
Abstract
In this study, two types of ZnO nanorods (NRs) were hydrothermally grown on Si substrates by controlling the location of the Ga dopant. The growth behavior of ZnO NRs grown on the Ga-doped ZnO (GZO) seed layer (NR1s) was affected by the particle size controlled by the Ga dopant, while that of ZnO NRs grown in a Ga-doped aqueous solution (NR2s) was affected by complexes caused by the Ga dopant in the aqueous solution. The photoresponsivity of the ZnO NR1s overall increased with increasing concentration of Ga dopant due to the large surface area related to the density of ZnO NR1s. However, in case of the ZnO NR2s, the photoresponsivity of the undoped ZnO (UZO) NR2s was larger than that of the GZO NR2s due to the larger surface area for oxygen molecule adsorption in the UZO NR2s. Finally, under a 3-mM Ga doping concentration, the photoresponsivity of the ZnO NR1s was approximately 11.5 times higher than that of the ZnO NR2s, indicating that the ZnO NR1s are promising candidates for use in ultraviolet (UV) photodetector applications. These results indicate that when the ZnO NRs are grown using the hydrothermal method, doping at the seed layer is more suitable for high-performance UV photodetectors.
Similar content being viewed by others

1 Introduction
In recent years, ultraviolet (UV) photodetectors based on semiconductors have attracted considerable attention in a wide range of fields including water purification systems, flame detection, and early missile plume detection [1]. Therefore, the fabrication of high-performance UV photodetectors is important. Among semiconductor materials, ZnO is considered one of the most suitable materials for the fabrication of semiconductor-based UV photodetectors, owing to its wide direct band gap of 3.37 eV, large exciton binding energy of 60 meV, and the ease of synthesizing various one dimensional (1D) nanostructures including nanowires, nanobelts, and nanorods (NRs) [2]. Those 1D nanostructures have unique characteristics, such as a large surface-to-volume ratio and quantum-confinement effect, which can enhance the performance of UV photodetectors [3]. Several growth methods are used to fabricate ZnO NR-based UV photodetectors, including chemical vapor deposition, electrodeposition, pulsed-laser deposition, and the hydrothermal method [4,5,6,7]. Among these methods, the hydrothermal method has several advantages, including a low growth temperature, low cost, and easy control of the dopant concentration [8]. However, a seed layer is necessary to grow well-aligned ZnO NRs when ZnO NRs are grown using the hydrothermal method [9]. In addition, structural defects, such as the formation of interstitial oxygen caused by the generation of excess oxygen ions, are formed in the ZnO lattice during the hydrothermal method, thus, deteriorating the crystallinity and reducing the electrical conductivity of those NRs [10]. This phenomenon reduces the mobility of electrons by capturing the electrons in a potential well, thus, deteriorating the performance of hydrothermally grown ZnO NR-based UV photodetectors. Various approaches, such as change of growth parameters, doping, and thermal annealing at high temperature, have been developed to overcome the limitations of hydrothermally grown ZnO NRs [11, 12]. Among these methods, the doping method is the most fundamental method and involves the addition of group III elements, such as Al, Ga, and In. Especially, the addition of Ga dopant to ZnO NRs enhances the electrical conductivity of those NRs by generating electrons from the substitution of Ga dopants for Zn atoms [13]. In addition, the lattice distortion of Ga-doped ZnO (GZO) is reduced in comparison to that of the Al and In-doped ZnO because the covalent bond length of Ga–O (1.92 Å) is similar to the covalent bond length of the Zn–O (1.97 Å) but not to those of Al–O (2.70 Å) and In–O (2.10 Å) [14]. Therefore, the Ga dopant is most effective at increasing the electrical conductivity and reducing the defect density of the hydrothermally grown ZnO NRs.
When the ZnO NRs are grown via the hydrothermal method, the Ga dopant can be added to the ZnO NRs by using two doping locations. Herein, the doping location means possible locations that the Ga dopant can occupy during the preparation process of ZnO NRs. In other words, the Ga dopant can be added to the sol–gel solution used for the seed layer deposition and the aqueous solution used for the NR growth. When the Ga dopant is added during the preparation process for the ZnO NRs, the Ga dopant is generally added to an aqueous solution. [15, 16]. In this case, the ZnO NRs are formed on the ZnO seed layer during the growth process. After that, the Ga3+ ions present in the aqueous solution substitute at the Zn sites of ZnO NRs; consequently, GZO NRs are synthesized. However, at this time, lattice distortion has occurred due to the substituted Ga dopant and the formation of Ga2O3 on the surfaces of the GZO NRs, thus, causing a deterioration in the crystallinity of GZO NRs. Another method of adding the Ga dopant to ZnO NRs using the hydrothermal method is by adding the Ga dopant to the seed layer. Generally, the seed layer is important when using the hydrothermal method to grow ZnO NRs [17], and the morphological and structural properties of the seed layer significantly influence the structural and the optical properties of ZnO NRs. Contrary to a Ga-doped aqueous solution, the defects caused by the addition of the Ga dopant to the seed layer are not formed in the ZnO NRs which are grown on the GZO seed layer; thus, high-quality ZnO NRs can be synthesized. Moreover, the addition of Ga dopant to the ZnO seed layer decreases the particle size of the GZO seed layer [18], thus increasing the density of ZnO NRs, indicating that the high-density ZnO NRs have highly active surfaces, which stimulates the oxygen adsorption and the desorption processes [19].
Although two approaches can be used for doping ZnO NRs grown using the hydrothermal method, what type of location is related to the enhancement in the photoresponse properties of the ZnO NRs is uncertain. Therefore, the optimal doping location for enhancing the photoresponse properties of ZnO NRs must be investigated by comparing the ZnO NRs grown using two doping locations. Thus, in this study, we hydrothermally grew two types of ZnO NRs by controlling the doping location of the Ga dopant. In addition, we fabricated a ZnO NR-based UV photodetector to investigate the effect of doping location on the photoresponse properties of ZnO NRs.
2 Experiments and discussion
GZO NRs and ZnO NRs were grown on the ZnO and GZO seed layers, respectively, using the hydrothermal method. Prior to the deposition of the seed layer by using a sol–gel spin coating method, Si (100) substrates (5 ~ 10 Ω·cm) were immersed in a piranha solution (H2SO4:H2O2 = 4:1) for 15 min to remove the organic matter on the Si substrate’s surface. The Si substrates were then immersed in hydrofluoric (HF:H2O = 1:9) for 1 min to remove residual material and subsequently rinsed with deionized (DI) water for 2 min. Finally, the Si substrates were sequentially cleaned for 10 min in acetone (CH3COCH3, ≥ 99.5%, CHEMICALS) and ethanol (C2H5OH, ≥ 94.5%, CHEMICALS) with ultrasonication, rinsed with DI water and blown dried with nitrogen gas. To prepare the precursor solution for the ZnO seed layer, we dissolved zinc acetate dihydrate [Zn(CH3COO)2·2H2O, ≥ 99.0%, Junsei] in 2-methoxyethanol (CH3OCH2CH2OH, ≥ 99.0%, Junsei) and monoethanolamine (MEA, C2H7NO, ACS reagent, ≥ 99.0%, Sigma-Aldrich). The concentration of the ZnO precursor solution was 0.5 M and the zinc acetate/MEA molar ratio was 1: 1. On the other hand, to prepare the precursor solutions for the GZO seed layers, we added gallium nitrate hydrate (GaN3O9·xH2O) at 0, 1, 2, 3, or 4 mM to the zinc acetate solution. The resultant ZnO and GZO sol–gel solutions were stirred using a magnetic stirrer at 60 °C for 2 h, resulting in a clear and homogeneous solution. After that, the prepared ZnO and GZO sol–gel solutions were spin-coated on the Si substrates at 2000 rpm for 20 s using the sol–gel spin-coating method, after which the spin-coated ZnO and GZO seed layers were pre-heated for 10 min. This process of the spin-coating and pre-heat thermal treatment was repeated four times after which the ZnO and GZO seed layers were post-heated at 600 °C for 1 h in a furnace in air.
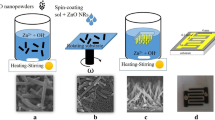
To grow the ZnO NRs on the heat-treated ZnO and GZO seed layers, respectively, using the hydrothermal method, we prepared two kinds of growth solutions. The growth solution used to grow ZnO NRs on the prepared GZO seed layer was obtained by dissolving zinc nitrate hexahydrate [Zn(NO3)2·6H2O, crystallized, ≥ 99.0%, Sigma-Aldrich] and hexamethylenetetramine (HMT, C6H12N4, ACS reagent, ≥ 99.0%, Sigma-Aldrich) at 0.025 M in 80 mL of DI water. The growth solution used to grow the ZnO NRs on the prepared ZnO seed layer was obtained by dissolving gallium nitrate hydrate (GaN3O9·xH2O) at 0, 1, 2, 3, or 4 mM in the precursor solution of 0.025 M. After that, the two types of ZnO NRs were grown on the ZnO and the GZO seed layers in a Teflon-lined autoclave at 140 °C for 4 h by using the resultant aqueous solutions. Herein, the two types of ZnO NRs grown on the GZO and the ZnO seed layers are named as ZnO NR1s and ZnO NR2s, respectively. As soon as the growths of the ZnO NR1s and ZnO NR2s had been completed, the hydrothermally grown ZnO NR1s and ZnO NR2s were rinsed with DI water and blown dry with nitrogen gas (Fig. 1).

Simple schematic of the growth processes for the two types of ZnO NRs
The morphological differences between the ZnO NR1s and the ZnO NR2s were observed by using field emission scanning electron microscopy (FE-SEM; TESCAN NIRA3LM), and the structural properties were validated by using X-ray diffraction (XRD) with Cu-kα radiation (λ = 0.15406 Å) at an accelerating voltage of 40 kV. The optical properties of the ZnO NR1s and the ZnO NR2s were analyzed using photoluminescence (PL) measurement with a He-Cd laser (325 nm) at an excitation power of 20 mW and a single grating monochromator of 0.75 m (HORIBA, Triax 550). In addition, indium electrodes were coated onto the Si substrates via a thermal evaporator, and the time-dependent photoresponsivity was measured under a bias voltage of 0.05 V with a 365-nm UV light at a power density of 10 mW/cm2.
Figure 2 shows top-view FE-SEM images of the ZnO NR1s with increasing Ga doping concentration from 0 to 4 mM. From the images, as the Ga doping concentration was increased, the density of the ZnO NR1s can be seen to increased gradually from 3.2 × 109 rods/cm2 to 5.1 × 109 rods/cm2. Generally, the morphology of the ZnO NRs grown using hydrothermal method is greatly affected by the seed layer, and the density of the ZnO NRs is determined by the number of particles on the seed layer [20]. The progress of the growth of ZnO NR1s can be expressed using the following reactions [21, 22]:

Top-view and cross-sectional FE-SEM images of ZnO NR1s with increasing Ga doping concentration: a 0 mM, b 1 mM, c 2 mM, d 3 mM, and e 4 mM
The sol–gel spin-coated GZO thin films consist of ZnO particles with positively charged (001)-Zn surfaces and negatively charged (00_1 \(\stackrel{-}{1}\) \(\stackrel{-}{1}\))-O surface, with the (001)-Zn surfaces having a higher chemical reactivity than the (00_1)-O surfaces [23]. At the initial stage of the ZnO NR1s growth, Zn2+ ions are generated from the zinc nitrate hexahydrate and OH– ions are generated from the dissociation of HMT (Eqs. 1 ~ 3). As soon as the Zn2+ and the OH− ions are generated, Zn(OH)42− complexes are formed through the combination of Zn2+ ions and OH− ions in the aqueous solution (Eq. 4). The generated negatively charged Zn(OH)42− complexes move to the positively charged (001)-Zn polar surface of the GZO seed layer and form ZnO nuclei that act as the growth units on the GZO seed layer (Eq. 5). After the formation of ZnO nuclei on the GZO seed layer, the remaining Zn2+ and OH− ions in the aqueous solution continue to form Zn(OH)2 complexes, and the generated Zn(OH)2 complexes serve as growth units for ZnO NRs (Eqs. 6, 7). From these results, the seed layer can be confirmed to play a crucial role in the growth of ZnO NRs because the ZnO nuclei, an essential factor in the ZnO NR growth, are formed on the seed layer [24]. In addition, the number of generated ZnO nuclei on the GZO seed layer increases with decreasing the particle size of the GZO seed layer due to the increased surface area [25]. We confirmed that the density of ZnO NR1s increased with increasing Ga doping concentration of the GZO seed layer; therefore, the morphological differences of the GZO seed layers with different Ga doping concentrations must be evaluated to confirm the effect of GZO seed layer on the growth of the ZnO NR1s.
Figure 3 shows the morphological differences of the GZO seed layers with different Ga doping concentrations. From the images, the particle size can be confirmed to decrease with increasing Ga doping concentration. This is because the addition of Ga dopant to the sol–gel solution interrupts the hydrolysis and condensation reactions during the preparation of the sol–gel solution, thus, decreasing the nucleation and grain growth of ZnO [26]. Thus, the particle size of the sol–gel spin-coated GZO seed layer was reduced with increasing Ga doping concentration. These results confirmed that the particle size of the GZO seed layer decreased with increasing Ga doping concentration and that the ZnO NR1s grown on the GZO seed layer with the highest Ga doping concentration of 4 mM had the highest density of all the ZnO NR1s. In addition, these results indicated that the particle size of the GZO seed layer influenced the density of ZnO NRs and that the density of ZnO NRs could be determined by controlling the Ga doping concentration in the GZO seed layer. The insets of Fig. 2 are cross-sectional FE-SEM images of the ZnO NR1s. The images show that no change in the length of the ZnO NR1s was observed up to a 2-mM Ga doping concentration and that as the Ga doping concentration was increased from 3 to 4 mM, the length of ZnO NR1s gradually increased from 1.92 to 2.14 μm. This is because the incorporation of the Ga dopant into the ZnO NRs improves the growth in the [001] direction and induces a reduction in the diameter of ZnO NRs [27]. In addition, the diameter of ZnO NR1s was observed to have been decreased from 0.18 to 0.10 μm with increasing Ga doping concentration to 4 mM. Therefore, the ZnO NR1s with the highest Ga doping concentration of 4 mM had the smallest diameter and the longest length compared to the others, thus, confirming that the diameter and length of the ZnO NRs can be controlled by controlling the Ga doping concentration.

FE-SEM images of a Ga-doped ZnO seed layer for various Ga doping concentrations: a 0 mM, b 1 mM, c 2 mM, d 3 mM, and e 4 mM
Figure 4 shows top-view FE-SEM images of ZnO NR2s with different Ga doping concentrations. In contrast to the ZnO NR1s grown on the GZO seed layer, the density of ZnO NR2s decreased with increasing Ga doping concentration from 2.8 × 109 rods/cm2 to 7 × 108 rods/cm2. The above results confirmed that the particle size of the GZO seed layer greatly influenced the density of the ZnO NR1s. However, the growth of the ZnO NR2s on the ZnO seed layer indicates that the ZnO NR2s are rarely affected by the particle size of the ZnO seed layer. The growth of ZnO NR2s can be expressed using the following equations [28, 29]:

Top-view and cross-sectional FE-SEM images of ZnO NR2s for various Ga doping concentrations: a 0 mM, b 1 mM, c 2 mM, d 3 mM, and e 4 mM
At the initial stage of the ZnO NR2s growth, the ZnO nuclei are generated on the ZnO seed layers and the remaining Zn2+ and OH− ions combine and form Zn(OH)2 complexes in the same way as the ZnO NR1s. However, when the Ga dopant was added into the aqueous solution, the Ga3+ ions from gallium nitrate hydrate reacted with OH− ions to form Ga(OH)3 complexes (Eq. 13), thus, reducing the formation of Zn(OH)2 complexes because the number of OH− ions that can react with Zn2+ ions is decreased. Therefore, under a high Ga doping concentration, the number of generated Ga(OH)3 complexes is increased and the number of generated Zn(OH)2 complexes is decreased. Consequently, the density of ZnO NR2s decreased with increasing the concentration of Ga dopant in the aqueous solution. The insets of Fig. 4 show the cross-sectional FE-SEM images of ZnO NR2s. The images show that the undoped ZnO (UZO) NR2s grew towards the [001] direction while the GZO NR2s gradually grew along the [100] and the [101] directions with increasing Ga doping concentration. The growth of the GZO NR2s toward a lateral direction is attributed to the formation Ga(OH)3 complexes in the aqueous solution with the Ga dopant added. In other words, the generated Ga(OH)3 complexes reduce heterogeneous nucleation, and then the reduced heterogeneous nucleation provides sufficient space to grow laterally rather than vertically during the growth of the ZnO NRs [30]. Thus, when the Ga dopant is added to the aqueous solution, the density of ZnO NR2s decreased with increasing Ga doping concentration, and the vertical growth direction of ZnO NR2s changed to a diagonal direction.
XRD analysis was conducted to investigate the crystal structure of hydrothermally grown ZnO NRs. Figure 5 shows XRD patterns of the ZnO NR1s with Ga doping concentrations of 0, 1, 2, 3, and 4 mM. A strong peak was observed in the patterns of all ZnO NR1s at 34.42°, which correspond to diffraction from the ZnO (002) plane. Two weak diffraction peaks were observed at 31.82° and 36.26°, which correspond to the diffraction from the ZnO (100) and the (101) planes, respectively, indicating that ZnO NR1s have a wurtzite structure [31]. Among the three ZnO diffraction peaks, the most prominent ZnO (002) peak indicates that the ZnO NRs are grown along a direction perpendicular to the substrate, which corresponds to the results of the FE-SEM measurements. Meanwhile, the ZnO (002) peak moved toward a higher angle from 34.42° to 34.48° with increasing Ga doping concentration. This phenomenon can be explained using Bragg’s law [32]:
where d is the interplanar distance, θ is the Bragg angle, n is the order of diffraction and is generally 1, and λ is the X-ray wavelength of the Cu-kα source (λ = 0.15406 nm). This is because the Ga3+ ions with a relatively small ionic radii (0.62 Å) replace the Zn2+ (0.74 Å) ions in the ZnO lattice [33]. Consequently, the interplanar distance is reduced, leading to a shift to higher Bragg angle with increasing Ga doping concentration. Interestingly, in the XRD pattern of the ZnO NR1s with the highest Ga doping concentration, an additional peak was observed at 37.46°, which corresponded to the formation of the Ga2O3 (401) plane as a secondary phase; Ga2O3 is generally formed in the grain boundary regions [34].

XRD patterns of ZnO NR1s with various Ga doping concentrations
Figure 6a shows the XRD patterns of the ZnO NR2s with different Ga doping concentrations. Three diffraction peaks were observed in the XRD patterns of all the ZnO NR2s: 31.78°, 34.42°, and 36.26°, corresponding to the ZnO (100), (002), and (101) peaks, respectively. This confirms the wurtzite structure of the ZnO NR2s without any secondary phases. Contrary to the XRD results for the ZnO NR1s, the intensity of the ZnO (002) peak gradually decreased with increasing Ga doping concentration. Generally, the intensity of the ZnO (002) peak is related to the number of the vertically grown ZnO NRs [35]. As shown in the FE-SEM results, as the Ga doping concentration was increase, the density of ZnO NR2s gradually decreased, and the growth direction of the ZnO NR2s is gradually inclined diagonally. Therefore, the number of the ZnO NR2s grown along the (002) direction gradually declined, which resulted in a reduction in the intensity of the ZnO (002) peak. To further understand the crystallographic preferred or random orientation of the ZnO NR2s with different Ga doping concentrations, we used the following equation to calculate the texture coefficient (TC) [36], and the results are shown in Fig. 6b:
where TC(hkl) is the texture coefficient of the (hkl) plane, I(hkl) is the relative intensity of (hkl) plane, Ir(hkl) is the intensity of oriented crystals, and n is the number of diffraction peaks. The TC represents the texture of a specific plane, namely, the deviation of a plane from the ideal structure. The UZO NR2s had the highest TC(002) value of 82.85%. The TC(002) value of the GZO NR2s decreased from 56.78 to 44.94%, whereas the TC(100) and the TC(101) values of the GZO NR2s increased as the Ga doping concentration was increased. These calculated TC values demonstrate that the UZO NR2s grew along the direction perpendicular to the substrate whereas the GZO NR2s grew toward the lateral direction with increasing Ga doping concentration.

a XRD patterns and b texture coefficients of ZnO NR2s with various Ga doping concentrations
Figure 7a shows the PL spectra of the ZnO NR1s with different Ga doping concentrations. The PL spectra of ZnO generally consist of two emissions, which are the near-band-edge (NBE) emission and the deep level (DL) emission [37]. The NBE emission, which is observed in the UV region, is generated by the recombination of free excitons, and the DL emission, which is observed in the visible region, is attributed to structural defects, such as oxygen vacancy, zinc vacancy, interstitial oxygen, and interstitial zinc [38, 39]. From the PL results, the ZnO NR1s grown on the GZO seed layer are observed to have lower NBE emission intensity compared to the ZnO NR1s grown on the UZO seed layer. This is caused by the occurrence of a lattice distortion in the ZnO NR1s due to the ionic radii of Ga3+ ions (0.62 Å) being smaller than that of Zn2+ ions (0.74 Å). Therefore, the Ga dopant acts as a scattering center and generates a Coulomb electric field effect [40], which causes the loss of exciton Coulomb interaction. Because of the presence of the weak Coulomb interaction, the free excitons that are the origin of NBE emission are ionized. As a result, the ZnO NR1s grown on the GZO seed layer have a lower intensity of NBE emission than ZnO NR1s grown on the UZO seed layer. In case of the DL emission, a broad yellow emission which is found in the hydrothermally grown nanostructures can be observed at approximately 558 nm and is attributed to interstitial oxygen defects [41]. The DL emission intensity of ZnO NR1s decreased with increasing Ga doping concentration of the GZO seed layer. This is because a higher number of ZnO nuclei are formed on the GZO seed layer than in the UZO seed layer. In other words, a larger number of oxygen atoms are required to grow ZnO NRs on the GZO seed layer due to the higher number of ZnO nuclei. This indicates that the interstitium of oxygen gradually decreased with increasing Ga doping concentration. Therefore, the ZnO NR1s grown on the GZO seed layer with the highest Ga doping concentration had the lowest intensity of the broad yellow emission. Figure 7b shows that the NBE emission peak of the ZnO NR1s was blue shifted from 378.0 to 376.5 nm with increasing Ga doping concentration. The blue shift of the NBE emission peak is generated by the Moss-Burstein effect, which can be explained by using the following equation [42]:
where ΔEg is the change in the bandgap, h is the Plank constant, m* is an effective mass, and ne is the carrier concentration. As the Ga doping concentration is increased, the carrier concentration (ne) increases due to the Ga dopant acting as donor. The increased charge concentration causes the Fermi level to shift into the conduction band, thus, widening the bandgap and finally blue shifting the NBE emission.

a PL spectra of ZnO NR1s and b the NBE peak position with increasing Ga doping concentration
Figure 8 shows the PL spectra of ZnO NR2s with increasing Ga doping concentration from 0 to 4 mM. The NBE emission intensities of the UZO NR2s was higher than that of the GZO NR2s. Interestingly, the intensity of the NBE emission increased with increasing Ga doping concentration up to 3 mM and then greatly decreased. Generally, the incorporation of Ga dopant into the ZnO lattice increases the intensity of NBE emission due to the increased number of electrons [43]. However, at high Ga doping concentration, a large amount of Ga substitution occurs, causing the lattice distortion due to the ionic radii of Ga3+ ions (0.62 Å) being smaller than that of Zn2+ ions (0.74 Å). Thus, when the Ga dopant at 4 mM was added into the aqueous solution, a larger amount of lattice distortion occurred in the GZO NR2s and the number of trapped excitons increased, indicating a reduction in the NBE emission intensity. The DL emission of the ZnO NR2s was centered at approximately 558 nm, and the intensity of the DL emission was remarkably reduced after addition of the Ga dopant into the ZnO NR2s. This is because the Ga dopant in the aqueous solution replaced Zn atoms in the ZnO lattice, forming GaO molecules. Consequently, the trivalent Ga3+ ions of the GaO molecules combine with more oxygen atoms than the bivalent Zn2+ ions of the ZnO molecules due to a larger number of coordinate linkage of the Ga dopant [44]. Therefore, a relatively a large number of O atoms are bonded to the GZO NR2s than to the UZO NR2s. Thus, the amount of combined oxygen increased, and the interstitial oxygen in the ZnO lattice decreased, causing a decrease in the intensity of the broad yellow emission.

PL spectra of ZnO NR2s with increasing Ga doping concentration
To investigate the effect of the Ga doping concentration on the time-dependent UV photoresponse of the ZnO NR1s, we fabricated a metal–semiconductor-metal UV photodetector based on ZnO NR1s with different Ga doping concentrations, and the measurement results are shown in Fig. 9a. All ZnO NR1s had low current values in dark conditions; however, the photocurrent value increased upon exposure to UV light. These behaviors can be explained using the following mechanism. Generally, when the ZnO NR-based UV photodetectors are exposed to air in the dark, oxygen molecules in air are adsorbed on the surfaces of the ZnO NRs, and the adsorbed oxygen molecules capture electrons in the ZnO NRs [O2 (g) + e− → O2−] [45]. Consequently, a depletion region is formed near the surfaces of the ZnO NRs, which reduces the conductivity. At this time, the current generated by an applied voltage is called the dark current, which has a relatively low value. However, upon exposure of the ZnO NRs to UV light, electron–hole pairs (EHPs) are generated [hν → e− + h+]. The photogenerated holes (h+) react with adsorbed oxygen ions (O2−), thus reducing the depletion region [h+ + O2− → O2 (g)], whereas the photogenerated electrons (e−) move along the electrodes, thus rapidly increasing the photocurrent value. The unpaired photogenerated electrons are accumulated until the oxygen desorption and re-adsorption processes reach an equilibrium state, which slowly increases the photocurrent. [46]. Conversely, when the UV light is turned off, the initial photocurrent sharply decreases due to the recombination of the EHPs. After the sharp photocurrent reduction, depletion regions form by the recombination of electrons existing in the NRs and oxygen molecules in air, thus inducing a slow photocurrent reduction. Thus, the photocurrent of the ZnO NR1s continuously increases from 1.43 to 7.61 μA with increasing Ga doping concentration up to 3 mM. Because the ZnO NR1s are grown on the GZO seed layer, the Ga dopant added to the GZO seed layer rarely influences the growth of the ZnO NR1s. Therefore, the photocurrent of the ZnO NR1 depends significantly on the shape of the ZnO NR1s and not the Ga dopant. According to the above mechanism, the photocurrent increases when the photogenerated holes react with oxygen ions adsorbed on the surfaces of the NRs. In other words, when the ZnO NR1s are exposed to UV light, the photocurrent can be controlled by using the surface areas of ZnO NR1s. The FE-SEM results confirmed that the GZO seed layer consisting of small-sized particles causes the formation in a large number of ZnO nuclei, which results in a high density of NRs. Thus, the photocurrent of the ZnO NR1s grown on the GZO seed layer with a high Ga content is improved due to large surface-to-volume ratio. However, interestingly, the photocurrent value of the ZnO NR1s with the highest Ga content was significantly reduced, which was attributed to the formation of Ga2O3 on the surface of the GZO seed layer, as shown in the XRD patterns. This is because Ga2O3 is nonconductive and acts as an electron trap.

a Photoresponse and b responsivity of ZnO NR1s with various Ga doping concentrations
Figure 9b shows the photoresponsivity of ZnO NR1-based UV photodetector. The photoresponsivity of the ZnO NR1s can be calculated using the following equation [47]:
where R is the responsivity, Iph is the photocurrent, Idark is the dark current, and Pop is the optical power of the UV light source. The photoresponsivity of the ZnO NR1s steadily increased from 1.40 to 10.3 μA/W up to 3 mM and is reduced slightly to 9.52 μA/W at 4 mM. This result indicates that the responsivity depends on the density of the ZnO NR1s. During the oxygen desorption and re-adsorption process, a depletion region near the surface of the NRs is formed and reduced. The depletion region reduced by oxygen desorption widens the width of the tunnel through which the electrons move, so the current flowing in the ZnO NRs is relatively increased. Because high-density ZnO NRs have a large surface area, adsorbable sites in dark conditions are many. Therefore, the presence of many adsorbed oxygens relatively increases the recombination rate with holes, so the depletion region is greatly reduced. Thus, when the photoresponsivity of ZnO NR1-based UV photodetectors is enhanced due to a rapid increase in the current, the photoresponsivity of the ZnO NR1s with the highest density is slightly reduced. This is because the photoresponsivity is not only affected by the ZnO-NR density, but also by the crystallinity. A UV sensor with poor crystallinity has low photoresponsivity because the trapping probability is large [48].
Figure 10a shows the time-dependent current-switching curves of the ZnO NR2-based UV photodetectors with a different Ga doping concentration in an aqueous solution. The UZO NR2s show large photocurrent in comparison with the GZO NR2s. This is because the Ga dopant present in the aqueous solution significantly influences the growth of ZnO NR2s. From the FE-SEM and the XRD results, the Ga dopant added in the aqueous solution was observed to reduce the density of the ZnO NR2s. Generally, ZnO has intrinsic n-type conductivity due to defects such as oxygen vacancies and interstitial zinc acting as electron donors. Therefore, the GZO NR2s exhibited a reduced photocurrent due to the small amount of electron concentration existing in the GZO NRs. Figure 10b shows the photoresponsivity of a ZnO NR2-based UV photodetector. The photoresponsivity can be observed to be reduced from 5.46 to 0.509 μA/W with increasing Ga doping concentration. This confirms that the photoresponsivity of ZnO NR-based UV photodetectors is also strongly related to the density of ZnO NRs. The density of ZnO NR2s significantly decreased with increasing Ga doping concentration, as shown in Figs. 4 and 6. Because UV photodetectors with low density have a small area in which the oxygen molecules can be adsorbed, the adsorption and the desorption processes of oxygen molecules progress slowly. Therefore, the ZnO NR2s with small surface area influenced the depletion region and the number of electrons in the ZnO NR2s; consequently, the photoresponsivity gradually decreased.

a Photoresponse and b responsivity of ZnO NR2s with various Ga doping concentrations
Figure 11a, b show the photoresponsivity of ZnO NR1- and ZnO NR2-based UV photodetectors with Ga doping concentrations of 3 mM, respectively. The photoresponsivity of ZnO NR1s was 10.3 μA/W and that of ZnO NR2s was 0.896 μA/W, which confirms that the photoresponsivity of the ZnO NR1s grown on the GZO seed layer was improved. The difference between the ZnO NR1s and the ZnO NR2s was where the Ga dopant was added. In case of the ZnO NR1s, the Ga dopant was added in the GZO seed layer by using the sol–gel solution with the Ga dopant whereas, in the case of ZnO NR2s, the Ga dopant was added in the ZnO NR2s by using an aqueous solution with the Ga dopant. Both the ZnO NR1s and the ZnO NR2s require the deposition of the ZnO seed layers, which are crystallized through the pre-heat treatment process. Consequently, the ZnO NRs could be grown on the ZnO seed layer with a more stable surface because atoms move toward the low-energy surface during crystallization [49]. Therefore, the ZnO NR1s grown on the GZO seed layer had good crystallinity because the Ga dopant was doped on a stable surface. To support this claim, we observed the dependence of the increasing photocurrent on time. As time goes on, the photoresponsivity of the ZnO NR1s increased 0.03 μA/W while the photoresponsivity of the ZnO NR2s increased 0.21 μA/W. The ZnO NR1s show a uniform current, whereas the ZnO NR2s show an overall increasing current. The increasing current indicates that the ZnO NRs have poor crystallinity and a slow recombination rate, which is attributed to the trapped carriers. In other words, because the Ga dopant added to the ZnO NR2s acts as a trap, the current increases with increasing time. Thus, the ZnO NR1s grown on the GZO seed layer had a higher photocurrent and photoresponsivity than the ZnO NR2s owing to the larger surface area and the addition site of the Ga dopant. Therefore, the difference between the photoresponsivity of the ZnO NR1s and the ZnO NR2s is stark.

a Photoresponse of ZnO NR1s and b ZnO NR2s at a Ga doping concentration of 3 mM
3 Conclusion
In summary, we grew two types of ZnO NRs by using the hydrothermal method at various Ga doping concentrations: ZnO NRs grown on the GZO seed layer (NR1s) and those grown using Ga-dopant-added-aqueous solution (NR2s). As the Ga doping concentration was increased from 0 to 4 mM, the density of ZnO NR1s gradually increased from 3.2 × 109 rods/cm2 to 5.1 × 109 rods/cm2 due to a reduction in the particle size caused by the Ga dopant while the density of ZnO NR2s was remarkably reduced from 2.8 × 109 rods/cm2 to 7 × 108 rods/cm2 due to the formation of complexes related to the Ga dopant. The XRD results revealed that the ZnO NR1s were grown along the direction perpendicular to the substrate whereas the ZnO NR2s were gradually grown toward the lateral direction with increasing Ga doping concentration. The PL spectra of the ZnO NR1s and the ZnO NR2s exhibited a broad yellow emission. Interestingly, the intensity of the DL emission from the ZnO NR1s gradually decreased with increasing Ga doping concentration, indicating that a larger number of oxygen atoms was used to grow the ZnO NR1s due to the higher number of ZnO nuclei. On the other hand, the intensities of the DL emission from the GZO NR2s exhibited a lower value than that from the UZO NR2s because a relatively larger number of oxygen atoms were bonded in the GZO NR2s than in the UZO NR2s due to coordinate linkage number of the of the Ga dopant being larger. With respect to the UV photoresponse properties, overall, the photoresponsivity of ZnO NR1s increased due to the large surface-to-volume ratio while the UZO NR2s showed a larger photoresponsivity in comparison to the GZO NR2s, which was related to the density of ZnO NR2s. The ZnO NR1s showed photosensitivity that were significantly more enhanced than those of the ZnO NR2s, owing to the larger surface area and improved crystallinity of GZO seed layer compared to the ZnO seed layer. Therefore, we can be concluded that the optimal doping location of the Ga dopant is the seed layer and that ZnO NR1s grown on a GZO seed layer are suitable for fabricating high-performance UV photodetectors.
References
Y. Xie, L. Wei, G. Wei, Q. Li, D. Wang et al., Nanoscale Res. Lett. 8, 133 (2013)
T.V. Cuong, H.N. Tien, V.H. Luan, J.S. Chung, E.W. Shin et al., J. Nanosci. Nanotechnol. 12, 3313 (2012)
V. Kumar, R.G. Singh, N. Singh, A. Kapoor, R.M. Mehra et al., Mater. Res. Bull. 48, 362 (2013)
D. Bekermann, A. Gasparotto, D. Barreca, A. Devi, R.A. Fischer et al., ChemPhysChem 11, 2337 (2010)
S.N. Sarangi, J. Appl. Phys. 49, 355103 (2016)
Y. Sun, G.M. Fuge, M.N.R. Ashfol, Superlattices Microstruct. 39, 33 (2006)
X. Jiaqiang, C. Yuping, L. Yadong, S. Jianian, J. Mater. Sci. 40, 2919 (2005)
D.-T. Phan, G.-S. Chung, Sens. Actuator B Chem. 187, 191 (2013)
G. Jia, Y. Wang, J. Yao, J. Dig, Nanomater. Biostruct. 7, 261 (2012)
D.-T. Phan, G.-S. Chung, J. Electroceram. 32, 353 (2014)
J.T. Chen, J. Wang, R.F. Zhuo, D. Yan, J.J. Feng et al., Appl. Surf. Sci. 255, 3959 (2009)
X.Q. Zhao, C.R. Kim, J.Y. Lee, J.H. Heo, C.M. Shin et al., Appl. Surf. Sci. 255, 4461 (2009)
H. Jung, D. Kim, H. Kim, Appl. Surf. Sci. 297, 125 (2014)
R.V. Muniswami Naidu, A. Subrahmanyam, A. Verger, M.K. Jain, S.V.N. Bhaskara Rao et al., Electron. Mater. Lett. 8(4), 457 (2012)
C.-H. Hsiao, C.-S. Huang, S.-J. Young, S.-J. Chang, J.-J. Guo et al., IEEE Trans. Electron Dev. 60(6), 1905 (2013)
S.-J. Young, C.-L. Chiou, Y.-H. Liu, L.-W. Ji, Invent. Math. 3, 1 (2016)
H. Ghayour, H.R. Rezaie, Sh. Mirdamadi, A.A. Nourbakh, Vacuum 86, 101 (2011)
J.B. Zhong, J.Z. Li, J. Zeng, X.Y. He, W. Hu et al., Mater. Res. Bull. 47, 3893 (2012)
S.-J. Young, IEEE, , J. Sel. Top. Quantum Electron. 20(6), 381204 (2014)
A. Ali, X. Zhao, A. Ali, L. Duan, H. Niu et al., Superlattices Microstruct. 83, 422 (2015)
C.P.T. Nguyen, P.P.H. La, T.T. Trinh, T.A.H. Le, S. Bong et al., J. Nanosci. Nanotechnol. 14, 6261 (2014)
N. Kiomarsipour, R.S. Razavi, Ceram. Int. 39, 813 (2013)
R. Shi, P. Yang, X. Dong, Q. Ma, A. Zhang, Appl. Surf. Sci. 264, 162 (2013)
Y. Qu, X. Huang, Y. Li, G. Lin, B. Guo et al., J. Alloys Compd. 698, 719 (2017)
H.-S. Hong, G.-S. Chung, Sens. Actuator B Chem. 195, 446 (2014)
H.S. Hassan, A.B. Kashyout, H.M.A. Soliman, M.A. Uosif, N. Afify, Appl. Surf. Sci. 277, 73 (2013)
S.Y. Bae, C.W. Na, J.H. Kang, J. Park, J. Phys. Chem. B 109, 2526 (2005)
C.-C. Yang, Y.-K. Su, M.-Y. Chuang, T.-H. Kao, H.-C. Yu et al., IEEE J. Sel. Top. Quantum Electron. 21(4), 3800707 (2015)
G.C. Park, S.M. Hwang, J.H. Lim, J. Joo, Nanoscale. 6, 1840 (2014)
H. Wang, S. Baek, J. Song, J. Lee, S. Lim, Nanotechnology 19, 075607 (2008)
N. Kumar, A. Katiyar, A. Srivastava, Mater. Today 5, 9089 (2018)
T.-C. Lin, W.-C. Huang, C.-H. Liu, S.-C. Chang, Appl. Surf. Sci. 258, 3302 (2012)
M. Saha, S. Ghosh, V.D. Ashok, S.K. De, Phys. Chem. Chem. Phys. 17, 16067 (2015)
M.-J. Lee, T.-I. Lee, J. Lim, J. Bang, W. Lee et al., Electron. Mater. Lett. 5, 127 (2009)
M.E. Fragla, Y. Aleeva, G. Malandrino, Superlattices Microstruct. 48, 408 (2010)
H. Colak, E. Karakose, G. Kartopu, J. Mater. Sci. Mater. Electron. 29, 11964 (2018)
M. Kim, J.-Y. Leem, J. Korean Phys. Soc. 68(5), 705 (2016)
D.L. Zhu, Q. Wang, S. Han, P.J. Cao, W.J. Liu et al., Appl. Surf. Sci. 298, 208 (2014)
S.K. Mohd Bakhoria, C.A. Ling, S. Mahmud, Adv. Mater. 501, 179 (2012)
G.C. Park, S.M. Hwang, J.H. Choi, Y.H. Kwon, H.K. Cho et al., Phys. Status Solidi A Appl. Mat. 210(8), 1552 (2013)
J. Xu, P. Liu, S. Shi, X. Zhang, L. Wang et al., Appl. Surf. Sci. 258, 7118 (2012)
A. Khalil, M.A. Gondal, M.A. Dastageer, Appl. Catal. A Gen. 402, 162 (2011)
K.A. Chernenko, E.I. Gorokhova, S. B. Eron’ko, A.V. Sandulenko, I. D. Venevtsev, et al., IEEE Trans. Nucl. Sci. 65(8), 2196 (2018)
J. Zhao, X. Yan, Y. Yang, Y. Huang, Y. Zhan, Mater. Lett. 64, 569 (2010)
W. Lee, J.-Y. Leem, J. Korean Phys. Soc. 72(5), 610 (2018)
W. Jeon, J.-Y. Leem, J. Korean Phys. Soc. 72(10), 1237 (2018)
R. Amiruddin and M. C. Santhosh Kumar, Phys. Status Solidi A Appl. Mater. 1600658 (2017).
T.H. Flemban, M.A. Haque, I. Ajia, N. Alwadai, S. Mitra et al., ACS Appl. Mater. Interfaces. 9, 37120 (2017)
M.S. Kim, K.G. Yim, D.-Y. Lee, J.S. Kim, J.S. Kim et al., Appl. Surf. Sci. 257, 9019 (2011)
Acknowledgements
This work was supported by a Center for Women In Science, Engineering and Technology (WISET) grant funded by the Ministry of Science and ICT (MSIT) under the team research program for female engineering students (WISET-2020-082) and the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (No. 2018R1D1A1B07050792).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations
Rights and permissions
About this article

Cite this article
Kim, S., Leem, JY. Improved photoresponse properties of hydrothermally grown ZnO nanorods by controlling the Ga doping location. J. Korean Phys. Soc. 78, 144–156 (2021). https://doi.org/10.1007/s40042-020-00006-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40042-020-00006-w