different modes of ubiquitination of the adaptor TRAF3 selectively activate the expression of type I interferons and proinflammatory cytokines. Nat. Immunol. 11, 70–75 (2010).
Hollig, A. et al. Experimental subarachnoid hemorrhage in rats: comparison of two endovascular perforation techniques with respect to success rate, confounding pathologies and early hippocampal tissue lesion pattern. PLoS ONE 10, e0123398 (2015).
Luo, C. et al. Paravascular pathways contribute to vasculitis and neuroinflammation after subarachnoid hemorrhage independently of glymphatic control. Cell Death Dis. 7, e2160 (2016).
Leclerc, J. L. et al. A comparison of pathophysiology in humans and rodent models of subarachnoid hemorrhage. Front. Mol. Neurosci. 11, 71 (2018).
Acknowledgements
Dingding Zhang was funded by the Natural Science Foundation of Jiangsu Province (No. BK20180126). Chunhua Hang, Wei Li, Zong Zhuang, Yue Lu, and Lingyun Wu were funded by the National Natural Science Foundation of China (No. 81771291 for Chunhua Hang, No. 81870922 for Wei Li, No. 81971127 for Zong Zhuang, No. 81901203 for Yue Lu, No. 81801166 for Lingyun Wu).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by A. Sawa
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhou, Y., Tao, T., Liu, G. et al. TRAF3 mediates neuronal apoptosis in early brain injury following subarachnoid hemorrhage via targeting TAK1-dependent MAPKs and NF-κB pathways. Cell Death Dis 12, 10 (2021). https://doi.org/10.1038/s41419-020-03278-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-020-03278-z
This article is cited by
-
The Role of Circular RNAs in Ischemic Stroke
Neurochemical Research (2023)
-
Cognitive Deficits Found in a Pro-inflammatory State are Independent of ERK1/2 Signaling in the Murine Brain Hippocampus Treated with Shiga Toxin 2 from Enterohemorrhagic Escherichia coli
Cellular and Molecular Neurobiology (2023)
-
Hypoxia and TNF-α Synergistically Induce Expression of IL-6 and IL-8 in Human Fibroblast-like Synoviocytes via Enhancing TAK1/NF-κB/HIF-1α Signaling
Inflammation (2023)
-
LncRNA XIST Exacerbates Oxygen-Glucose Deprivation/Reoxygenation-Induced Cerebral Injury Through the miR-25-3p/TRAF3 Axis
Molecular Neurobiology (2023)
-
miR-452-3p Targets HDAC3 to Inhibit p65 Deacetylation and Activate the NF-κB Signaling Pathway in Early Brain Injury after Subarachnoid Hemorrhage
Neurocritical Care (2022)
