The Role of the Unfolded Protein Response on Renal Lipogenesis in C57BL/6 Mice

 , ,
, ,  and
and
Abstract
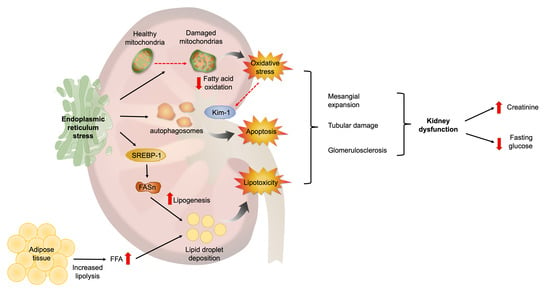
:
1. Introduction
2. Materials and Methods
2.1. Mice and Experimental Protocol
2.2. Renal Function Parameters
2.3. Histological Analysis
2.4. Apoptosis Analysis
2.5. Electron Microscopy
2.6. Biochemical Determinations
2.7. RNA Extraction and mRNA Quantitation
2.8. Protein Extraction, SDS/PAGE and Immunoblotting
2.9. Kidney Lipid Content
2.10. Statistical Analysis
3. Results
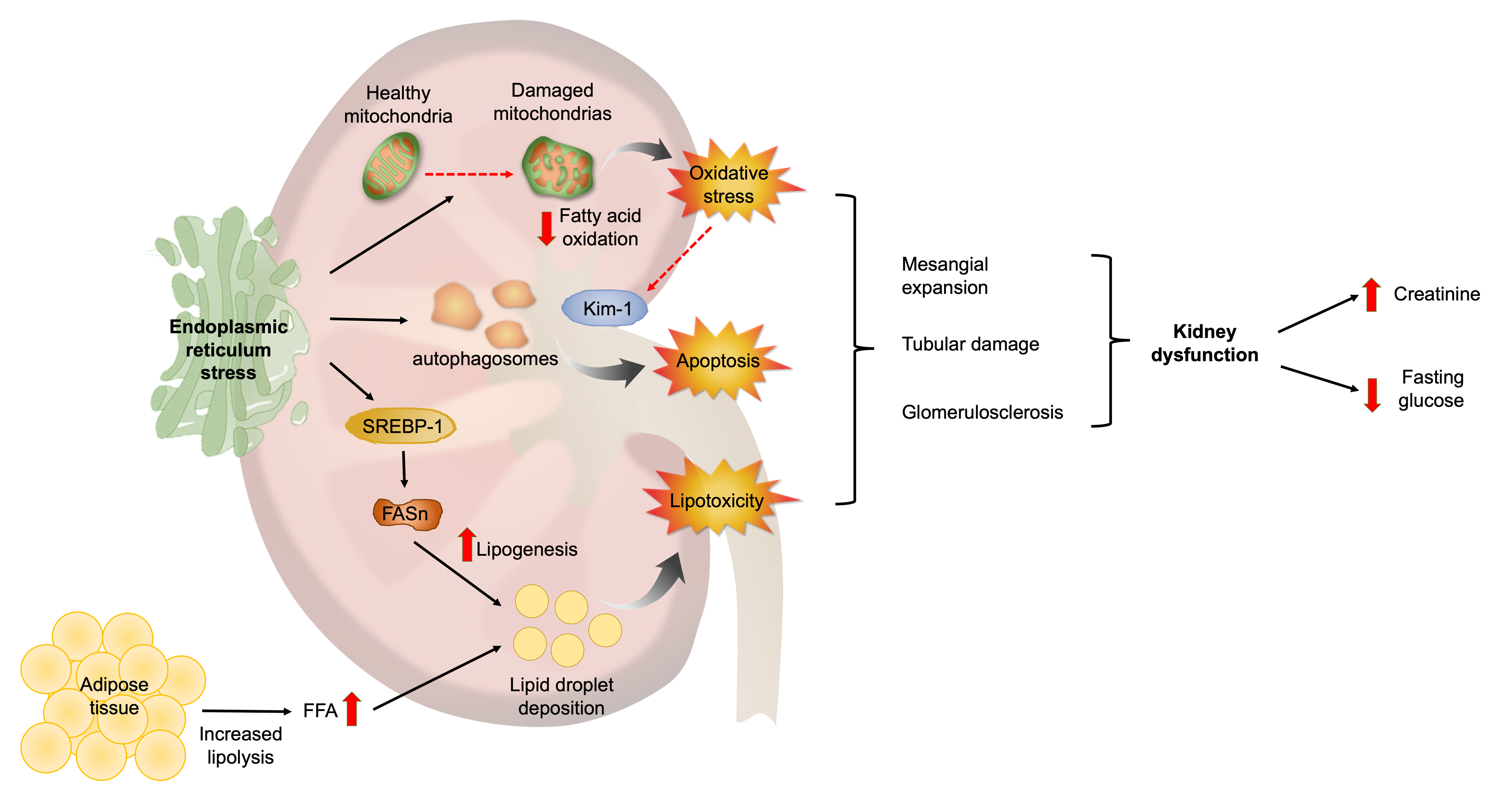
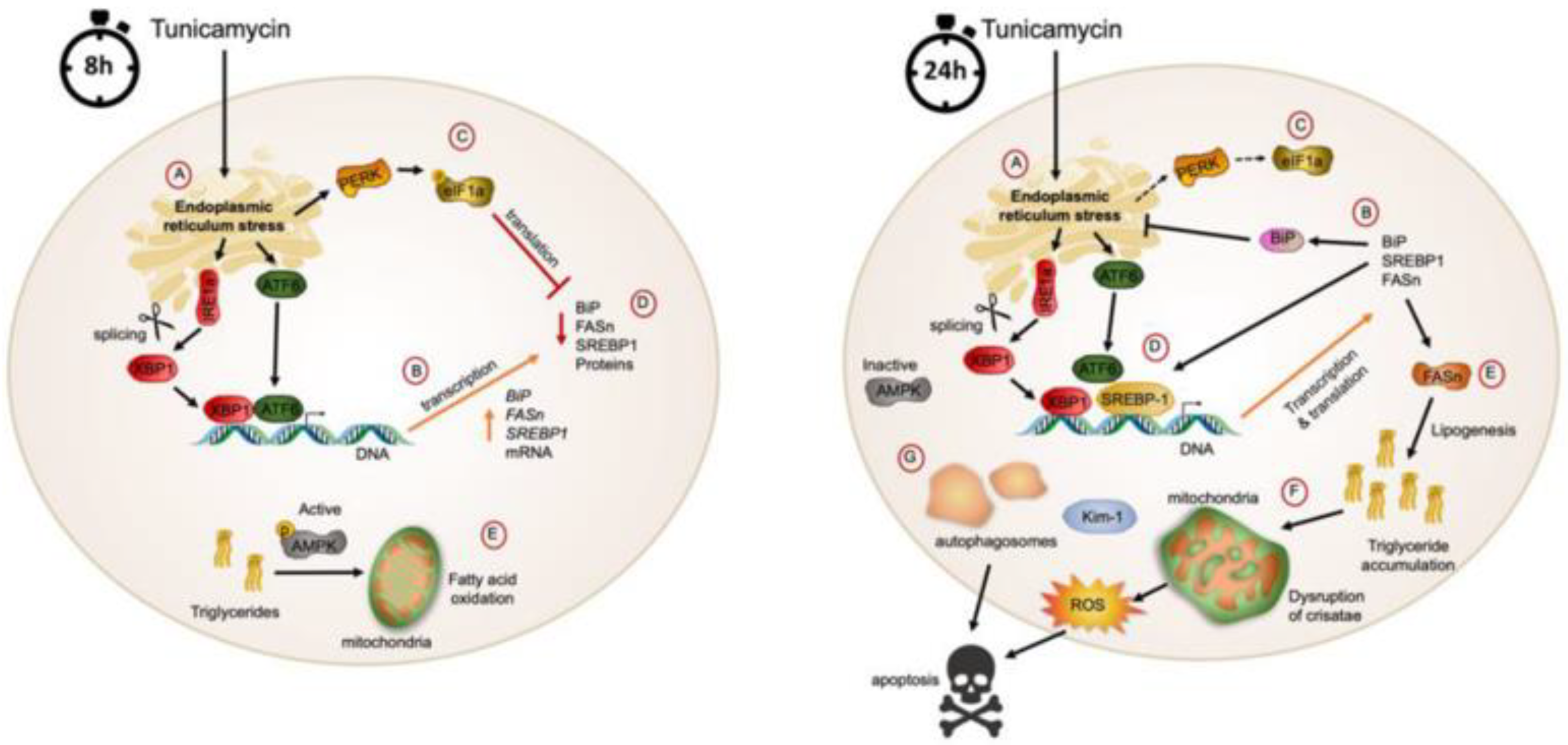
3.1. ER Stress Is Associated with Alterations in Kidney Function and Structure
3.2. ER Stress Alters the Metabolic and Endocrine Parameters of Mice Treated with Tunicamycin
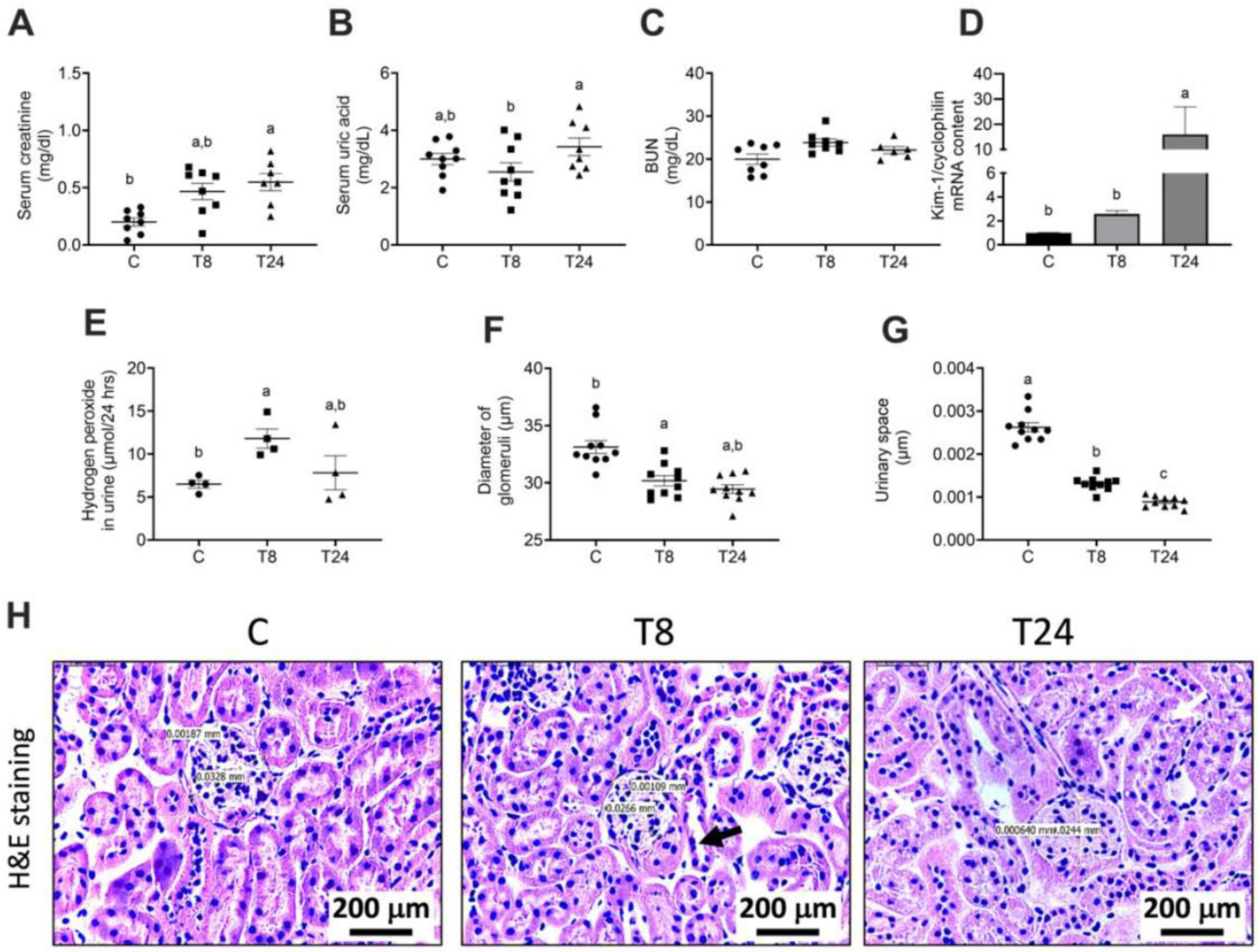
3.3. Tunicamycin Causes ER Stress in the Kidney
3.4. ER Stress Associates with Renal Lipogenesis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mende, C.W.; Einhorn, D. Fatty Kidney Disease: A New Renal and Endocrine Clinical Entity? Describing the Role of the Kidney in Obesity, Metabolic Syndrome, and Type 2 Diabetes. Endocr. Pract. 2019, 25, 854–858. [Google Scholar] [CrossRef]
- Kume, S.; Uzu, T.; Araki, S.; Sugimoto, T.; Isshiki, K.; Chin-Kanasaki, M.; Sakaguchi, M.; Kubota, N.; Terauchi, Y.; Kadowaki, T.; et al. Role of altered renal lipid metabolism in the development of renal injury induced by a high-fat diet. J. Am. Soc. Nephrol. 2007, 18, 2715–2723. [Google Scholar] [CrossRef] [Green Version]
- Bagby, S.P. Obesity-initiated metabolic syndrome and the kidney: A recipe for chronic kidney disease? J. Am. Soc. Nephrol. 2004, 15, 2775–2791. [Google Scholar] [CrossRef] [Green Version]
- Gyebi, L.; Soltani, Z.; Reisin, E. Lipid nephrotoxicity: New concept for an old disease. Curr. Hypertens. Rep. 2012, 14, 177–181. [Google Scholar] [CrossRef]
- Mascali, A.; Franzese, O.; Nistico, S.; Campia, U.; Lauro, D.; Cardillo, C.; Di Daniele, N.; Tesauro, M. Obesity and kidney disease: Beyond the hyperfiltration. Int. J. Immunopathol. Pharmacol. 2016, 29, 354–363. [Google Scholar] [CrossRef] [Green Version]
- Izquierdo-Lahuerta, A.; Martinez-Garcia, C.; Medina-Gomez, G. Lipotoxicity as a trigger factor of renal disease. J. Nephrol. 2016, 29, 603–610. [Google Scholar] [CrossRef]
- Lhotak, S.; Sood, S.; Brimble, E.; Carlisle, R.E.; Colgan, S.M.; Mazzetti, A.; Dickhout, J.G.; Ingram, A.J.; Austin, R.C. ER stress contributes to renal proximal tubule injury by increasing SREBP-2-mediated lipid accumulation and apoptotic cell death. Am. J. Physiol. Renal Physiol. 2012, 303, F266–F278. [Google Scholar] [CrossRef] [Green Version]
- Hall, M.E.; do Carmo, J.M.; da Silva, A.A.; Juncos, L.A.; Wang, Z.; Hall, J.E. Obesity, hypertension, and chronic kidney disease. Int. J. Nephrol. Renovasc. Dis. 2014, 7, 75–88. [Google Scholar] [CrossRef] [Green Version]
- Simon, N.; Hertig, A. Alteration of Fatty Acid Oxidation in Tubular Epithelial Cells: From Acute Kidney Injury to Renal Fibrogenesis. Front. Med. (Lausanne) 2015, 2, 52. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, J.M. Lipotoxicity. Kidney Int. 2006, 70, 1560–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herman-Edelstein, M.; Scherzer, P.; Tobar, A.; Levi, M.; Gafter, U. Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J. Lipid. Res. 2014, 55, 561–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Jiang, T.; Li, J.; Proctor, G.; McManaman, J.L.; Lucia, S.; Chua, S.; Levi, M. Regulation of renal lipid metabolism, lipid accumulation, and glomerulosclerosis in FVBdb/db mice with type 2 diabetes. Diabetes 2005, 54, 2328–2335. [Google Scholar] [CrossRef] [PubMed]
- Proctor, G.; Jiang, T.; Iwahashi, M.; Wang, Z.; Li, J.; Levi, M. Regulation of renal fatty acid and cholesterol metabolism, inflammation, and fibrosis in Akita and OVE26 mice with type 1 diabetes. Diabetes 2006, 55, 2502–2509. [Google Scholar] [CrossRef] [Green Version]
- Guebre-Egziabher, F.; Alix, P.M.; Koppe, L.; Pelletier, C.C.; Kalbacher, E.; Fouque, D.; Soulage, C.O. Ectopic lipid accumulation: A potential cause for metabolic disturbances and a contributor to the alteration of kidney function. Biochimie 2013, 95, 1971–1979. [Google Scholar] [CrossRef]
- Sieber, J.; Jehle, A.W. Free Fatty acids and their metabolism affect function and survival of podocytes. Front. Endocrinol. (Lausanne) 2014, 5, 186. [Google Scholar] [CrossRef] [Green Version]
- Tesauro, M.; Canale, M.P.; Rodia, G.; Di Daniele, N.; Lauro, D.; Scuteri, A.; Cardillo, C. Metabolic syndrome, chronic kidney, and cardiovascular diseases: Role of adipokines. Cardiol. Res. Pract. 2011, 2011, 653182. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Zhao, Z.; Xia, J.; Xin, L.; Chen, Y.; Yang, S.; Li, K. A Long-Term High-Fat/High-Sucrose Diet Promotes Kidney Lipid Deposition and Causes Apoptosis and Glomerular Hypertrophy in Bama Minipigs. PLoS ONE 2015, 10, e0142884. [Google Scholar] [CrossRef] [Green Version]
- Koh, E.S.; Lim, J.H.; Kim, M.Y.; Chung, S.; Shin, S.J.; Choi, B.S.; Kim, H.W.; Hwang, S.Y.; Kim, S.W.; Park, C.W.; et al. Anthocyanin-rich Seoritae extract ameliorates renal lipotoxicity via activation of AMP-activated protein kinase in diabetic mice. J. Transl. Med. 2015, 13, 203. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Halaihel, N.; Zhang, W.; Rogers, T.; Levi, M. Role of sterol regulatory element-binding protein 1 in regulation of renal lipid metabolism and glomerulosclerosis in diabetes mellitus. J. Biol. Chem. 2002, 277, 18919–18927. [Google Scholar] [CrossRef] [Green Version]
- de Vries, A.P.; Ruggenenti, P.; Ruan, X.Z.; Praga, M.; Cruzado, J.M.; Bajema, I.M.; D’Agati, V.D.; Lamb, H.J.; Pongrac Barlovic, D.; Hojs, R.; et al. Fatty kidney: Emerging role of ectopic lipid in obesity-related renal disease. Lancet Diabetes Endocrinol. 2014, 2, 417–426. [Google Scholar] [CrossRef]
- Wang, H.; Zhu, L.; Hao, J.; Duan, H.; Liu, S.; Zhao, S.; Liu, Q.; Liu, W. Co-regulation of SREBP-1 and mTOR ameliorates lipid accumulation in kidney of diabetic mice. Exp. Cell Res. 2015, 336, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Inagi, R. Endoplasmic reticulum stress in the kidney as a novel mediator of kidney injury. Nephron Exp. Nephrol. 2009, 112, e1–e9. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H. ER stress and diseases. FEBS J. 2007, 274, 630–658. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Vaziri, N.D. Sterol regulatory element-binding proteins, liver X receptor, ABCA1 transporter, CD36, scavenger receptors A1 and B1 in nephrotic kidney. Am. J. Nephrol. 2009, 29, 607–614. [Google Scholar] [CrossRef]
- Sethi, G.; Shanmugam, M.K.; Kumar, A.P. SREBP-1c as a molecular bridge between lipogenesis and cell cycle progression of clear cell renal carcinoma. Biosci. Rep. 2017, 37, BSR20171270. [Google Scholar] [CrossRef] [Green Version]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef] [Green Version]
- Katsoulieris, E.; Mabley, J.G.; Samai, M.; Sharpe, M.A.; Green, I.C.; Chatterjee, P.K. Lipotoxicity in renal proximal tubular cells: Relationship between endoplasmic reticulum stress and oxidative stress pathways. Free Radic. Biol. Med. 2010, 48, 1654–1662. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Bravo, R.; Parra, V.; Gatica, D.; Rodriguez, A.E.; Torrealba, N.; Paredes, F.; Wang, Z.V.; Zorzano, A.; Hill, J.A.; Jaimovich, E.; et al. Endoplasmic reticulum and the unfolded protein response: Dynamics and metabolic integration. Int. Rev. Cell Mol. Biol. 2013, 301, 215–290. [Google Scholar] [CrossRef] [Green Version]
- Cunard, R. Endoplasmic Reticulum Stress in the Diabetic Kidney, the Good, the Bad and the Ugly. J. Clin. Med. 2015, 4, 715–740. [Google Scholar] [CrossRef] [Green Version]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Bravo, R.; Gutierrez, T.; Paredes, F.; Gatica, D.; Rodriguez, A.E.; Pedrozo, Z.; Chiong, M.; Parra, V.; Quest, A.F.; Rothermel, B.A.; et al. Endoplasmic reticulum: ER stress regulates mitochondrial bioenergetics. Int. J. Biochem. Cell Biol. 2012, 44, 16–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christianson, J.C.; Ye, Y. Cleaning up in the endoplasmic reticulum: Ubiquitin in charge. Nat. Struct. Mol. Biol. 2014, 21, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Cunard, R.; Sharma, K. The endoplasmic reticulum stress response and diabetic kidney disease. Am. J. Physiol. Renal Physiol. 2011, 300, F1054–F1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Gorgun, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Yang, F.; Jiang, S.; Sun, X.; Xu, J. Induction of Liver Steatosis in BAP31-Deficient Mice Burdened with Tunicamycin-Induced Endoplasmic Reticulum Stress. Int. J. Mol. Sci. 2018, 19, 2291. [Google Scholar] [CrossRef] [Green Version]
- Winnay, J.N.; Boucher, J.; Mori, M.A.; Ueki, K.; Kahn, C.R. A regulatory subunit of phosphoinositide 3-kinase increases the nuclear accumulation of X-box-binding protein-1 to modulate the unfolded protein response. Nat. Med. 2010, 16, 438–445. [Google Scholar] [CrossRef] [Green Version]
- Sato, N.; Urano, F.; Yoon Leem, J.; Kim, S.H.; Li, M.; Donoviel, D.; Bernstein, A.; Lee, A.S.; Ron, D.; Veselits, M.L.; et al. Upregulation of BiP and CHOP by the unfolded-protein response is independent of presenilin expression. Nat. Cell Biol. 2000, 2, 863–870. [Google Scholar] [CrossRef]
- Yan, W.; Frank, C.L.; Korth, M.J.; Sopher, B.L.; Novoa, I.; Ron, D.; Katze, M.G. Control of PERK eIF2alpha kinase activity by the endoplasmic reticulum stress-induced molecular chaperone P58IPK. Proc. Natl. Acad. Sci. USA 2002, 99, 15920–15925. [Google Scholar] [CrossRef] [Green Version]
- National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals, 8th ed.; National Academies Press: Washington, DC, USA, 2011. [CrossRef]
- Mohanty, J.G.; Jaffe, J.S.; Schulman, E.S.; Raible, D.G. A highly sensitive fluorescent micro-assay of H2O2 release from activated human leukocytes using a dihydroxyphenoxazine derivative. J. Immunol. Methods 1997, 202, 133–141. [Google Scholar]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [PubMed]
- Ozcan, L.; Tabas, I. Role of endoplasmic reticulum stress in metabolic disease and other disorders. Annu. Rev. Med. 2012, 63, 317–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, M.; Yoshida, H. Endoplasmic reticulum stress in kidney function and disease. Curr. Opin. Nephrol. Hypertens. 2015, 24, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Nishi, H.; Higashihara, T.; Inagi, R. Lipotoxicity in Kidney, Heart, and Skeletal Muscle Dysfunction. Nutrients 2019, 11, 1664. [Google Scholar] [CrossRef] [Green Version]
- Cirrik, S.; Hacioglu, G.; Ayyildiz, S.N.; Tezcan, B.; Abidin, I.; Aydin-Abidin, S.; Noyan, T. Renal response to tunicamycin-induced endoplasmic reticulum stress in BDNF heterozygous mice. Adv. Clin. Exp. Med. 2019, 28, 1161–1170. [Google Scholar] [CrossRef]
- Woo, C.W.; Cui, D.; Arellano, J.; Dorweiler, B.; Harding, H.; Fitzgerald, K.A.; Ron, D.; Tabas, I. Adaptive suppression of the ATF4-CHOP branch of the unfolded protein response by toll-like receptor signalling. Nat. Cell Biol. 2009, 11, 1473–1480. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhang, R.; Huang, L.; Zheng, Z.; Vlassara, H.; Striker, G.; Zhang, X.; Guan, Y.; Zheng, F. Excessive Oxidative Stress Contributes to Increased Acute ER Stress Kidney Injury in Aged Mice. Oxid. Med. Cell. Longev. 2019, 2019, 2746521. [Google Scholar] [CrossRef]
- Choi, Y.J.; Shin, H.S.; Choi, H.S.; Park, J.W.; Jo, I.; Oh, E.S.; Lee, K.Y.; Lee, B.H.; Johnson, R.J.; Kang, D.H. Uric acid induces fat accumulation via generation of endoplasmic reticulum stress and SREBP-1c activation in hepatocytes. Lab. Investig. 2014, 94, 1114–1125. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Zhao, F.; Cheng, S.; Wang, X.; Hao, Y. Uric acid-induced endoplasmic reticulum stress triggers phenotypic change in rat glomerular mesangial cells. Nephrology (Carlton) 2013, 18, 682–689. [Google Scholar] [CrossRef]
- Liu, H.; Xiong, J.; He, T.; Xiao, T.; Li, Y.; Yu, Y.; Huang, Y.; Xu, X.; Huang, Y.; Zhang, J.; et al. High Uric Acid-Induced Epithelial-Mesenchymal Transition of Renal Tubular Epithelial Cells via the TLR4/NF-kB Signaling Pathway. Am. J. Nephrol. 2017, 46, 333–342. [Google Scholar] [CrossRef]
- Rashdan, N.A.; Pattillo, C.B. Hydrogen peroxide in the ER: A tale of triage. Redox Biol. 2020, 28, 101358. [Google Scholar] [CrossRef]
- Marchewka, Z.; Plonka, J. The diagnostic importance of the new marker KIM-1 in kidney damage. Postepy Hig. Med. Dosw. (Online) 2013, 67, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Johno, H.; Nakajima, S.; Kato, H.; Yao, J.; Paton, A.W.; Paton, J.C.; Katoh, R.; Shimizu, F.; Kitamura, M. Unfolded protein response causes a phenotypic shift of inflamed glomerular cells toward redifferentiation through dual blockade of Akt and Smad signaling pathways. Am. J. Pathol. 2012, 181, 1977–1990. [Google Scholar] [CrossRef]
- Brewer, J.W.; Diehl, J.A. PERK mediates cell-cycle exit during the mammalian unfolded protein response. Proc. Natl. Acad. Sci. USA 2000, 97, 12625–12630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torre-Villalvazo, I.; Bunt, A.E.; Aleman, G.; Marquez-Mota, C.C.; Diaz-Villasenor, A.; Noriega, L.G.; Estrada, I.; Figueroa-Juarez, E.; Tovar-Palacio, C.; Rodriguez-Lopez, L.A.; et al. Adiponectin synthesis and secretion by subcutaneous adipose tissue is impaired during obesity by endoplasmic reticulum stress. J. Cell. Biochem. 2018, 119, 5970–5984. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Wustefeld, T.; Horn, R.; Luedde, T.; Srinivas Rao, A.; Manns, M.P.; Trautwein, C.; Brabant, G. High adiponectin in chronic liver disease and cholestasis suggests biliary route of adiponectin excretion in vivo. J. Hepatol. 2005, 42, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Heidari, M.; Nasri, P.; Nasri, H. Adiponectin and chronic kidney disease; a review on recent findings. J. Nephropharmacol. 2015, 4, 63–68. [Google Scholar] [PubMed]
- Luke, R.G. Chronic renal failure—A vasculopathic state. N. Engl. J. Med. 1998, 339, 841–843. [Google Scholar] [CrossRef]
- Zoccali, C. Cardiovascular risk in uraemic patients-is it fully explained by classical risk factors? Nephrol. Dial. Transpl. 2000, 15, 454–457. [Google Scholar] [CrossRef] [Green Version]
- Morris, D.L.; Rui, L. Recent advances in understanding leptin signaling and leptin resistance. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1247–E1259. [Google Scholar] [CrossRef] [Green Version]
- Munusamy, S.; do Carmo, J.M.; Hosler, J.P.; Hall, J.E. Obesity-induced changes in kidney mitochondria and endoplasmic reticulum in the presence or absence of leptin. Am. J. Physiol. Renal Physiol. 2015, 309, F731–F743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkinson, L.L.; Fischer, M.A.; Lopaschuk, G.D. Leptin activates cardiac fatty acid oxidation independent of changes in the AMP-activated protein kinase-acetyl-CoA carboxylase-malonyl-CoA axis. J. Biol. Chem. 2002, 277, 29424–29430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozcan, U.; Ozcan, L.; Yilmaz, E.; Duvel, K.; Sahin, M.; Manning, B.D.; Hotamisligil, G.S. Loss of the tuberous sclerosis complex tumor suppressors triggers the unfolded protein response to regulate insulin signaling and apoptosis. Mol. Cell 2008, 29, 541–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosoi, T.; Sasaki, M.; Miyahara, T.; Hashimoto, C.; Matsuo, S.; Yoshii, M.; Ozawa, K. Endoplasmic reticulum stress induces leptin resistance. Mol. Pharmacol. 2008, 74, 1610–1619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, S.; Watkins, S.M.; Hotamisligil, G.S. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab. 2012, 15, 623–634. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Vera, L.; Fischer, W.H.; Montminy, M. The CREB coactivator CRTC2 links hepatic ER stress and fasting gluconeogenesis. Nature 2009, 460, 534–537. [Google Scholar] [CrossRef] [Green Version]
- Cano, N. Bench-to-bedside review: Glucose production from the kidney. Crit. Care 2002, 6, 317–321. [Google Scholar] [CrossRef]
- Feng, B.; Huang, X.; Jiang, D.; Hua, L.; Zhuo, Y.; Wu, D. Endoplasmic Reticulum Stress Inducer Tunicamycin Alters Hepatic Energy Homeostasis in Mice. Int. J. Mol. Sci. 2017, 18, 1710. [Google Scholar] [CrossRef]
- Yu, L.; Strandberg, L.; Lenardo, M.J. The selectivity of autophagy and its role in cell death and survival. Autophagy 2008, 4, 567–573. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Syed, T.W.; Liu, R.; Yu, J. Role of Endoplasmic Reticulum Stress, Autophagy, and Inflammation in Cardiovascular Disease. Front. Cardiovasc. Med. 2017, 4, 29. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Control | Tunicamycin 8 h | Tunicamycin 24 h |
|---|---|---|---|
| Body weight, (g) | 29.0 ± 1.6 | 32.3 ± 1.4 | 29.0 ± 0.8 |
| Kidney weight, (g) | 0.196 ± 0.02 | 0.230 ± 0.01 | 0.205 ± 0.01 |
| Kidney weight/body weight (%) | 0.67 | 0.71 | 0.72 |
| Glucose, mg/dl | 232 ± 9.7 a | 205 ± 15.0 a,b | 170.8 ± 13.4 b |
| Insulin (ng/mL) | 2.01 ± 0.12 c | 6.00 ± 0.22 b | 8.44 ± 0.51 a |
| NEFA (mM) | 1.81 ± 0.1 c | 3.79 ± 0.1 a | 2.89 ± 0.3 b |
| Glycerol (mM) | 0.15 ± 0.004 b | 0.26 ± 0.001 a | 0.30 ± 0.002 a |
| Leptin (ng/mL) | 2.00 ± 0.17 b | 11.02 ± 0.63 a | 8.80 ± 0.80 a |
| Adiponectin (µg/mL) | 12.34 ± 2.1 b | 21.14 ± 4.4 a | 17.52 ± 2.5 a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Figueroa-Juárez, E.; Noriega, L.G.; Pérez-Monter, C.; Alemán, G.; Hernández-Pando, R.; Correa-Rotter, R.; Ramírez, V.; Tovar, A.R.; Torre-Villalvazo, I.; Tovar-Palacio, C. The Role of the Unfolded Protein Response on Renal Lipogenesis in C57BL/6 Mice. Biomolecules 2021, 11, 73. https://doi.org/10.3390/biom11010073
Figueroa-Juárez E, Noriega LG, Pérez-Monter C, Alemán G, Hernández-Pando R, Correa-Rotter R, Ramírez V, Tovar AR, Torre-Villalvazo I, Tovar-Palacio C. The Role of the Unfolded Protein Response on Renal Lipogenesis in C57BL/6 Mice. Biomolecules. 2021; 11(1):73. https://doi.org/10.3390/biom11010073
Chicago/Turabian StyleFigueroa-Juárez, Elizabeth, Lilia G. Noriega, Carlos Pérez-Monter, Gabriela Alemán, Rogelio Hernández-Pando, Ricardo Correa-Rotter, Victoria Ramírez, Armando R. Tovar, Iván Torre-Villalvazo, and Claudia Tovar-Palacio. 2021. "The Role of the Unfolded Protein Response on Renal Lipogenesis in C57BL/6 Mice" Biomolecules 11, no. 1: 73. https://doi.org/10.3390/biom11010073