
Abstract
Acute kidney injury (AKI) is an extremely common medical affliction affecting both adult and pediatric patients resulting from hypoxic, nephrotoxic, and septic insults affecting approximately 20% of all hospital patients and up to 50% of patients in the intensive care unit. There are currently no therapeutics for patients who suffer AKI. Much recent work has focused on designing and implementing therapeutics for AKI. This review focuses on a family of enzymes known as sirtuins that play critical roles in regulating many cellular and biological functions. There are 7 mammalian sirtuins (SIRT1–7) that play roles in regulating the acylation of a wide variety of pathways. Furthermore, all but one of the mammalian sirtuins have been shown to play critical roles in mediating AKI based on preclinical studies. These diverse enzymes show exciting potential for therapeutic manipulation. This review will focus on the specific roles of each of the investigated sirtuins and the potential for manipulation of the various sirtuins and their effector pathways in mediating kidney injury.
Similar content being viewed by others
AKI clinical findings
Acute kidney injury (AKI) is a significant health care concern associated with high morbidity and mortality [1, 2]. Approximately 20% of hospitalized patients have AKI, and 20–60% of adult critically ill patients have AKI [3]. Recent observational studies in pediatric populations including the Assessment of Worldwide Acute Kidney Injury, Renal Angina, and Epidemiology (AWARE)—critically ill children and Assessment of Worldwide Acute Kidney injury Epidemiology in Neonates (AWAKEN) studies have shown similar alarming trends related to morbidity and mortality in this population [4]. AKI is characterized by an abrupt decline of kidney function, resulting in an inability to maintain electrolyte, acid-base, and fluid homeostasis [5]. AKI is a complex and multifactorial disease typically occurring as a mixed etiology of ischemia, nephrotoxicity, and sepsis. Furthermore, a recent landmark study found that COVID-19 patients presented with increased prevalence of AKI and with a distinct pathophysiology, signifying a new risk factor for AKI [6]. Many groups are working to identify the underlying mechanisms involved in AKI, including apoptosis, dysregulation of metabolism, autophagy, inflammation, and the cell cycle [7,8,9,10,11]. However, despite considerable improvements to our understanding of the pathophysiology of AKI, the exact mechanisms are still poorly understood, and no specific therapy exists.
AKI pathophysiology
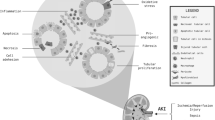
Multiple models of ischemic and toxic injury to the kidneys show a special susceptibility to injury of the renal tubular epithelial cells (RTECs). The proximal tubular epithelial cells are especially sensitive to injury because they require more active transport mechanisms than other kidney cell types. During AKI, there are 3 major types of changes that are observed and they are related to nuclear/DNA, cytoplasmic, or immune response. These are discussed below.
Nuclear/DNA changes
Significant work in the AKI field has focused on the role of nuclear/DNA alterations including DNA damage which directly effects DNA repair, cell cycle arrest, senescence, and/or cell death in RTECs [12]. Much work has also been done related to the role of increased mitosis/proliferative mechanisms and how the proximal tubule has an amazing ability to repair through dedifferentiation and expression of developmental markers followed by redifferentiation [13]. Finally, recent work has characterized how epigenetic changes including deacetylation/acetylation and demethylation/methylation affect the RTECs [14].
Cytoplasmic changes
Cytoplasmic alterations of RTECs during AKI is a growing field with recent studies focusing on many different aspects of cellular homeostasis, such as changes in expression levels of mitochondrial enzymes, protein acylation/deacylation, peroxisomal function, energy utilization (particularly fatty acid oxidation in the mitochondrial rich RTECs), upregulation of pro-apoptotic pathways, elevated pro-oxidative pathways, reactive oxygen species (ROS) accumulation, and cytoskeletal component changes, among others [15].
Immune response
Although intracellular signaling and mechanisms of RTECs play a critical role in driving the injury and repair response, the role of extra-tubular and in particular recruitment of pro-inflammatory, pro-fibrotic, and immune factors following RTEC damage can be paramount to tissue recovery/repair [10]. The ability of the RTECs to mount an extracellular response and recruit in the factors to clean up and repair the damaged cells can make a significant impact upon whether the injury progresses towards a fibrotic response and subsequent future injury that can culminate in chronic kidney disease or whether the injury resolves.
Role of sirtuins in RTECs during AKI
Sirtuins are a multidimensional group of genes that may provide insight into protective mechanisms during AKI. Sirtuins are a family of NAD+-dependent class III histone deacylases. The first identified sirtuin, Sir2, was found in budding yeast Saccharomyces cerevisiae and described as a regulator of transcriptional silencing of mating-type loci [16]. The discovery of Sir2’s dependence on NAD+ revealed a role for sirtuins as both energy sensors and as transcriptional effectors regulating the acetylation state of histones [17]. Subsequently, many homologs of Sir2 have been discovered across all domains of life, establishing a highly conserved class of enzymes [18,19,20].
In mammals, there are seven sirtuins, SIRT1–7, which function to regulate metabolism and other diverse physiologic processes through direct enzymatic action on target proteins. Sirtuins act in different cellular compartments and exhibit broad enzymatic activity as deacetylases, mono-ADP ribosyltransferases, demalonylases, deglutarylases, and desuccinylases [21,22,23]. Apart from the classic role as histone deacetylases, a diverse set of protein targets have also been identified in the cytoplasm and mitochondria [24]. Although sirtuins have notably been studied for their role in caloric restriction and the prevention of aging-related diseases such as cardiovascular disease and diabetes, their diverse substrates and role as sensors of cellular energy balance make them a critical player to restoring cellular homeostasis following injury [25]. The kidney is one organ majorly susceptible to age-related diseases, and sirtuins have been implicated in the pathophysiology of chronic and acute kidney diseases [26, 27].
In this review, we will focus on the functions of mammalian sirtuins and address their role in kidney physiology and AKI.
Sirtuin mechanism of action in the nucleus/DNA of damaged RTECs
There are 3 nuclear sirtuins (SIRT1, 6, and 7) that have been shown to play key roles in AKI. SIRT1 and SIRT6 are found in the nucleus, while SIRT7 is uniquely localized to the nucleolus (Table 1). SIRT1 is the most studied sirtuin. Originally described as a histone deacetylase, it was soon discovered that SIRT1 deacetylates many other proteins [22, 28, 29]. Following DNA damage, SIRT1 deacetylates and represses p53 to reduce cell apoptosis and senescence [31, 32]. Similarly, SIRT1 regulates the acetylation of the forkhead box type O (FOXO) transcription factors to attenuate FOXO-induced apoptosis and cell cycle arrest [33]. SIRT1 also regulates both members of the PGC-1α/ERR-α complex, essential metabolic transcription factors which control mitochondrial biogenesis and gluconeogenesis (Table 1) [34,35,36]. Mitochondrial dysfunction plays a critical role in the pathogenesis of AKI, particularly in relation to maladaptive induction of apoptosis. When an injury or stress exceeds the mitochondria’s ability to sense and respond to changes in nutrient availability, apoptosis is commonly initiated [37]. Apoptosis can activate intrinsically when the mitochondria fragments in response to decreased energy supply or when cytochrome C is released following outer mitochondrial membrane permeabilization [38]. Apoptosis of the tubular epithelial cells is further mediated by a network of factors including tumor suppressor protein p53, BCL2 family proteins, and caspases [39]. Given SIRT1’s strong role in regulating apoptosis, it is not surprising there is a wealth of literature describing the renoprotective effects of SIRT1. In its renoprotective capacity, SIRT1 was shown to protect against oxidative stress-induced apoptosis via deacetylation of FOXO3 in proximal tubular epithelial cells [40]. Further to this, in proximal tubular-specific SIRT1 transgenic mice, cisplatin-induced injury was attenuated by maintaining peroxisome number with concomitant upregulation of catalase and reduction of renal oxidative stress [41]. In another study, it was shown that SIRT1 activates PGC-1α, resulting in renoprotection by activating mitochondrial biogenesis and improved respiration via oxidative phosphorylation [42]. Finally, SIRT1 also showed a protective effect by regulating apoptosis through deacetylating p53 and inhibiting p53-dependent transcription during cellular stress [43]. Thus, the mechanism of Sirt1 protection is mediated by suppression of apoptosis likely downstream of metabolic signaling pathways (Fig. 1). SIRT6 and SIRT7 primarily have been shown to affect the inflammatory response following AKI and are discussed below.

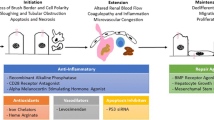
Sirtuin roles in limiting kidney injury. Sirtuins are expressed in different subcellular compartments and regulate different cellular and biological functions to impact kidney injury. SIRT3 and SIRT5 are mitochondrial sirtuins. SIRT3 maintains mitochondrial dynamics and energy homeostasis and promotes autophagy. SIRT5 is involved in fatty acid oxidation and energy metabolism. SIRT1, SIRT6, and SIRT7 are nuclear sirtuins. SIRT1 has a strong role in maintaining mitochondria as well as in regulating apoptosis. SIRT6 is important for the maintenance of podocyte and glomerular function. SIRT 7 is involved in inflammation through regulation of NF-κB. SIRT2 shuttles between the nucleus and cytoplasm. Nuclear SIRT2 regulates MKP-1 to promote inflammation. Sirtuins in green font are renoprotective, whereas those in red font were renoprotective when deleted. Only Sirt4 has not been studied in the context of kidney injury
Sirtuin mechanism of action in cytoplasmic components/mitochondria of damaged RTECs
There are 3 known mitochondrial sirtuins (SIRT3, 4, and 5) (Table 1). While SIRT3 is responsible for global protein deacetylation in mitochondria, SIRT4 exists as a mitochondrial ADP-ribosyltransferase and SIRT5 exhibits enzymatic activities as a deacetylase, desuccinylase, and demalonylase (Table 1). The roles of SIRT3 and SIRT5 have been elucidated in kidney injury and are discussed below, while the role of SIRT4 is currently unknown in this context.
SIRT2 is localized primarily to the cytoplasm, and there is growing evidence for additional roles in the nucleus (Table 1). Like SIRT1, SIRT2 also regulates PGC-1α and FOXO transcription factors [44, 45]. SIRT2 also deacetylates α-tubulin to affect mitotic progression [46, 47]. During the G2/M transition, nuclear SIRT2 deacetylates histone H4K20 to regulate cell cycle progression and genome stability [48]. Moreover, SIRT2’s activity mediates caspase-3 levels to affect apoptosis and oxidative stress (Table 1) [49].
As the major mitochondrial deacetylase, SIRT3 is responsible for the regulation of several metabolic enzymes and components of oxidative phosphorylation. SIRT3 deacetylates and activates mitochondrial acetyl-CoA synthetase (AceCS2), an enzyme involved in acetate utilization [50]. Similarly, SIRT3 has been shown to deacetylate long-chain acyl-CoA dehydrogenase (LCAD) to regulate fatty acid oxidation [51]. SIRT3 has also been demonstrated to deacetylate mitochondrial ribosome subunit MRPL10 to inhibit mitochondrial protein synthesis [52]. In addition, SIRT3 regulates various components of the electron chain, such as complex II and ATP synthase, to enhance ATP levels [53, 54]. These studies reflect only part of SIRT3’s wide range of functions in mitochondrial ATP production, fatty acid oxidation, mitochondrial homeostasis, and ROS management (Table 1). SIRT3 plays a significant role in the kidney, especially in the proximal and distal tubular compartments which contain abundant mitochondria. Several studies have described a renoprotective role for SIRT3 due to its role in maintaining mitochondrial dynamics and energy homeostasis. Indeed, Sirt3 knockout (KO) mice administered cisplatin have more severe AKI and compromised mitochondrial dynamics [55]. SIRT3 overexpression in mice has the effect of promoting autophagy through regulation of the AMPK/mTOR pathway and protecting against a model of sepsis-AKI [56]. SIRT3 also regulates FAO in mice by deacetylating liver kinase B1 and activating AMPK with the effect of reducing ROS and lipid peroxidation (Fig. 1) [57].
Mitochondrial SIRT5 was initially identified as a deacetylase targeting carbamoyl phosphate synthetase (CPS1) to regulate the urea cycle in liver [58]. However, it has since been discovered that SIRT5 functions as a demalonylase, deglutarylase, and desuccinylase rather than as a deacetylase [59]. SIRT5 has been shown to bind cardiolipin in the inner mitochondrial membrane and desuccinylate electron transport enzymes complex I, complex II, and ATP synthase [60]. SIRT5 desuccinylation targets identified via large-scale profiling studies suggest SIRT5 has a significant role in energy metabolism (Table 1) [61]. Recently, Sirt5 KO mice were shown to have significantly improved kidney function and less tubular damage following both ischemic and cisplatin challenge [62]. SIRT5 deficiency appears to be protective by reducing mitochondrial-derived ROS and driving peroxisomal FAO. Modifications to this mitochondrial-peroxisomal axis are significant to the pathogenesis of AKI. It is generally accepted that an underlying basis of kidney injury is impaired energetics in the highly metabolically active nephron segments (Fig. 1).
Sirtuin mechanism of action in recruiting inflammatory/ fibrotic factors to damaged RTECs
Several sirtuins (including Sirt2, Sirt3, Sirt6, and SIRT7) have been shown to play roles in the recruitment of inflammatory/fibrotic factors to the kidney after injury.
Previously, a lack of SIRT2 in mice reduces lipopolysaccharide (LPS)-induced increases in neutrophil gelatinase-associated lipocalin (NGAL) [63]. SIRT2 deficiency also reduced infiltration of renal neutrophils and macrophages and reduced expression of inflammatory chemokines CXCL2 and CCL2 [63]. The same group recently showed that SIRT2 affects mitogen-activated protein kinase-1 (MAPK-1), and Sirt2 KO mice and Sirt2 transgenic mice show amelioration and aggravation, respectively, of kidney injury, apoptosis, and inflammation induced by cisplatin (Fig. 1) [64].
SIRT3 overexpression promotes autophagy, upregulates p-AMPK and downregulates p-mTOR in cecal ligation and puncture mice, attenuating sepsis-induced AKI, tubular cell apoptosis, and inflammatory cytokine accumulation in the kidneys [65]. The blockage of autophagy induction largely abolished the protective effect of SIRT3 in sepsis-induced AKI. These findings indicate that SIRT3 protects against sepsis-induced AKI by inducing autophagy through regulation of the AMPK/mTOR pathway.
SIRT6 plays a significant role in genomic DNA stability and repair. Sirt6 knockout mice present with severe progeria and typically only live for 3 months [66]. SIRT6 deacetylates histone H3 at various lysine sites to maintain genome integrity and telomere function [67]. In response to DNA damage, it has been shown that SIRT6 promotes DNA repair under oxidative stress by activating poly[adenosine diphosphate (ADP)-ribose] polymerase 1 (PARP1; Table 1) [68]. In the context of the kidney, SIRT6 appears to be important for podocyte function and maintenance of glomerular function, as Sirt6 deletion in mice induces podocyte injury and decreased slit diaphragm protein expression [69]. Sirt6 overexpression in HK-2 kidney epithelial cells inhibits apoptosis induced by LPS and promotes autophagy while SIRT6 silencing promotes the secretion of cytokines tumor necrosis factor α (TNF-α) and interleukin-6 (IL-6) [70]. SIRT6 also deacetylates histone 3 to effectively inhibit extracellular signal-regulated kinase 1/2 (ERK1/2) expression and reduce inflammation and apoptosis caused by cisplatin (Fig. 1) [71].
SIRT7 uniquely activates RNA polymerase I (RNA Pol I) by deacetylating upstream binding factor (UBF) [72]. Others have shown that SIRT7 regulates RNA Pol I transcription by deacetylating PAD53, a component of RNA Pol I (Table 1) [73]. Interestingly, the lack of SIRT7 in mice shows protection against cisplatin-induced AKI. By regulating the nuclear expression of transcription factor nuclear factor kappa B (NF-κB), SIRT7 deficiency ameliorates cisplatin-induced AKI [74]. NF-κB is a potent stimulator of the immune system and the inflammatory response following AKI. Further to this, studies have shown that the inhibition of NF-κB can attenuate the inflammatory response and reduce the amount of injury following AKI [75]. Both the innate and adaptive immune systems are involved in the pathogenesis of AKI and virtually every immune cell has been implicated in AKI [76,77,78,79]. The protective phenomenon seen in SIRT7 deficient mice is perhaps due to SIRT7 deficiency reducing the expression of TNF-α, which normally enhances ROS production through the NADPH oxidase complex (Fig. 1) [74].
Potential therapies and clinical implications
The study of sirtuins has revealed a number of protein targets involved in the pathogenesis of AKI, and some sirtuins have been shown to exert strong renoprotective effects. As such, sirtuin-activating compounds (STACS) represent a clinically relevant approach to treat kidney diseases. Considerable efforts have been put towards finding small molecules to modulate the activity of sirtuins for pharmaceutical purposes. Resveratrol was among the first STACs identified in 2003 for its ability to significantly increase SIRT1. However, while in vitro studies indicate resveratrol activates SIRT1 [80], its mechanism was quickly disputed as this effect might instead be a downstream result from its immediate biological targets [81, 82]. Nevertheless, several clinical trials of resveratrol and SRT2104, another SIRT1 activator, have shown promising results in diabetes and cardiovascular disease [83]. NAD+ boosters as sirtuin activators represent another emerging therapeutic area of interest as NAD+ depletion is a major contributor to the pathogenesis of kidney diseases. NAD+ repletion through the pharmacological manipulation of nicotinamide phosphoribosyltransferase (the rate-limiting enzyme in the NAD+ salvage pathway) has been shown to have therapeutic potential as a means to improve kidney function and decrease tubular injury [55].
Given that deletion of SIRT5 and SIRT7 is renoprotective, another therapeutic avenue exists for selective sirtuin inhibitors. In the case of SIRT5, which uniquely targets succinyl modifications, inhibitors targeting the succinyl substrate are currently under development [84, 85]. The development of either STACs or sirtuin inhibitors holds some risk as sirtuins are involved in myriad pathways, and whether modulating sirtuins will have a beneficial or deleterious effect in humans is unclear. The compromise to this double-edged sword is the development of therapeutics targeting the pathways shown to be regulated by sirtuins rather than the sirtuins themselves.
Conclusion
The study of sirtuins in the kidney has led to impressive advances in our understanding of sirtuin targets involved in renoprotection and in the development of a number of different pharmacological interventions that are effective in ameliorating injury in animal models of AKI. However, the promise of these developments is generally tempered by the results of clinical trials in patients with AKI. Therapies effective in animal models of AKI have translated to little or no effectiveness in humans, and such therapies are yet to be explored in pediatric populations. However, this failing might stem from deficiencies in preclinical models of AKI and an ability to design the clinical trial itself [86]. Although many auspicious sirtuin targets have been identified, the failure to effectively translate animal data to an effective human intervention highlights the importance of studying AKI in multiple model systems. In addition to the multiple in vivo models of AKI available, including the ischemia-reperfusion model, cisplatin-induced AKI, and sepsis-associated AKI, multiple in vitro models are used to study AKI [87, 88]. The mechanistic differences between these various models add to the complexity of AKI pathogenesis, and elucidating the role that sirtuins play in each model will further the understanding and therapeutic application of the sirtuins.
Data availability
Not applicable.
References
Mehta RL, Cerdá J, Burdmann EA, Tonelli M, García-García G, Jha V, Susantitaphong P, Rocco M, Vanholder R, Sever MS, Cruz D, Jaber B, Lameire NH, Lombardi R, Lewington A, Feehally J, Finkelstein F, Levin N, Pannu N, Thomas B, Aronoff-Spencer E, Remuzzi G (2015) International Society of Nephrology’s 0by25 initiative for acute kidney injury (zero preventable deaths by 2025): a human rights case for nephrology. Lancet 385:2616–2643
Coca SG, Yusuf B, Shlipak MG, Garg AX, Parikh CR (2009) Long-term risk of mortality and other adverse outcomes after acute kidney injury: a systematic review and meta-analysis. Am J Kidney Dis 53:961–973
Susantitaphong P, Cruz DN, Cerda J, Abulfaraj M, Alqahtani F, Koulouridis I, Jaber BL, Acute Kidney Injury Advisory Group of the American Society of Nephrology (2013) World incidence of AKI: a meta-analysis. Clin J Am Soc Nephrol 8:1482–1493
Ciccia E, Devarajan P (2017) Pediatric acute kidney injury: prevalence, impact and management challenges. Int J Nephrol Renov Dis 10:77–84
Basile DP, Anderson MD, Sutton TA (2012) Pathophysiology of acute kidney injury. Compr Physiol 2:1303–1353
Batlle D, Soler MJ, Sparks MA, Hiremath S, South AM, Welling PA, Swaminathan S, COVID-19 and ACE2 in Cardiovascular, Lung, and Kidney Working Group (2020) Acute kidney injury in COVID-19: emerging evidence of a distinct pathophysiology. J Am Soc Nephrol 31:1380–1383
Bonegio R, Lieberthal W (2002) Role of apoptosis in the pathogenesis of acute renal failure. Curr Opin Nephrol Hypertens 11:301–308
Wei Q, Xiao X, Fogle P, Dong Z (2014) Changes in metabolic profiles during acute kidney injury and recovery following ischemia/reperfusion. PLoS One 9:e106647
Kaushal GP, Shah SV (2016) Autophagy in acute kidney injury. Kidney Int 89:779–791
Rabb H, Griffin MD, McKay DB, Swaminathan S, Pickkers P, Rosner MH, Kellum JA, Ronco C, Acute Dialysis Quality Initiative Consensus XIII Work Group (2016) Inflammation in AKI: current understanding, key questions, and knowledge gaps. J Am Soc Nephrol 27:371–379
Price PM, Safirstein RL, Megyesi J (2009) The cell cycle and acute kidney injury. Kidney Int 76:604–613
Yan M, Tang C, Ma Z, Huang S, Dong Z (2016) DNA damage response in nephrotoxic and ischemic kidney injury. Toxicol Appl Pharmacol 313:104–108
Humphreys BD (2014) Kidney injury, stem cells and regeneration. Curr Opin Nephrol Hypertens 23:25–31
Guo C, Dong G, Liang X, Dong Z (2019) Epigenetic regulation in AKI and kidney repair: mechanisms and therapeutic implications. Nat Rev Nephrol 15:220–239
Ralto KM, Parikh SM (2016) Mitochondria in acute kidney injury. Semin Nephrol 36:8–16
Klar AJ, Fogel S, Macleod K (1979) MAR1-a regulator of the HMa and HMalpha loci in Saccharomyces cerevisiae. Genetics 93:37–50
Imai S, Armstrong CM, Kaeberlein M, Guarente L (2000) Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403:795–800
Brachmann CB, Sherman JM, Devine SE, Cameron EE, Pillus L, Boeke JD (1995) The SIR2 gene family, conserved from bacteria to humans, functions in silencing, cell cycle progression, and chromosome stability. Genes Dev 9:2888–2902
Derbyshire MK, Weinstock KG, Strathern JN (1996) HST1, a new member of the SIR2 family of genes. Yeast 12:631–640
Michan S, Sinclair D (2007) Sirtuins in mammals: insights into their biological function. Biochem J 404:1–13
Sauve AA (2010) Sirtuin chemical mechanisms. Biochim Biophys Acta 1804:1591–1603
Sauve AA, Wolberger C, Schramm VL, Boeke JD (2006) The biochemistry of sirtuins. Annu Rev Biochem 75:435–465
Carafa V, Rotili D, Forgione M, Cuomo F, Serretiello E, Hailu GS, Jarho E, Lahtela-Kakkonen M, Mai A, Altucci L (2016) Sirtuin functions and modulation: from chemistry to the clinic. Clin Epigenetics 8:61
Verdin E, Hirschey MD, Finley LW, Haigis MC (2010) Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends Biochem Sci 35:669–675
Kupis W, Pałyga J, Tomal E, Niewiadomska E (2016) The role of sirtuins in cellular homeostasis. J Physiol Biochem 72:371–380
Morigi M, Perico L, Benigni A (2018) Sirtuins in renal health and disease. J Am Soc Nephrol 29:1799–1809
Kitada M, Kume S, Koya D (2014) Role of sirtuins in kidney disease. Curr Opin Nephrol Hypertens 23:75–79
Zhang T, Kraus WL (2010) SIRT1-dependent regulation of chromatin and transcription: linking NAD(+) metabolism and signaling to the control of cellular functions. Biochim Biophys Acta 1804:1666–1675
Rahman S, Islam R (2011) Mammalian Sirt1: insights on its biological functions. Cell Commun Signal 9:11
Liszt G, Ford E, Kurtev M, Guarente L (2005) Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. J Biol Chem 280:21313–21320
Langley E, Pearson M, Faretta M, Bauer UM, Frye RA, Minucci S, Pelicci PG, Kouzarides T (2002) Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J 21:2383–2396
Vaziri H, Dessain SK, Eaton EN, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA (2001) hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 107:149–159
Giannakou ME, Partridge L (2004) The interaction between FOXO and SIRT1: tipping the balance towards survival. Trends Cell Biol 14:408–412
Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P (2005) Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 434:113–118
Wilson BJ, Tremblay AM, Deblois G, Sylvain-Drolet G, Giguère V (2010) An acetylation switch modulates the transcriptional activity of estrogen-related receptor alpha. Mol Endocrinol 24:1349–1358
Nemoto S, Fergusson MM, Finkel T (2005) SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha}. J Biol Chem 280:16456–16460
Havasi A, Borkan SC (2011) Apoptosis and acute kidney injury. Kidney Int 80:29–40
Waterhouse NJ, Goldstein JC, von Ahsen O, Schuler M, Newmeyer DD, Green DR (2001) Cytochrome c maintains mitochondrial transmembrane potential and ATP generation after outer mitochondrial membrane permeabilization during the apoptotic process. J Cell Biol 153:319–328
Kinsey GR, Okusa MD (2011) Pathogenesis of acute kidney injury: foundation for clinical practice. Am J Kidney Dis 58:291–301
Wang YQ, Cao Q, Wang F, Huang LY, Sang TT, Liu F, Chen SY (2015) SIRT1 protects against oxidative stress-induced endothelial progenitor cells apoptosis by inhibiting FOXO3a via FOXO3a ubiquitination and degradation. J Cell Physiol 230:2098–2107
Hasegawa K, Wakino S, Yoshioka K, Tatematsu S, Hara Y, Minakuchi H, Sueyasu K, Washida N, Tokuyama H, Tzukerman M, Skorecki K, Hayashi K, Itoh H (2010) Kidney-specific overexpression of Sirt1 protects against acute kidney injury by retaining peroxisome function. J Biol Chem 285:13045–13056
Tran MT, Zsengeller ZK, Berg AH, Khankin EV, Bhasin MK, Kim W, Clish CB, Stillman IE, Karumanchi SA, Rhee EP, Parikh SM (2016) PGC1alpha drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 531:528–532
Kim JY, Jo J, Kim K, An HJ, Gwon MG, Gu H, Kim HJ, Yang AY, Kim SW, Jeon EJ, Park JH, Leem J, Park KK (2019) Pharmacological activation of Sirt1 ameliorates cisplatin-induced acute kidney injury by suppressing apoptosis, oxidative stress, and inflammation in mice. Antioxidants (Basel) 8:322
Jing E, Gesta S, Kahn CR (2007) SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metab 6:105–114
Krishnan J, Danzer C, Simka T, Ukropec J, Walter KM, Kumpf S, Mirtschink P, Ukropcova B, Gasperikova D, Pedrazzini T, Krek W (2012) Dietary obesity-associated Hif1alpha activation in adipocytes restricts fatty acid oxidation and energy expenditure via suppression of the Sirt2-NAD+ system. Genes Dev 26:259–270
Skoge RH, Dolle C, Ziegler M (2014) Regulation of SIRT2-dependent alpha-tubulin deacetylation by cellular NAD levels. DNA Repair (Amst) 23:33–38
Dryden SC, Nahhas FA, Nowak JE, Goustin AS, Tainsky MA (2003) Role for human SIRT2 NAD-dependent deacetylase activity in control of mitotic exit in the cell cycle. Mol Cell Biol 23:3173–3185
Serrano L, Martínez-Redondo P, Marazuela-Duque A, Vazquez BN, Dooley SJ, Voigt P, Beck DB, Kane-Goldsmith N, Tong Q, Rabanal RM, Fondevila D, Muñoz P, Krüger M, Tischfield JA, Vaquero A (2013) The tumor suppressor SirT2 regulates cell cycle progression and genome stability by modulating the mitotic deposition of H4K20 methylation. Genes Dev 27:639–653
Nie H, Hong Y, Lu X, Zhang J, Chen H, Li Y, Ma Y, Ying W (2014) SIRT2 mediates oxidative stress-induced apoptosis of differentiated PC12 cells. Neuroreport 25:838–842
Hallows WC, Lee S, Denu JM (2006) Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc Natl Acad Sci U S A 103:10230–10235
Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, Stevens RD, Li Y, Saha AK, Ruderman NB, Bain JR, Newgard CB, Farese RV Jr, Alt FW, Kahn CR, Verdin E (2010) SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464:121–125
Yang Y, Cimen H, Han MJ, Shi T, Deng JH, Koc H, Palacios OM, Montier L, Bai Y, Tong Q, Koc EC (2010) NAD+-dependent deacetylase SIRT3 regulates mitochondrial protein synthesis by deacetylation of the ribosomal protein MRPL10. J Biol Chem 285:7417–7429
Finley LW, Haas W, Desquiret-Dumas V, Wallace DC, Procaccio V, Gygi SP, Haigis MC (2011) Succinate dehydrogenase is a direct target of sirtuin 3 deacetylase activity. PLoS One 6:e23295
Rahman M, Nirala NK, Singh A, Zhu LJ, Taguchi K, Bamba T, Fukusaki E, Shaw LM, Lambright DG, Acharya JK, Acharya UR (2014) Drosophila Sirt2/mammalian SIRT3 deacetylates ATP synthase beta and regulates complex V activity. J Cell Biol 206:289–305
Morigi M, Perico L, Rota C, Longaretti L, Conti S, Rottoli D, Novelli R, Remuzzi G, Benigni A (2015) Sirtuin 3–dependent mitochondrial dynamic improvements protect against acute kidney injury. J Clin Invest 125:715–726
Zhao WY, Zhang L, Sui MX, Zhu YH, Zeng L (2016) Protective effects of sirtuin 3 in a murine model of sepsis-induced acute kidney injury. Sci Rep 6:33201
Li M, Li CM, Ye ZC, Huang J, Li Y, Lai W, Peng H, Lou TQ (2020) Sirt3 modulates fatty acid oxidation and attenuates cisplatin-induced AKI in mice. J Cell Mol Med 24:5109–5121
Nakagawa T, Lomb DJ, Haigis MC, Guarente L (2009) SIRT5 Deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell 137:560–570
Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, Kim J, Woo J, Kim JH, Choi BH, He B, Chen W, Zhang S, Cerione RA, Auwerx J, Hao Q, Lin H (2011) Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 334:806–809
Zhang Y, Bharathi SS, Rardin MJ, Lu J, Maringer KV, Sims-Lucas S, Prochownik EV, Gibson BW, Goetzman ES (2017) SIRT5 binds to cardiolipin and regulates the electron transport chain. J Biol Chem 292:10239–10249
Park J, Chen Y, Tishkoff DX, Peng C, Tan M, Dai L, Xie Z, Zhang Y, Zwaans BM, Skinner ME, Lombard DB, Zhao Y (2013) SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol Cell 50:919–930
Chiba T, Peasley KD, Cargill KR, Maringer KV, Bharathi SS, Mukherjee E, Zhang Y, Holtz A, Basisty N, Yagobian SD, Schilling B, Goetzman ES, Sims-Lucas S (2019) Sirtuin 5 regulates proximal tubule fatty acid oxidation to protect against AKI. J Am Soc Nephrol 30:2384–2398
Jung YJ, Lee AS, Nguyen-Thanh T, Kim D, Kang KP, Lee S, Park SK, Kim W (2015) SIRT2 regulates LPS-induced renal tubular CXCL2 and CCL2 expression. J Am Soc Nephrol 26:1549–1560
Jung YJ, Park W, Kang KP, Kim W (2020) SIRT2 is involved in cisplatin-induced acute kidney injury through regulation of mitogen-activated protein kinase phosphatase-1. Nephrol Dial Transplant 35:1145–1156
Zhao W, Zhang L, Chen R, Lu H, Sui M, Zhu Y, Zeng L (2018) SIRT3 protects against acute kidney injury via AMPK/mTOR-regulated autophagy. Front Physiol 9:1526
Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, Liu P, Mostoslavsky G, Franco S, Murphy MM, Mills KD, Patel P, Hsu JT, Hong AL, Ford E, Cheng HL, Kennedy C, Nunez N, Bronson R, Frendewey D, Auerbach W, Valenzuela D, Karow M, Hottiger MO, Hursting S, Barrett JC, Guarente L, Mulligan R, Demple B, Yancopoulos GD, Alt FW (2006) Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 124:315–329
Tennen RI, Chua KF (2011) Chromatin regulation and genome maintenance by mammalian SIRT6. Trends Biochem Sci 36:39–46
Mao Z, Hine C, Tian X, Van Meter M, Au M, Vaidya A, Seluanov A, Gorbunova V (2011) SIRT6 promotes DNA repair under stress by activating PARP1. Science 332:1443–1446
Huang W, Liu H, Zhu S, Woodson M, Liu R, Tilton RG, Miller JD, Zhang W (2017) Sirt6 deficiency results in progression of glomerular injury in the kidney. Aging (Albany NY) 9:1069–1083
Zhang Y, Wang L, Meng L, Cao G, Wu Y (2019) Sirtuin 6 overexpression relieves sepsis-induced acute kidney injury by promoting autophagy. Cell Cycle 18:425–436
Li Z, Xu K, Zhang N, Amador G, Wang Y, Zhao S, Li L, Qiu Y, Wang Z (2018) Overexpressed SIRT6 attenuates cisplatin-induced acute kidney injury by inhibiting ERK1/2 signaling. Kidney Int 93:881–892
Grob A, Roussel P, Wright JE, McStay B, Hernandez-Verdun D, Sirri V (2009) Involvement of SIRT7 in resumption of rDNA transcription at the exit from mitosis. J Cell Sci 122:489–498
Chen S, Seiler J, Santiago-Reichelt M, Felbel K, Grummt I, Voit R (2013) Repression of RNA polymerase I upon stress is caused by inhibition of RNA-dependent deacetylation of PAF53 by SIRT7. Mol Cell 52:303–313
Miyasato Y, Yoshizawa T, Sato Y, Nakagawa T, Miyasato Y, Kakizoe Y, Kuwabara T, Adachi M, Ianni A, Braun T, Komohara Y, Mukoyama M, Yamagata K (2018) Sirtuin 7 deficiency ameliorates cisplatin-induced acute kidney injury through regulation of the inflammatory response. Sci Rep 8:5927
Zhang H, Sun SC (2015) NF-kappaB in inflammation and renal diseases. Cell Biosci 5:63
Bolisetty S, Agarwal A (2009) Neutrophils in acute kidney injury: not neutral any more. Kidney Int 75:674–676
Lee S, Huen S, Nishio H, Nishio S, Lee HK, Choi BS, Ruhrberg C, Cantley LG (2011) Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol 22:317–326
Li L, Huang L, Sung SJ, Lobo PI, Brown MG, Gregg RK, Engelhard VH, Okusa MD (2007) NKT cell activation mediates neutrophil IFN-gamma production and renal ischemia-reperfusion injury. J Immunol 178:5899–9115
Renner B, Strassheim D, Amura CR, Kulik L, Ljubanovic D, Glogowska MJ, Takahashi K, Carroll MC, Holers VM, Thurman JM (2010) B cell subsets contribute to renal injury and renal protection after ischemia/reperfusion. J Immunol 185:4393–4400
Alcain FJ, Villalba JM (2009) Sirtuin activators. Expert Opin Ther Pat 19:403–414
Beher D, Wu J, Cumine S, Kim KW, Lu SC, Atangan L, Wang M (2009) Resveratrol is not a direct activator of SIRT1 enzyme activity. Chem Biol Drug Des 74:619–624
Pacholec M, Bleasdale JE, Chrunyk B, Cunningham D, Flynn D, Garofalo RS, Griffith D, Griffor M, Loulakis P, Pabst B, Qiu X, Stockman B, Thanabal V, Varghese A, Ward J, Withka J, Ahn K (2010) SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J Biol Chem 285:8340–8351
Bonkowski MS, Sinclair DA (2016) Slowing ageing by design: the rise of NAD(+) and sirtuin-activating compounds. Nat Rev Mol Cell Biol 17:679–690
Kalbas D, Liebscher S, Nowak T, Meleshin M, Pannek M, Popp C, Alhalabi Z, Bordusa F, Sippl W, Steegborn C, Schutkowski M (2018) Potent and selective inhibitors of human sirtuin 5. J Med Chem 61:2460–2471
Rajabi N, Auth M, Troelsen KR, Pannek M, Bhatt DP, Fontenas M, Hirschey MD, Steegborn C, Madsen AS, Olsen CA (2017) Mechanism-based inhibitors of the human sirtuin 5 deacylase: structure-activity relationship, biostructural, and kinetic insight. Angew Chem Int Ed Eng 56:14836–14841
Skrypnyk NI, Siskind LJ, Faubel S, de Caestecker MP (2016) Bridging translation for acute kidney injury with better preclinical modeling of human disease. Am J Physiol Ren Physiol 310:F972–F984
Bao YW, Yuan Y, Chen JH, Lin WQ (2018) Kidney disease models: tools to identify mechanisms and potential therapeutic targets. Zool Res 39:72–86
Faria J, Ahmed S, Gerritsen KGF, Mihaila SM, Masereeuw R (2019) Kidney-based in vitro models for drug-induced toxicity testing. Arch Toxicol 93:3397–3418
Funding
This work was supported by an NIH R56 and R01 (SSL DK121758). Further, the work was supported by the University of Pittsburgh Dean’s Faculty Advancement award (SSL). The work was further supported by the Children’s Hospital of Pittsburgh RK Mellon foundation (TC).
Author information
Authors and Affiliations
Contributions
Kevin Peasley wrote the first draft of the manuscript and was involved in the editing process. Dr. Sunder Sims-Lucas wrote the manuscript, edited the paper, and is the corresponding author. Drs. Takuto Chiba and Eric Goetzman were heavily involved in editing the manuscript and making critical intellectual contributions.
Corresponding author
Ethics declarations
Conflict of interest
Not applicable.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
All authors have reviewed the manuscript and agreed to its publication.
Code availability
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Peasley, K., Chiba, T., Goetzman, E. et al. Sirtuins play critical and diverse roles in acute kidney injury. Pediatr Nephrol 36, 3539–3546 (2021). https://doi.org/10.1007/s00467-020-04866-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-020-04866-z