
Abstract
As the fossil fuel reserves are depleting rapidly, there is a need for alternate fuels to meet the day to day mounting energy demands. As fossil fuel started depleting, a quest for alternate forms of fuel was initiated and biofuel is one of its promising outcomes. First-generation biofuels are made from edible sources like vegetable oils, starch, and sugars. Second-generation biofuels (SGB) are derived from lignocellulosic crops and the third-generation involves algae for biofuel production. Technical challenges in the production of SGB are hampering its commercialization. Advanced molecular technologies like metagenomics can help in the discovery of novel lignocellulosic biomass-degrading enzymes for commercialization and industrial production of SGB. This review discusses the metagenomic outcomes to enlighten the importance of unexplored habitats for novel cellulolytic gene mining. It also emphasizes the potential of different metagenomic approaches to explore the uncultivable cellulose-degrading microbiome as well as cellulolytic enzymes associated with them. This review also includes effective pre-treatment technology and consolidated bioprocessing for efficient biofuel production.
Similar content being viewed by others

1 Introduction
The cognizance of fossil fuel depletion started in the early 1970s. Engineers then suggested that the consumption of more fuel than extracted will ultimately lead to its exhaustion. To prevent this, the demand for these fuels should be moderated along with the quest for alternate forms of energy that are capable of replacing fossil fuels. For a decade, there has been a constant increase in primary energy consumption (PEC) around the world. The average PEC growth rate (PECGR) for the years 2008–2018 globally was 1.6%, and remarkably in 2019, it decreased to 1.3%. The reasons for this decline are many and one of the chief reasons is the feeble economic growth of nations like Russia, the USA, and India. The decline of PECGR was observed in many nations except China. China stands as the highest individual country contributing about 24.3% of global PECGR, followed by the USA, India, and Russia with 16.2%, 5.8%, and 5.1% respectively. Though the global PECGR in 2019 has decreased, the individual PECGR by countries like China and India has increased from 135.33 and 33.30 exajoules (EJ) respectively to 141.70 and 34.06 EJ when compared to 2018 [1].
To control the surge in energy consumption, emerging countries like India and China have started to use the available fuels more efficiently. The oil consumption of countries like China and India has increased by 5% and 2.9% respectively. On the one hand, oil production is going down drastically, and on the other hand, oil consumption has been increasing [1,2,3]. Due to the prevailing COVID-19 pandemic, global energy review suggests that during the lockdown phase economies over the globe can expect a 20 to 40 % decline in economic output. In India, the national lockdown has reduced the energy demand by 30% [4]. Once this pandemic phase is over, it is expected the global energy demand would eventually increase and the statistics of the past 10 years suggest the need for alternate fuel like biofuels.
In India, there is a 9.8% increase in the production of energy from renewable sources when compared with the previous year 2018. Global biofuel production increased from 1787 thousand barrels of oil equivalents per day (tboe/d) to 1842 tboe/d which accounts for about a 3% increase in global biofuel production. India has increased its biofuel production by 24.9% when compared with 2018 and stands in the second position after Indonesia (37.5%) in the Asian-Pacific countries [1,2,3]. The demand for bioenergy is expected to increase by up to 11% by 2040 [5]. In India, the contribution of bioenergy to total energy demand is gradually increasing. This increase can be accelerated as India has abundant reserves of biomass, which can be the raw material for producing various forms of biofuel. The pursuit is for efficient technology that can convert the biomass into bioenergy. According to the United Nations Food and Agriculture Organization, in India, there is an increase in the total area covered by forests in recent times. This signifies that there is no insufficiency of lignocellulosic raw material in India [6]. Plant biomass is a reliable source for sugars and when subjected to fermentation will yield biofuel. So, lignocellulose is a very significant source for the production of various biofuels [7].
Lignocellulose is chiefly composed of cellulose, hemicellulose, and lignin. Cellulose contributes about 40–50%, hemicellulose contributes 25–30% of lignocellulose and the rest is lignin [8]. Consequently, keeping present inferior lignocellulose separating approaches [9] in view, humongous attention has been given to improve the methods of lignocellulose hydrolysis facilitated by the novel, efficient, and engineered enzymes [10]. Combinations of different glycosyl hydrolases are necessary for the comprehensive breakdown of lignocellulose into a blend of different sugars. In a lignocellulose-degrading habitat, the microbiome produces different mixtures of glycosyl hydrolases which aid in the thorough degradation of lignocellulose [11, 12]. So the quest for novel and efficient approaches for biofuel applications is still going on and one such new approach is metagenomics. Metagenomics is a novel approach for studying genomes of the entire microbiome residing at a given habitat. It helps in understanding the microbial composition of the habitat and gives a way to explore and exploit many novel genes from uncultivable/cultivable microbiome [13]. The increasing demand for steadfast and efficient lignocellulases and hemicellulases targeting biofuels may be met by this novel approach of metagenomics. In this review, the emphasis is to analyze most of the reported metagenomic-derived cellulases (endoglucanases in specific).
Lignocellulosic biomass which is available as agricultural, industrial, and municipal solid waste and forest residues around the globe is a prospective raw material for bioethanol as well as other value-added biochemical production [14,15,16]. Production of bioethanol from lignocellulosic biomass consists of three important steps. (i) The pre-treatment process reduces the recalcitrant nature of lignin, thus allowing enzymatic hydrolysis of biomass converting it to fermentable sugars. Pre-treatment techniques can be categorized into physical/mechanical, physicochemical, chemical, and biological. Each pre-treatment method has its advantages and disadvantages (Table 1). Based on the composition of biomass and economics, appropriate pre-treatment method is employed. (ii) Saccharification and fermentation, the leftover biomass (rich in cellulose) after pre-treatment will be converted to monomeric glucose by using cellulase enzymes. Bacteria, fungi, and actinomycetes are major cellulase-producing microorganisms at the laboratory scale, for industrial and bioenergy applications (Table 2). (iii) Recovery, to separate/extract the bioethanol produced from the raw fermentation broth to obtain high-purity bioethanol. Even though several separation methods are available, either distillation or in combination with other processes remains the primary approach for bioethanol purification.
In the orthodox method of producing bio-alcohol, processes like saccharification as well as fermentation are performed as a distinct individual process, involving their respective optimum parameters. This process is referred to as separate hydrolysis and fermentation (SHF) [18]. The chief limitation of SHF is the cellulase enzyme’s feedback inhibition, implicated by sugars liberated by the hydrolysis of the substrate [19,20,21]. To overcome this issue, simultaneous saccharification and fermentation (SSF) was recommended, which enhanced the enzyme consumption and efficiency of the process [22,23,24,25]. The major drawback of this process is incompatible temperatures of hydrolysis (45–60 °C) and fermentation (30 °C) [22,23,24, 26]. To alleviate this issue, non-isothermal simultaneous saccharification and fermentation (NSSF) has been proposed, involving partial enzymatic hydrolysis at optimum temperature, and as soon as the culture media is inoculated, the optimum temperature for the microbial growth is set [20, 27]. In the process of simultaneous saccharification, filtration, and fermentation (SSFF), membranes are used to obtain a clear sugar-rich filtrate from the hydrolysis liquid. The filtrate contains hydrolyzed sugars along with partially hydrolyzed lignocellulosic biomass. After glucose, xylose is the next abundant saccharide in many lignocellulosic materials. It would be apt to use the simultaneous saccharification and co-fermentation (SSCF) process to efficiently use the xylose part of the filtrate. In this process, xylose and glucose utilizing wild-type or engineered microorganisms are employed for ethanol production [28,29,30,31]. However, it is essential to gaze for new alternatives to the SSCF process, and one such alternative is consolidated bioprocessing (CBP) (Fig. 1).

Various strategies of bioprocessing for converting lignocellulosic biomass to biofuel. (CBP means consolidated bioprocessing; SSCF means simultaneous saccharification and co-fermentation; SSF means simultaneous saccharification and fermentation; SHCF means separate hydrolysis and co-fermentation; SHF means separate hydrolysis and fermentation)
Consolidated bioprocessing (CBP) has been designed to evade the setbacks and expenses of orthodox biofuel production from lignocellulosic biomass. This involves the application of either pure culture or consortia depending on the output and the process parameters. CBP aims to associate the processes like production of enzyme, hydrolysis, and fermentation into a single step and also try to combine the pentose sugar utilization process into the same. This is expected to improve the efficacy of the processes by eliminating the dependency on various hydrolytic enzymes that are being supplemented exogenously and decreasing the cellulase feedback inhibition by sugars [32, 33]. This leads to the reduction of unit operations involved in the total process, thus decreasing the process inclusive capital costs [32]. Further advances in CBP can evade the pre-treatment process of biomass by producing the biofuel from raw biomass [18].
2 Structure of lignocellulose
The major constituents of plant biomass mainly include cellulose, hemicellulose, and lignin. Apart from these, there are also minor volumes of ash, extractives, protein, and pectin. Yet, the configuration of these components differs among different species of plants and can also vary in the same plant concerning its stage of development, age, and other conditions. The degree of association of polymers with each other in the heteromatrix depends on the source of the biomass, its species, and its type [34,35,36]. To determine the most appropriate energy conversion path for specific biomass, the most important factor that has to be considered is the relative composition of the three main polymers in the lignocellulosic biomass [37]. To liberate sugars for fermentation, lignocellulosic biomass needs to undergo an aggressive pre-treatment process, to produce a substrate that is further broken down either using commercially available cellulolytic enzymes or employing microbes that are capable of producing such enzymes [38].
The rigidity of the plant cell wall is due to the presence of structural polymers, e.g., lignin and cellulose. Along with them, there are also minor volumes of ash, extractives, protein, and pectin. Apart from plants, there are other sources of cellulose; few microbes are also capable of producing cellulose like bacteria (Gluconacetobacter xylinum) [39] and algae (Cladophora green algae) [40]. In its unbranched homopolymer form, cellulose is made up of β-d-glucopyranose units, which are connected by β-(1,4)-glycosidic linkages. Reiterating units of cellobiose form the polymer chains of cellulose [34, 41]. Twenty to three hundred cellulose chains are connected by hydrogen bonds and van der Waals interactions to give a microfibril, this is concealed by hemicellulose and lignin, and these microfibrils are further grouped to give cellulose fibers. The sugar d-glucose can be liberated from cellulose by treating with acid or enzymes that break β-(1,4)-glycosidic bonds. In any given biomass, cellulose is observed to be in two forms—amorphous and crystalline cellulose. Most of the cellulose exists in crystalline form, whereas only a small proportion of it is amorphous. The unorganized cellulose chains existing in the amorphous form are easier to degrade using enzymes [42]. Cellulose elementary fibril (CEF) is a thin fibrillary product of cellulose synthase and a bundle of CEFs is often referred to as macrofibril. A macrofibril should not be confused with microfibril; microfibril may have a minor part of either macrobfibril or sometimes a CEF too as given in Fig. 2 [43].

Structural architecture of lignocellulosic biomass
The second most abundantly found polymer in lignocellulosic biomass, making up about 20–50% of it, is hemicellulose. It has a backbone that can be either a heteropolymer or a homopolymer. Unlike cellulose, which is a chemically homogeneous polymer, hemicellulose has short branches containing different sugars such as pentoses, hexoses, and uronic acids. The branches are connected by β-(1,4)-glycosidic linkages, and sometimes also by β-(1,3)-glycosidic linkages [34, 44]. Hemicelluloses have a special virtue of being the most heat-labile and chemically sensitive among all the major components of lignocellulosic biomass [45, 46]. As hemicellulose coats the fibrils of cellulose in the cell wall of plants, it has been proposed that to improve the digestibility of cellulose, it is required to get rid of 50% or more of the hemicellulose present [46]. Therefore, severity conditions during the pre-treatment process are generally compromised to maximize sugar recovery. The fraction of hemicellulose acquired after pre-treatment can be obtained in solid state, or a mixture of solid and liquid states, depending on the chosen method of pre-treatment [36].
The next in abundance with respect to lignocellulosic biomass as polymer is lignin. It is primarily observed in the cell wall of plants. The composition of lignin is very complex with many cross-linked polymers. This provides mechanical support as well as structural integrity to overcome microbial attacks and oxidative stress. The chief components of this polymer are sinapyl alcohol (syringyl alcohol), coniferyl alcohol (guaiacyl propanol), and coumaryl alcohol (p-hydroxyphenyl propanol) linked by different ether bonds [45]. Lignin glues together the different constituents of lignocellulosic biomass [46]. The intimate association of lignin with cellulose microfibrils is the main cause of hindrance for both microbial degradation of biomass through enzymes [47]. In 2000, Chang and Holtzapple showed that, upon increasing lignin removal, the digestibility of biomass was enhanced [48]. Delignification causes engorgement of the biomass and disrupts the structure of lignin. It also increases the internal surface area and makes cellulose more accessible to cellulolytic enzymes. During the pre-treatment process, lignin liquefies and then solidifies once cooled, which allows for it to be precipitated out [33, 46, 49].
3 Enzymes involved in hydrolysis
It is very difficult to completely hydrolyze cellulosic biomass due to the presence of microfibrils and its high degree of self-association. As part of the ecological carbon cycle, cellulose degradation with the help of microbial cellulosomes and cellulases drifts the carbon to environmentally available CO2 from various fixed biomasses [50]. To fulfill their nutritional requirements, microorganisms are capable of performing enzymatic degradation of cellulose. A group of cellulase enzymes, which are members of the glycosyl hydrolase (GHase) family (EC: 3.2.1), are responsible for the complete hydrolysis of cellulose [51,52,53]. These enzymes not only cleave the glycosidic bonds which are observed between carbohydrates but also cleave the glycosidic bonds between a carbohydrate and a non-carbohydrate compound. According to the International Union of Biochemistry and Molecular Biology (IUBMB) nomenclature, glycosidases (glycosyl hydrolases) were classified based on the characters like molecular mechanism and specificity towards substrates. However, there are diverse varieties of polysaccharides existing in nature [54]. To overcome this problem, glycosyl hydrolases were proposed to be classified based on their similarities in the amino acid sequences [55]. By using this classification, we can account for structural and functional relationships, mechanistic models, and substrate specificity [56].
The mechanism of glycosyl hydrolase catalysis is classified broadly into two types [57], one is the inverting mechanism and the other is the retaining mechanism. In the inverting mechanism, an anomeric position is shifted from β to α through a single-displacement mechanism, whereas in the retaining mechanism, anomeric carbon remains in the same positions as it is mediated by the double-displacement mechanism Fig. 3 [59]. In both the mechanisms, there will be no change in the proton donor’s position and it remains in the distance within which it can form hydrogen bonding. In the inverting mechanism, the catalytic base is distantly placed from the anomeric position to accommodate the water molecule between the sugar molecule and the base. As the retaining mechanism does not involve water molecules, it remains in the vicinity of the sugar molecule’s anomeric position [51]. Intermediate states like the epoxides [60] and oxacarbenium-ion like states [61] are observed in both the mechanisms (inverting, retaining mechanisms).

Mechanisms of glycosidic bond cleavage (adapted from [58])
For instance, the endoglucanase from Nasutitermes takasagoensis (a termite) belonging to the family glycosyl hydrolase 9 possesses three conserved catalytic residues, out of which two are aspartic acid and one is glutamic acid. Both the aspartic acid residues deprotonate water molecule (acts as a base) creating a nucleophile, which is capable of attacking the anomeric carbon. This breaks the glycosidic linkage and inverts the anomeric position. Glutamate residue acts as a proton donor/acid and protonates the sessile oxygen in the glycosidic bond [58]. Most of the endoglucanases belonging to GH-5 family have a conserved dyad of glutamic acid residues which take part in catalysis. Tripti et al. reported that the enzyme Cel-1, which was isolated from a buffalo rumen, possessed a conserved dyad of glutamic acid residues (E314 and E179) in its active site [62]. It is observed that many families of GH do not possess catalytic proton donor/acceptor and/or nucleophile [63, 64]. So, alternate catalytic mechanisms like substrate-assisted catalysis, proton transferring network, non-carboxylate residues, and exogenous base/nucleophile [65] also exist.
Cellulase enzymes can be classified into three types: endoglucanases (EC 3.2.1.4), cellobiohydrolases (EC 3.2.1.91 or 3.2.1.176), and β-glucosidases (EC 3.2.1.21) [11].
3.1 Endoglucanase
Endo-1,4-β-d-glucanases attack the cellulose chain randomly, thus leading to the formation of more free ends, which serve as a substrate for exoglucanases. Thus, the two enzymes work together to accelerate the hydrolysis process [66].
3.2 Cellobiohydrolase
The tunnel-like active site present on cellobiohydrolase or β-1,4-exoglucanase binds to the reducing or non-reducing ends of microfibrils, thus allowing the enzyme to cut the cellulose polymer into shorter chains and release either glucose or cellulose as the product [67].
3.3 β-Glucosidase
The β-glucosidase (β-d-glucoside gluco-hydrolase) enzyme facilitates the transfer of a glycosyl moiety among nucleophilic oxygens. This enzyme performs the essential task of hydrolyzing cellobiose, which is the key factor that defines the rate of reaction. The product of cellobiose hydrolysis is glucose and increased glucose inhibits the β-glucosidase by feedback regulation [68, 69]. This enzyme can thus regulate the entire process of cellulose hydrolysis [70, 71]. Apart from participating in glycosyl bond hydrolysis, this enzyme can also catalyze the reverse process of hydrolysis under certain definite conditions [72]. The schematic representation of the mode of action for all three enzymes (endoglucanase, exoglucanase, β-glucosidase) and their products are represented in Fig. 4.

Schematic representation of the mode of action for all three enzymes (endoglucanase, exoglucanase, β-glucosidase) and their products
4 Carbohydrate-binding modules
Carbohydrate-binding modules (CBMs) were originally referred to as cellulose-binding domains (CBDs) as the domains discovered in initial times were all associated with cellulose binding. Later, when a lot of other carbohydrate-binding moieties were also discovered, the group emerged as CBMs [73]. A CBM is an adjoining polypeptide chain to some carbohydrate-active enzymes and it is capable of folding independently. It tends to bind to carbohydrates but does not modify its chemical structure. CBMs perform two important actions, one is to channelize appropriate carbohydrate to its catalytic fragment and the other is to hold the catalytic fragment at its vicinity to facilitate easy substrate binding (Fig. 5) [74]. Aromatic amino acids in ligand-binding sites play a very vital role in the binding of the substrate, especially the amino acids tyrosine and tryptophan [75]. To date, there are about 180,143 modules classified into 85 families and 559 modules yet to be classified (CAzy database).

Schematic representation of the carbohydrate-binding module
All the CBMs have been grouped into three categories based on the topographical anatomy of the ligand-binding site. The first is type A CBM which has a hydrophobic and planar surface as a salient feature. This gives type A CBM the capability of binding to polysaccharides like chitin and cellulose. Examples of type A include CBM families 1, 2, 3, 5, 63, 64, and 79. Though the binding of the substrate in both types A, B, and C is due to the same aromatic amino acids, the topology of type B CBM is different. In type B, the substrate can bind to two different sites of the same protein, one is a variable loop site (VLS) and the other a concave face site (CFS). The VLS is present at the tail of the protein whereas CFS is present on the concave portion of protein. Type B can bind to an extensive range of glycans (like galactans, xylans, and mannans) but cannot bind to substrates like cellulose. Examples of type B include CBM families 4, 16, 22, 31, 48, 58, 61, 75, and 80. The topological anatomy of type C is entirely different from the other two with a pocket-like substrate binding site which allows only small saccharides (like mono-, di-, or tri-saccharides) to interact with it. Examples of type C include CBM families 13, 14, 32, 42, 62, 66, and 71 [75].
5 Culture-independent as well as culture-dependent approaches
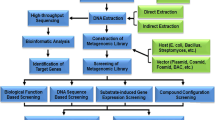
Microbes possess huge metabolic diversity which helps them to adapt to varied climatic conditions, increasing the eventual range of colonizing environments [76]. However, about 99% of bacteria existing in any given tellurian habitat cannot be cultivated. Correspondingly, this cultivable percentage is still low in marine habitats and it is about 0.001 to 0.1% only [77]. Hence, with the help of cultivable approaches, only < 1% of the microbes existing in any habitat can be explored [78]. Therefore, microbial enzymes derived from pure cultures which are currently being used for biofuel production are not an apt indication of the full potential for bio-catalysis possessed by microorganisms. Taking this into consideration, in recent years, some metagenomics-based and culture-independent-based methods have been established to survey the different bio-diversities in varied environments. Metagenomics not only provides the diversity profiling of microbial communities but also allows the quest for identifying novel genes and proteins/enzymes of industrial as well as biotechnological importance (like lignocellulose-degrading enzymes) overcoming the uncultivable nature of the microbes [78]. In a study conducted by Fang Z et al., a β-glucosidase (Bgl1A) was isolated from a marine microbial metagenomic library. Bgl1A was observed to be very stable even at highly saline conditions. When the concentration of glucose was increased, the enzyme activity of Bgl1A has slowly decreased. There are huge numbers of enzymes like Bgl1A that need to be explored in varied extreme environments with high potential [79]. Omics-based methods are also being used nowadays to analyze microbial metabolic diversity. The diversified approaches regarding both the cultivable and uncultivable microbes are represented in the form of a flow chart in Fig. 6.

Schematic representation of culture-dependent and culture-independent techniques (adapted from [78])
6 Exploring lignocellulosic biomass with the help of functional and structural metagenomics
Metagenomics has two major objectives, the first objective is to know about the taxonomic composition of the entire microbiome (structural) and the next objective is to know about the total genomes and associated genes of the microbiome (functional). Structural metagenomics is about identifying the major genera and species of organisms inhabiting a particular ecosystem to study and understand their roles in environmental interactions, evolutionary aspects, and biogeochemical cycles, whereas functional metagenomics is about the study of genomic diversity in an environmental sample with the purpose of isolating new genes and pathways which can encode functional enzymes or synthesize new biomolecules [80]. The steps involved in both of them are represented in the form of a flow chart in Fig. 7a, b. Functional metagenomics has been successfully used in isolating and identifying new protein families, especially lignocellulolytic enzymes such as cellulases, esterases, lipases, and xylanases [82,83,84].

a Schematic representation of steps involved in structural and functional metagenomics. b Process flowsheet of steps involved in metagenomic library construction (adapted from [81])
Generally, in metagenomics, two most common methods are used to understand a given microbiome. The first one is amplicon-based metagenomics and the second one is shotgun metagenomics. Amplicon-based metagenomics involves 16S ribosomal RNA for bacterial identification, internal transcribed spacer (ITS), and 18S region for fungal and eukaryote identification. The 16S/18S rRNA genes contain both hypervariable and conserved sequences. 16S sequencing targets hypervariable V1–V9 regions of the 16S ribosomal RNA gene to identify the diverse bacterial communities in the microbiome. While analyzing the 16S rDNA sequencing data, sequences with greater than 97% identity are grouped into an operational taxonomical unit (OTU) and each OTU is considered as taxa [85]. While analyzing the data if any two organisms have identical 16S rRNA sequence, they are considered as one species even though they are from two different species. This is the major drawback of this method. In the case of closely related species like Shigella flexneri and Escherichia coli (E. coli), this method cannot be used as it cannot differentiate them [86]. A great amount of sequence variation is observed in the ITS region located between 5.8S and 18S rRNA genes. ITS region sequences help in analyzing the fungal diversity of the sample whereas 18S rRNA sequences help in studying fungal taxonomies [87]. The fungal taxonomic analysis depends on the ribosomal genes like large subunit (LSU) or 28S, small subunit (SSU) or 18S, and 5.8S subunit rRNA genes. The 16S/18S rRNA metagenomics does not justify the title metagenomics as rRNA sequencing gives information about specific communities among microbiomes and not the entire microbiome existing in the habitat [88]. For analyzing all the microbes existing in the habitat, an untargeted shotgun sequencing approach has been designed. Shotgun metagenomics involves processes sample collection, processing, and sequencing, quality filtering the obtained sequencing reads, assembly of reads, binning contigs, and analyzing the obtained data [88, 89].
Before a shotgun study, there is a need for estimating the amount of microbial diversity of the given habitat to identify the relative abundance of species. This can be accomplished by 16S/18S rRNA metagenomic data analysis of the habitat. Species abundance is expected to be more in a soil sample when compared to a gut sample of an organism. So there is a need to generate more sequencing data for a soil sample than for a gut sample. Unless the sequencing depth is increased, there are fewer chances of identifying rare taxa. So, as the species abundance increases, the sequencing depth should also be increased for better insight into the habitat [89]. For instance, the shotgun metagenomic analysis of a carboxymethylcellulose (as sole carbon source) enriched bacterial consortia revealed the reconstruction of six complete genomes and four out of which were novel. Three of them were Bacillus thermozeamaize, Geobacillus thermoglucosidasius, and Caldibacillus debilis. The CAZy analysis revealed the presence of several genes associated with the degradation of lignocellulosic material. Out of all the genomes, the genome of Bacillus thermozeamaize had abundant glycosyl hydrolases (GHs) [90].
The metagenomic approach has been used to study diverse environments, but out of these, only a few are lignocellulosic-rich ecosystems [78, 91,92,93]. Although the chemical composition and the structural complexity of lignocellulosic biomass may make it difficult for microorganisms to colonize these environments and also hinder the primary and essential high-quality DNA extraction step, the best option to study lignocellulosic microbes and investigate the catabolic potential of related non-cultivable organisms is by using lignocellulosic materials. Therefore, environments that contain sugarcane bagasse, wheat straw, corn stover, rice straw, etc. are ideal for identifying new lignocellulolytic enzymes from uncharacterized microbial populations using functional metagenomics [93].
7 Process of metagenomic library construction
There exist a few technical limitations in the process of construction and functional streaming of metagenomic libraries which are specific to lignocellulose rich ecosystems. High-quality DNA is a critical requirement for library construction, but during its extraction from lignocellulosic material, it is often contaminated by acids, furan derivatives, and phenolic compounds [94]. These contaminants can cause denaturation of nucleic acids, interfere with DNA transformation, and also act as inhibitors for several enzymes required for library preparation [95]. The quality of DNA extracted from plant biomass can also be compromised by the presence of fertilizers, preservatives, and stabilizers from industrial processes. Due to limited microbial colonization of lignocellulosic biomass, metagenomic DNA (mgDNA) yield from this material is low and the presence of the above-mentioned contaminants will further decrease the yield of mgDNA. Variations in sample granularity have also been observed to affect yields [91, 94]. Consequently, no standardized methods exist to date to extract mgDNA from lignocellulosic materials. So, for the reason stated above, there are no optimized mgDNA extraction protocols from any lignocellulose-rich sample. However, to date, only amended protocols of existing procedures are being used for mgDNA extraction [91, 93].
The problem of low mgDNA yields due to low microbial load in lignocellulosic-rich materials can be solved by using enrichment strategies. In the case of cellulolytic organisms, the sample is supplemented with additional cellulose to enrich the sample with only cellulolytic organisms. Due to this, the target organisms would mostly increase in their population and eventually lead to an increase in the yield of mgDNA [96]. If the target organisms are prokaryotes, there is a need to separate them from eukaryotes to avoid DNA contamination and vice versa. Considering the advantage of small size, prokaryotes can be separated using size-selective filters and centrifugation to avoid eukaryotic genome contamination [97, 98]. When microbial populations are subjected to enrichment, the natural biomass ecosystem undergoes external modifications, thus making structural studies lose their relevance. One of the strategies is to use silica gel in the process of mgDNA extraction after the lysis of cells; this will reduce the shearing of mgDNA and increase the yield as well as the quality of mgDNA. The other strategy is to separate the large particulate matter from the sample (before the mgDNA extraction) by gently centrifuging at 3000 rpm (645 times gravity) to obtain a translucent pellet (containing microbes) and then using lysis buffer (1% cetyltrimethylammonium bromide (CTAB), 100 mM ethylenediaminetetraacetic acid (EDTA), 1.5 M NaCl, 100 mM Tris-HCl) and proteinase to lyse the microbes. These strategies increase the chances of isolating novel genes encoding cellulases, xylanases, and lipases/esterases.
Following the purification of mgDNA, it is size-fractionated and then cloned into plasmids (< 20 kb insert size), cosmids and fosmids (< 40 kb insert size), or bacterial artificial chromosomes (BACs) (> 40 kb insert size), depending on the target of functional screening of mgDNA libraries. Generally, genes involved in related metabolic pathways are present in clusters (such as operons or super-operonic clusters) in the microorganism’s genome. These clusters are more frequently observed in prokaryotes than in eukaryotes. Thus, it is preferable to clone the mgDNA into cosmids or fosmids for functional screening [99, 100]. On the other hand, when short inserts are cloned into plasmids, large gene clusters cannot be recovered, thus reducing the productivity of functional metagenomics [101]. However, plasmids containing promoters located on both sides of a multiple cloning site facilitate bidirectional transcription, which can increase the number of positive clones in plasmid-based libraries [102]. Furthermore, since gene expression is greatly host-dependent, broad host range systems should be used to maximize the chances of successful expression and detection of target genes [103]. E. coli is frequently used as a host for economical, effective, and high-level production of several heterologous proteins [104]. All the steps involved in the process of fosmid library construction are represented in the form of a flow chart in Fig. 7b. However, the use of E. coli hosts may have limited the number of lignocellulolytic enzymes that have been isolated from metagenomic libraries [105]. For example, the use of bacterial host systems such as E. coli significantly reduces the chances of identifying and isolating lignocellulolytic enzymes of fungal origin. This is due to a variety of factors such as differences in codon usage, promoter regulation and activation, and RNA processing and translation, which hinder the functional expression of eukaryotic genes in prokaryotic systems. Furthermore, essential post-translational modifications (such as glycosylation of eukaryotic cellulases and xylanases to facilitate secretion) are lacking in prokaryotic hosts [105,106,107]. Due to these limitations, the majority of lignocellulolytic enzymes identified to date belong to prokaryotic proteins [105]. Therefore, the use of alternate host expression systems should be considered instead of E. coli (e.g., Pseudomonas putida, Burkholderia graminis, Bacillus subtilis, Ralstonia metallidurans, Caulobacter vibrioides, Thermus thermophilus, Sulfolobus solfataricus, and Streptomyces) [108,109,110]. For example, T. thermophilus has been successfully used as a metagenomic library host for the detection of esterases and recombinant expression of xylanases, giving a higher yield of active clones than E. coli [111, 112]. The ongoing development of eukaryotic host systems will also contribute significantly to metagenomic studies of biomass-degrading enzymes.
8 Metagenomic 16S rDNA sequencing
After unleashing the potential of 16S rDNA for the phylogenetic analysis of bacteria in the early 1990s, it has been applied extensively. The metagenomic 16S rDNA sequencing gives details of the entire bacterial species surviving in the habitat. Furthermore, many researchers have even used sequential metagenomics for the identification of putative genes. Few examples are presented in Table 3.
9 Metagenomic-derived functional cellulases
Several cellulases have been isolated from different microorganisms and analyzed, but common practices for isolation and characterization by cultivable methods lead to the biased selection of microorganisms. Thus, commercial cellulases produced by Trichoderma or Aspergillus strains lack accessory enzyme activities and are unable to perform efficient saccharification of untreated biomass [136]. The development of more efficient cellulases using the metagenomic approach could increase the possibility of improving cocktails of enzymes used in lignocellulose conversion. Cellulase enzyme systems required for industrial conversion of cellulosic biomass should be able to function under robust physiochemical conditions.
9.1 Endoglucanase
Endoglucanase initiates the cellulose degradation process; thus, they are crucial for the cellulolytic action of microorganisms. The complete genes found in environmental samples such as insect gut, soil, spill water, and feces belonged to the GH5, GH9, GH12, GH44, and GH45 families of glycosyl hydrolases. The source of these genes may be from a varied group of microbes such as Bacillus, Cellulomonas, Cellvibrio, Clostridium, Vibrio, and other unknown microbes.
Endoglucanases exhibit differences in the amino acids located at positions 244 to 1005 in the protein sequence. Characterization was also done after performing heterologous expression and purification of endoglucanases. Upon performing kinetic analysis of the enzyme as well as studying its structure-function relationships, it was discovered that the functional properties of the endoglucanase are mainly dependant on the environment that the genes are isolated from. For example, a majority of cellulases from biogas digester [137], sugarcane bagasse compost [138], or rice straw compost [139] were found to be thermophilic. Similarly, cellulases from soda lake [140] or mangrove soil [141] were capable of withstanding high concentrations of salt. However, there are also a few enzymes which do not correspond in nature to the environment that they were isolated from, which can be observed in the case of the endoglucanase isolated from compost soil [142], which remains active even at a low temperature range of around 10–40 °C and has optimum activity at 25 °C. In 2016, Maruthamuthu performed functional metagenomics-based screening for hemicellulases and cellulases in two groups of wheat straw-degrading microorganisms using a multi-substrate approach revealing new thermo-alkaliphilic enzymes [143]. In 2016, Cheng et al. isolated and characterized nonspecific endoglucanase from the metagenomic library of goat rumen [144].
There are about 165 glycosyl hydrolase families (GH-1 to GH-165). Some families like GH-5, GH-13, GH-30, and GH-43 have 56, 42, 9, and 37 sub-families respectively. To date, there are about 664,285 modules classified into 165 families and 10520 modules yet to be classified (CAzy database). Since there is a continuous effort for identifying novel GHs from the past few decades, a great number of GHs were discovered and characterized. A large portion of GHs identified and grouped under the GH-5 family were cellulose-degrading and GH-93 were arabinan-degrading [145]. From Table 4, it has been observed that about 50% of listed endoglucanases belong to the GH-5 family and the rest of the endoglucanases belong to 9 different GH families like GH-5, GH-6, GH-8, GH-9, GH-16, GH-44, GH-45, and GH-74.
Out of all the considered metagenome-derived endoglucanases, only three had carbohydrate-binding domains (CBMs). As the enzymes are all proved beyond doubt about their capability for cellulose degradation, they can be grouped under type A CBMs. It is observed that all three had the CBM at the c-terminal only. A few more details of the metagenomic-derived endoglucanases associated with CBMs are presented in Table 5.
As the majority of the enzymes derived through metagenomics originate from uncultivable bacteria, their characteristics may be differing from the regular enzymes. Their optimum pH and temperature depend on the habitat from which it has been isolated. Various characters of enzyme-like co-factors (different metal ions), the effect of different types of detergents and organic solvents would not only help to enhance the enzyme’s activity but also support in further steps like bioprocessing and fermentation, at industrial scale.
9.2 The pH and temperature optima of metagenome-derived endoglucanases
As discussed earlier, the major drawback of second-generation biofuels is the lack of efficient, thermostable enzymes to liberate fermentable sugars from biomass. So, industries require robust, acid/base-tolerant, thermostable cellulases to increase biofuel production. Generally, every enzyme has an optimum temperature and pH, at which it attains maximum activity. The habitat of the organism and its intracellular environment decide the optimum temperature and pH of the enzymes in that organism. pH and temperature play a major role in both the folding and unfolding of a protein. Table 6 presents the information about the optimum pH and temperature for various endoglucanases reported in the literature so far.
Out of the 44 endoglucanases listed (in Table 6), 28 were observed to be in the range of 20–50 °C, and 9 were observed to be in the range of 50–60 °C. Only 7 were observed to be in the range of 60–90 °C, these thermostable enzymes have industrial importance. About 40% of listed endoglucanases were observed to be between 45 and 50 °C and 85% are active above 45 °C. All the animal gut-derived endoglucanases were in the range of 45 to 55 °C except PersiCel4 which had 85 °C. All the listed endoglucanases are in the pH range of 4 to 8.5. About 70% (31 enzymes) of the listed endoglucanases are acidic, 20% (9 enzymes) were neutral, and 10% (4 enzymes) were observed to be basic. The majority of the animal gut-derived endoglucanases were observed to be acidic, which might be due to the acidic environment of the gut [181].
9.3 Metal ions and their effect
Metal ions associate/dissociate with proteins to activate or inactivate them by interacting with amino groups, carboxylic groups, and other functional groups like sulfhydryl groups of amino acids [182]. They can also act as electron donors and acceptors. Though, the effect of various other divalent ions on cellulases has been variable depending on the structural linage of the protein. For an ion, the two characters which decide its potential are the ionic radius and charge [183]. Ionic radius and the potential to attract charged amino acids are inversely proportional. The lesser the ionic radius, the greater the potential to attract charged amino acids; this plays a key role in disturbing the residues of the catalytic site [182]. Water also plays a crucial role in metal ion-macromolecule interactions. Depending on the extent of the ion’s interaction with water by sharing the electrons with its adjacent molecules of bulk water portrays ion’s soft or hard nature [184]. Metal ions generally used for cellulose characterization are of different valences like mono-, di-, and tri-valent (Ca2+, Co2+, Cu2+, Fe3+, Fe2+, Hg2+, Mn2+, Mg2+, Ni2+, and Zn2+) and their effects are reported in Table 7.
Out of many metal ions, Co2+ is considered as the activator for cellulases [185]. From the tabulated data (Table 7), it is observed that many metagenome-derived cellulases can enhance their catalytic ability in the presence of Co2+, Ca2+, and Mn2+ ions. When compared to Co2+ ions, Ca2+ and Mn2+ ions are observed to enhance more number of listed endoglucanases. The literature suggests that Cu2+ and Fe2+ have always been inhibitory towards cellulases [186], whereas the inhibitory effect of Fe2+ was observed to be less on metagenomic-derived endoglucanases. Cu2+, Hg2+, Ag+, and Zn2+ appear to be potent inhibitors. Though the literature available regarding the effect of Hg2+ and Ag+ ions on metagenome-derived endoglucanases is less, the available studies suggest that both the ions almost inhibit the enzyme’s activity. According to Ani Tejirian et al. [186], Hg2+ is a lethal inhibitor for cellulases; the reason might be the interaction of Hg2+ with sulfur in the amino acids like methionine and cysteine. In the same study, they have also reported that the effect of Fe3+ was much higher than that of Fe2+ [186]. Similar results were observed by Juan Liu et al. in the case of metagenome-derived endoglucanase Cel5G [149]. Even if Fe2+ is supplemented in the reaction, it voluntarily oxidizes to Fe3+ in the presence of water. The carbohydrates which have hemiacetal reducing ends are prone to undergo oxidation in the presence of Fe3+. This oxidation can reduce the availability of cellulase degradable cellulose to the enzyme thereby leading to a reduction in the product yield. Hence, comprehensive knowledge of metal ions that can enhance/inhibit the enzyme’s activity is essential while scaling up the process to the industrial level.
9.4 Detergents and their effect
In recent times, cellulases are being employed in various fields and one such field is the laundry detergent industry. Many detergent industries are following new methods by supplementing detergents with compatible enzymes to ensure ease stain removal without losing the smoothness of the fabric. For this application, the compatibility of cellulases with different types of detergents is analyzed. Detergents are compounds with amphipathicity. They are structurally well-defined with a head associated with a tail. The head group is polar and the tail is hydrophobic. They can be mainly grouped into four major clans. They are ionic detergents, non-ionic detergents, bile salts, and zwitterionic detergents.
Ionic detergents have a charged head group, maybe anionic or cationic, and the hydrophobic tail may be a hydrocarbon chain or sometimes steroidal support too, e.g., SDS (sodium dodecyl sulfate) and CTAB (cetyltrimethylammonium bromide). SDS has inhibited the majority of the listed endoglucanases (Table 8) and CTAB has also drastically reduced the activity of endoglucanases suggesting that these endoglucanases are incompatible with ionic detergents (SDS, CTAB). Non-ionic detergents have hydrophilic head groups that are uncharged. They are mild and do not denature proteins like ionic detergents as they do not act on protein-protein interactions, Examples are Triton X-100, Tween 20, Tween 40, and Tween 80. Two cases reported by Chang-Muk Lee et al. [157] and Puneet Gupta et al. [167] were observed to enhance the endoglucanase activity in the presence of Tween (20, 40, 80) (Table 8). Most of the listed endoglucanases have reduced their endoglucanase activity in the presence of Triton X-100 except one enzyme, which is reported by Marjolaine Martin et al. They reported that Triton X-100 has enhanced the endoglucanase activity of Cel 5.1_3 [156]. These reports suggest that the above-stated enzymes are compatible with Tween and Triton X-100. Bile salts have a strong steroidal backbone which results in having an indistinct head group; example is sodium cholate. Zwitterionic detergents have a combination of both ionic and non-ionic detergent properties; examples are CHAPS (3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate) [187]. None of the studies has used bile salts and zwitterionic detergents for characterizing their enzymes.
9.5 Organic solvents
In the available data, only a few endoglucanases were studied for the effect of organic solvents on their enzyme activity. Isopropanol had a deteriorating effect on all the reported metagenome-derived endoglucanases (Table 9). Xinxin hu et al. have reported the enhancement of activity due to isopropanol. In their study, it has been observed that the trypsin’s hydrophobic portion formed a hydrogen bond with the isopropanol molecule. The predicted amino acid of the hydrophobic region in the bond formation with isopropanol is serine in position 214. Due to this, there was a change in the secondary structure of the protein which in turn changed the tertiary structure and total symmetry of the amino acids. This effect has slacked the basic structure of the protein, exposing the active sites. As a result of this, the availability of active sites increased which might increase or decrease product formation [188].
In most cases, dimethyl sulfoxide (DMSO) has partially reduced the enzyme’s activity; in some cases, it has not shown any effect whereas Cel 5.1_3 reported by Marjolaine Martin et al. has been completely inhibited [156]. DMSO can alter protein characteristics, which might lead to either degradation or aggregation. In meagre amounts, it might not affect the activity of the enzyme but can influence its binding properties [189]. Conversely, at higher concentrations, DMSO unfolds the proteins [190]. Surprisingly, the En1 enzyme reported by xing yan et al. has enhanced its activity in the presence of DMSO [155]. Many enzymes listed in Table 9 had a deteriorating effect in the presence of ethanol and few cases like Umcel9y-1 reported by Yu Zhou et al. [160] and Cel 5.1_3 reported by Marjolaine Martin et al. [156]; the enzyme was completely inhibited. Many studies have reported that the protein denaturation by ethanol reaches its peak between 20 and 50% of ethanol [191].
Most of the reported enzymes had reducing effects on the enzyme’s activity in the presence of acetone, and surprisingly in the case of Cel5R reported by Narender Kumar et al., acetone has enhanced its activity [173]. Acetone is generally used for precipitating protein and sometimes for concentrating them also. Deborah M. Simpson et al. in 2010 reported that acetone which may sometimes remain as residual contamination in purified proteins can selectively alter amino acids which might lead to protein’s conformational changes. This modification might sometimes increase or decrease the activity of the enzyme [192]. Cel5R reported by Narender Kumar et al. [173] has enhanced its activity in the presence of methanol, and in most of the reported cases, it has partially reduced the enzyme’s activity whereas in the case of Cel5G reported by Juan Liu et al. [149] the activity was completely inhibited. In 2011, Soyoun Hwang et al. reported that the presence of methanol expands the structure of the protein, probably by reducing protein’s hydrophobic properties. This expansion might increase access to the active site and increase the activity or sometimes even decrease the activity if the expansion disrupts the active site integrity [193]. Chloroform is considered a potent protein denaturant. It is regularly used in combination with phenol and isoamyl alcohol for the process of DNA extraction to separate proteins from nucleic acids [194]. Table 9 represents the effect of a few organic solvents discussed above on metagenomic-derived endoglucanases.
9.6 Other chemical compounds
Cel01 reported by Heiko nacke [151] has enhanced its activity in the presence of glycerol and the rest of the reported enzymes had a declining effect. Generally, proteins are very stable in an aqueous environment with the help of co-solvents and one such co-solvent is glycerol [195]. Glycerol has the capability to swift the native protein to a more compressed structure. It also prevents the aggregation of proteins in the process of protein refolding [195].
In most of the reported enzymes, EDTA had a deteriorating effect, and in the case of Umcel9y-1 reported by Yu Zhou et al. [160], it has completely inhibited the enzyme activity. Few enzymes like C67-1, Cel5G, CS10, and PHS remained unaffected in the presence of EDTA [147, 149, 157, 167]. EDTA is a popular protease and metal chelating agent from the ages past, which can reduce the enzyme activity by chelating the metal ions that act as co-factors for activating many enzymes. But Gajendra S. Naika et al. explained the enhancing effect of EDTA on endoglucanases. According to them, EDTA is capable of opening the protein molecule partially to create a transitional state, which might improve the enzyme-substrate interaction. Thus, the EDTA exhumes a portion of a protein that facilitates a more appropriate structure for better interaction of substrate [196]. Among the listed endoglucanases, only two enzymes were evaluated for their activity in the presence of dimethylformamide (DMF) (Umcel9y-1, CS10) and both had a deteriorating effect [157, 160]. In small concentrations, DMF can easily remove the interacting water molecules from the protein’s surface and intensely compete for hydrogen bonds. This process denatures the protein structure leading to protein unfolding [197]. Table 10 represents the effect of few such chemical compounds on metagenomic-derived endoglucanases.
Dithiothreitol (DTT) has a special character in reducing disulfide bonds. Apart from that, in higher concentrations, it might create steric hindrance at the ligand binding site leading to decreased ligand binding and may bring conformational changes which would decrease the ligand binding further [198]. Surprisingly, three of the reported enzymes (C67-1, nmGH45, p4818Cel5_2A) had increased their activity in the presence of DTT and one (CelRH5) had a deteriorating effect [147, 170, 174, 177]. Only one (PHS) enzyme was checked for the activity in the presence of polyethylene glycol (PEG) and it has enhanced the enzyme’s activity [167]. PEG is a polymer that is hydrophilic and non-ionic, which has the capability of precipitating proteins [199]. Only one (EndoG) enzyme was checked for the activity in the presence of phenylmethylsulfonyl fluoride (PMSF) and it has partially reduced the enzyme’s activity [162]. PMSF has the capability of inhibiting a lot of proteases. The inhibition is mainly due to the interaction of serine residue in the active site of protein with PMSF [200]. NmGH45 reported by Junqi Zhao et al. [170] has enhanced its activity in the presence of β-mercaptoethanol. It is most commonly used for reducing disulfide bonds. Apart from that, it can also act as a chelating agent [201]. Sometimes, it might even activate some enzymes by capturing the metal ions which act as co-factors for inhibitors, thereby inhibiting the inhibitors from acting on their target proteins [182].
10 Significance of metagenome-derived cellulases in CBP
Stimulating the expression of cellulases and the involved secretion systems are the most puzzling phases in scheming cellulolytic organisms on an industrial scale. Cellulases exist as either bound to the surface (cellulosomes) or soluble extracellular (free cellulases). In 2014, Parisutham et al. have reported that each cellulolytic organism has over 400 different cellulose-related enzymes like glycoside hydrolases, glycoside esterases, carbohydrate esterase, cellulose-binding enzymes, endoglucanases, exoglucanases, and hemicellulases. Most of these enzymes can be introduced into their suitable hosts and utilized for CBP to depolymerize the cellulosic material into fermentable sugars as well as the subsequent conversion of obtained sugars into required biofuel [202]. Further, to make the CBP process efficient and cost-effective, metagenomics offers prospects to screen for novel cellulase genes from the pool of uncultivable microbes. A robust cellulase enzyme with high activity, wide substrate specificity, thermos-tolerance, and chemical and lignin tolerance can be obtained through metagenomics approach from natural and extreme environmental samples [202].
Metagenome-derived cellulases further may be improved by using protein engineering strategies for inhibitor tolerance, thermal, pH stability, enzymes with multi-functionality, synthetic cellulosome with a combination of active and robust cellulases, and plummeting product inhibition inside the cell [203,204,205,206,207]
11 Metagenome-derived libraries and their improved traits
To successfully grow various recombinant microbes on lignocellulosic biomass directly, support from various cellulases is required. These supporting enzymes can be either free enzymes from extracellular secretions or surface strapped like cellulosomes to accomplish biomass hydrolysis proficiently [202]. For acquiring ideal microbes for CBP, advanced approaches like metagenomics, metatranscriptomics, and even protein engineering can be employed. As the above-stated approaches have the potential in identifying novel cellulases with specificities towards diverse substrates along with efficient activity, chemical, and thermal tolerance. All the efforts for designing the best strategies compiled above aim to develop a futuristic design involving synthetic microbial consortia, to enable the engineering of robust microbial communities targeting highly efficient cellulose conversion industrially (Table 11).
12 Conclusion and future prospects
Despite being rich in lignocellulosic biomass, India still lags in the discovery of industrially viable lignocellulolytic enzymes for effective production of second-generation biofuels (SGB) at an industrial scale. In the last few years, advanced sequencing strategies like next-generation sequencing (NGS) have come up with the capability of generating large amounts of sequence data, unlike conventional methods. With the help of NGS and metagenomics, a lot of unexplored, uncultivable, novel lignocellulases can be identified and exploited for the acceleration of SGB production in India. In this review, few metagenomic outcomes were discussed to enlighten the importance of many unexplored habitats for novel cellulolytic gene mining. This review also highlights the potential of different metagenomics approaches as most of the uncultivable cellulose-degrading microbiome and their efficient enzymes remain unexploited. It is observed that the degree of success depends on the methodology opted for, as every methodology has its drawbacks. In the future, these drawbacks can be overcome and even different strategies can be developed in combination with various methodologies described in this review.
References
British Petroleum reports (2020) BP Statistical Review of World Energy 2020
BP Statistical Review of World Energy (2018) BP Statistical Review of World Energy 67:1–53
BP Statistical Review of World Energy (2019) BP Statistical Review of World Energy. 68:1–61
IEA I energy agency (2020) Global Energy Review 2020. Glob Energy Rev 2020. https://doi.org/10.1787/a60abbf2-en
IEA IEA (2017) World Energy Outlook-2017
Outlook IE (2015) India Energy Outlook. World Energy Outlook:1–191
Sørensen A, Lübeck M, Lübeck PS, Ahring BK (2013) Fungal beta-glucosidases: A bottleneck in industrial use of lignocellulosic materials. Biomolecules 3:612–631. https://doi.org/10.3390/biom3030612
Gray KA, Zhao L, Emptage M (2006) Bioethanol. Curr Opin Chem Biol 10:141–146. https://doi.org/10.1016/j.cbpa.2006.02.035
Mohanram S, Amat D, Choudhary J et al (2013) Novel perspectives for evolving enzyme cocktails for lignocellulose hydrolysis in biorefineries. Sustain Chem Process 1:15. https://doi.org/10.1186/2043-7129-1-15
Gowen CM, Fong SS (2010) Exploring biodiversity for cellulosic biofuel production. Chem Biodivers 7:1086–1097. https://doi.org/10.1002/cbdv.200900314
Bayer EA, Chanzy H, Lamed R, Shoham Y (1998) Cellulose, cellulases and cellulosomes. Curr Opin Struct Biol 8:548–557. https://doi.org/10.1016/S0959-440X(98)80143-7
Ljungdahl LG (2008) The cellulase/hemicellulase system of the anaerobic fungus Orpinomyces PC-2 and aspects of its applied use. Ann N Y Acad Sci 1125:308–321. https://doi.org/10.1196/annals.1419.030
Dash HR, Das S (2018) Molecular Methods for Studying Microorganisms From Atypical Environments, 1st edn. Elsevier Ltd.
Mekala NK, Potumarthi R, Baadhe RR, Gupta VK (2014) Current Bioenergy Researches: Strengths and Future Challenges. Elsevier
Zhou Z, Lei F, Li P, Jiang J (2018) Lignocellulosic biomass to biofuels and biochemicals: A comprehensive review with a focus on ethanol organosolv pretreatment technology
Ahorsu R, Medina F, Constantí M (2018) Significance and challenges of biomass as a suitable feedstock for bioenergy and biochemical production: A review. Energies 11. https://doi.org/10.3390/en11123366
Maurya DP, Singla A, Negi S (2015) An overview of key pretreatment processes for biological conversion of lignocellulosic biomass to bioethanol. 3. Biotech 5:597–609. https://doi.org/10.1007/s13205-015-0279-4
Jouzani GS, Taherzadeh MJ (2015) Advances in consolidated bioprocessing systems for bioethanol and butanol production from biomass: A comprehensive review. Biofuel Res J 2:152–195. 10.18331/BRJ2015.2.1.4
Tengborg C, Galbe M, Zacchi G (2001) Reduced inhibition of enzymatic hydrolysis of steam-pretreated softwood. Enzyme Microb Technol 28:835–844. https://doi.org/10.1016/S0141-0229(01)00342-8
Goshadrou A, Lefsrud M (2013) Enhanced NSSF for ethanol production by phosphoric acid pretreatment. Can Soc Bioeng CSBE13:4–9
Ishola MM, Jahandideh A, Haidarian B et al (2013) Simultaneous saccharification, filtration and fermentation (SSFF): A novel method for bioethanol production from lignocellulosic biomass. Bioresour Technol 133:68–73. https://doi.org/10.1016/j.biortech.2013.01.130
Brethauer S, Wyman CE (2010) Review: Continuous hydrolysis and fermentation for cellulosic ethanol production. Bioresour Technol 101:4862–4874. https://doi.org/10.1016/j.biortech.2009.11.009
Bhalla A, Bansal N, Kumar S et al (2013) Improved lignocellulose conversion to biofuels with thermophilic bacteria and thermostable enzymes. Bioresour Technol 128:751–759. https://doi.org/10.1016/j.biortech.2012.10.145
Kumagai A, Kawamura S, Lee SH et al (2014) Simultaneous saccharification and fermentation and a consolidated bioprocessing for Hinoki cypress and Eucalyptus after fibrillation by steam and subsequent wet-disk milling. Bioresour Technol 162:89–95. https://doi.org/10.1016/j.biortech.2014.03.110
Moshi AP, Crespo CF, Badshah M et al (2014) High bioethanol titre from Manihot glaziovii through fed-batch simultaneous saccharification and fermentation in Automatic Gas Potential Test System. Bioresour Technol 156:348–356. https://doi.org/10.1016/j.biortech.2013.12.082
Arijana B, Mardetko N, Kundas S, et al (2018) Bioethanol production from renewable raw materials and its separation and purification: A review. Food Technol Biotechnol 56:289–311. 10.17113/ftb.56.03.18.5546
Wu Z, Lee YY (1998) Nonisothermal simultaneous saccharification and fermentation for direct conversion of lignocellulosic biomass to ethanol. Appl Biochem Biotechnol - Part A Enzym Eng Biotechnol 70–72:479–492. https://doi.org/10.1007/BF02920161
Teixeira LC, Linden JC, Schroeder HA (1999) Optimizing peracetic acid pretreatment conditions for improved simultaneous saccharification and co-fermentation (SSCF) of sugar cane bagasse to ethanol fuel. Renew Energy 16:1070–1073. https://doi.org/10.1016/s0960-1481(98)00373-5
Teixeira LC, Linden JC, Schroeder HA (2000) Simultaneous saccharification and cofermentation of peracetic acid- pretreated biomass. Appl Biochem Biotechnol - Part A Enzym Eng Biotechnol 84–86:111–127. https://doi.org/10.1385/abab:84-86:1-9:111
Zhang J, Lynd LR (2010) Ethanol production from paper sludge by simultaneous saccharification and co-fermentation using recombinant xylose-fermenting microorganisms. Biotechnol Bioeng 107:235–244. https://doi.org/10.1002/bit.22811
Kang L, Wang W, Lee YY (2010) Bioconversion of kraft paper mill sludges to ethanol by SSF and SSCF. Appl Biochem Biotechnol 161:53–66. https://doi.org/10.1007/s12010-009-8893-4
Olson DG, McBride JE, Joe Shaw A, Lynd LR (2012) Recent progress in consolidated bioprocessing. Curr Opin Biotechnol 23:396–405. https://doi.org/10.1016/j.copbio.2011.11.026
Lynd LR, Weimer PJ, van Zyl WH, Pretorius IS (2002) Microbial Cellulose Utilization: Fundamentals and Biotechnology Lee R. Lynd, Paul J. Weimer, Willem H. van Zyl and Isak S. Pretorius Microbiol. Mol. Biol. Rev. 2002, 66(3):506. DOI: 10.1128/MMBR.66.3.506-577.2002. REFERENCES. Microbiol Mol Biol Rev 66:506–577. https://doi.org/10.1128/MMBR.66.3.506
Fengel VD, Wegener G (1985) Wood: Chemistry, Ultrstructure, Reactions. Acta Polym 35:295
Carere CR, Sparling R, Cicek N, Levin DB (2008) Third generation biofuels via direct cellulose fermentation. Int J Mol Sci 9:1342–1360. https://doi.org/10.3390/ijms9071342
Chandra RP, Bura R, Mabee WE et al (2007) Substrate pretreatment: The key to effective enzymatic hydrolysis of lignocellulosics? Adv Biochem Eng Biotechnol 108:67–93. https://doi.org/10.1007/10_2007_064
McKendry P (2002) Energy production from biomass (part 1): overview of biomass. Bioresour Technol 83:37–46. https://doi.org/10.1016/S0960-8524(01)00118-3
Kucharska K, Rybarczyk P, Hołowacz I et al (2018) Pretreatment of lignocellulosic materials as substrates for fermentation processes. Molecules 23:1–32. https://doi.org/10.3390/molecules23112937
Fortunati E, Kenny JM, Torre L (2019) Lignocellulosic materials as reinforcements in sustainable packaging systems: Processing, properties, and applications. Biomass, Biopolym Mater Bioenergy Constr Biomed other Ind Appl 87–102. https://doi.org/10.1016/B978-0-08-102426-3.00005-9
Ek R, Gustafsson C, Nutt A et al (1998) Cellulose powder from Cladophora sp. algae. J Mol Recognit 11:263–265. https://doi.org/10.1002/(SICI)1099-1352(199812)11:1/6<263::AID-JMR437>3.0.CO;2-G
Desvaux M (2005) Clostridium cellulolyticum: Model organism of mesophilic cellulolytic clostridia. FEMS Microbiol Rev 29:741–764. https://doi.org/10.1016/j.femsre.2004.11.003
Laureano-Pérez L, Teymouri F, Alizadeh H, Dale BE (2005) Understanding Factors that Limit Enzymatic Hydrolysis of Biomass Understanding Factors that Limit Enzymatic Hydrolysis of Biomass H ASAN A LIZADEH , AND B RUCE E . D ALE *. Appl Biochem Biotechnol 121–124. https://doi.org/10.1007/978-1-59259-991-2
Ding SY, Zhao S, Zeng Y (2014) Size, shape, and arrangement of native cellulose fibrils in maize cell walls. Cellulose 21:863–871. https://doi.org/10.1007/s10570-013-0147-5
Saha BC (2003) Hemicellulose bioconversion. J Ind Microbiol Biotechnol 30:279–291. https://doi.org/10.1007/s10295-003-0049-x
Hendriks ATWM, Zeeman G (2009) Pretreatments to enhance the digestibility of lignocellulosic biomass. Bioresour Technol 100:10–18. https://doi.org/10.1016/j.biortech.2008.05.027
Bajpai P (2016) Structure of Lignocellulosic Biomass. Pretreatment of Lignocellulosic Biomass for Biofuel Production, In, pp 7–12
Avgerinos GC, Wang DIC (1983) Selective solvent delignification for fermentation enhancement. Biotechnol Bioeng 25:67–83. https://doi.org/10.1002/bit.260250107
Chang VS, Holtzapple MT (2000) Fundamental Factors Affecting Biomass Enzymatic Reactivity. Appl Biochem Biotechnol 84–86:5–38. https://doi.org/10.1385/ABAB:84-86:1-9:5
Brownell HH, Saddler JN (1987) Steam pretreatment of lignocellulosic material for enhanced enzymatic hydrolysis. Biotechnol Bioeng 29:228–235. https://doi.org/10.1002/bit.260290213
Meng X, Ragauskas AJ (2014) Recent advances in understanding the role of cellulose accessibility in enzymatic hydrolysis of lignocellulosic substrates. Curr Opin Biotechnol 27:150–158. https://doi.org/10.1016/j.copbio.2014.01.014
Henrissat B, Daviest G (1997) Structural and sequence-based classification of glycoside hydrolases. Curr Opin Struct Biol 7:637–644
Franková L, Fry SC (2013) Biochemistry and physiological roles of enzymes that “cut and paste” plant cell-wall polysaccharides. J Exp Bot 64:3519–3550. https://doi.org/10.1093/jxb/ert201
Elleuche S, Schröder C, Sahm K, Antranikian G (2014) Extremozymes-biocatalysts with unique properties from extremophilic microorganisms. Curr Opin Biotechnol 29:116–123. https://doi.org/10.1016/j.copbio.2014.04.003
Naumoff DG (2011) Hierarchical classification of glycoside hydrolases. Biochem 76:622–635. https://doi.org/10.1134/S0006297911060022
Henrissat B (1991) A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem J 280:309–316. https://doi.org/10.1042/bj2800309
Lombard V, Golaconda Ramulu H, Drula E et al (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res 42:490–495. https://doi.org/10.1093/nar/gkt1178
Mccarter JD, Withers SG (1994) McCarter and Withers 1994.pdf. Curr Opin Struct Biol 4:885–892
Linton SM (2020) Review: The structure and function of cellulase (endo-β-1,4-glucanase) and hemicellulase (β-1,3-glucanase and endo-β-1,4-mannase) enzymes in invertebrates that consume materials ranging from microbes, algae to leaf litter. Comp Biochem Physiol Part - B Biochem Mol Biol 240:110354. https://doi.org/10.1016/j.cbpb.2019.110354
Thuan NH, Sohng JK (2013) Recent biotechnological progress in enzymatic synthesis of glycosides. J Ind Microbiol Biotechnol 40:1329–1356. https://doi.org/10.1007/s10295-013-1332-0
Sobala LF, Speciale G, Zhu S et al (2020) An Epoxide Intermediate in Glycosidase Catalysis. ACS Cent Sci 6:760–770. https://doi.org/10.1021/acscentsci.0c00111
Rye C, Withers SG (2002) Glycosidase mechanisms. Curr Opin Chem Biol 6:619–629. https://doi.org/10.1016/S1367-5931(02)00380-0
Dadheech T, Shah R, Pandit R et al (2018) Cloning, molecular modeling and characterization of acidic cellulase from buffalo rumen and its applicability in saccharification of lignocellulosic biomass. Int J Biol Macromol 113:73–81. https://doi.org/10.1016/j.ijbiomac.2018.02.100
Dennis RJ, Taylor EJ, MacAuley MS et al (2006) Structure and mechanism of a bacterial β-glucosaminidase having O-GlcNAcase activity. Nat Struct Mol Biol 13:365–371. https://doi.org/10.1038/nsmb1079
Hidaka M, Kitaoka M, Hayashi K et al (2006) Structural dissection of the reaction mechanism of cellobiose phosphorylase. Biochem J 398:37–43. https://doi.org/10.1042/BJ20060274
Vuong TV, Wilson DB (2010) Glycoside hydrolases: Catalytic base/nucleophile diversity. Biotechnol Bioeng 107:195–205. https://doi.org/10.1002/bit.22838
Dashtban M, Maki M, Leung KT et al (2010) Cellulase activities in biomass conversion: measurement methods and comparison. Crit Rev Biotechnol 30:302–309. https://doi.org/10.3109/07388551.2010.490938
Tiwari R, Nain L, Labrou NE, Shukla P (2018) Bioprospecting of functional cellulases from metagenome for second generation biofuel production: a review. Crit. Rev. Microbiol. 44:244–257
Mariano DCB, Leite C, Santos LHS et al (2017) Characterization of glucose-tolerant β-glucosidases used in biofuel production under the bioinformatics perspective: A systematic review. Genet Mol Res:16. https://doi.org/10.4238/gmr16039740
Srivastava N, Rathour R, Jha S et al (2019) Microbial beta glucosidase enzymes: Recent advances in biomass conversation for biofuels application. Biomolecules 9:1–23. https://doi.org/10.3390/biom9060220
Singhania RR, Patel AK, Sukumaran RK et al (2013) Role and significance of beta-glucosidases in the hydrolysis of cellulose for bioethanol production. Bioresour Technol 127:500–507. https://doi.org/10.1016/j.biortech.2012.09.012
Tiwari R, Singh S, Shukla P, Nain L (2014) Novel cold temperature active β-glucosidase from Pseudomonas lutea BG8 suitable for simultaneous saccharification and fermentation. RSC Adv 4:58108–58115. https://doi.org/10.1039/c4ra09784j
Bhatia Y, Mishra S, Bisaria VS (2002) Microbial β-glucosidases: Cloning, properties, and applications. Crit Rev Biotechnol 22:375–407. https://doi.org/10.1080/07388550290789568
Shoseyov O, Shani Z, Levy I (2006) Carbohydrate Binding Modules: Biochemical Properties and Novel Applications. Microbiol Mol Biol Rev 70:283–295. https://doi.org/10.1128/mmbr.00028-05
Abbott DW, Boraston AB (2012) Quantitative approaches to the analysis of carbohydrate-binding module function, 1st edn. Elsevier Inc.
Armenta S, Moreno-Mendieta S, Sánchez-Cuapio Z et al (2017) Advances in molecular engineering of carbohydrate-binding modules. Proteins Struct Funct Bioinforma 85:1602–1617. https://doi.org/10.1002/prot.25327
Falkowski PG, Fenchel T, Delong EF (2008) The microbial engines that drive earth’s biogeochemical cycles. Science (80- ) 320:1034–1039. https://doi.org/10.1126/science.1153213
Kennedy J, Marchesi JR, Dobson AD (2008) Marine metagenomics: strategies for the discovery of novel enzymes with biotechnological applications from marine environments. Microb Cell Fact 7:27. https://doi.org/10.1186/1475-2859-7-27
Batista-García RA, del Rayo S-CM, Talia P et al (2016) From lignocellulosic metagenomes to lignocellulolytic genes: trends, challenges and future prospects. Biofuels, Bioprod Biorefining 10:864–882. https://doi.org/10.1002/bbb.1709
Fang Z, Fang W, Liu J et al (2010) Cloning and characterization of a β-glucosidase from marine microbial metagenome with excellent glucose tolerance. J Microbiol Biotechnol 20:1351–1358. https://doi.org/10.4014/jmb.1003.03011
Simon C, Daniel R (2011) Metagenomic analyses: Past and future trends. Appl Environ Microbiol 77:1153–1161. https://doi.org/10.1128/AEM.02345-10
Batista-garcía RA, Casasanero R (2016) Alvárez-castillo A, et al. Prokaryotic diversity from the culture independent taxonomic analysis of a sugarcane bagasse metagenome. 4:22–38
Alvarez TM, Paiva JH, Ruiz DM et al (2013) Structure and function of a novel cellulase 5 from sugarcane soil metagenome. PLoS One 8:1–9. https://doi.org/10.1371/journal.pone.0083635
Coughlan LM, Cotter PD, Hill C, Alvarez-Ordóñez A (2015) Biotechnological applications of functional metagenomics in the food and pharmaceutical industries. Front Microbiol 6:1–22. https://doi.org/10.3389/fmicb.2015.00672
Couto GH, Glogauer A, Faoro H et al (2010) Isolation of a novel lipase from a metagenomic library derived from mangrove sediment from the south Brazilian coast. Genet Mol Res 9:514–523. https://doi.org/10.4238/vol9-1gmr738
Morgan XC, Huttenhower C (2012) Chapter 12: Human Microbiome Analysis. PLoS Comput Biol 8. https://doi.org/10.1371/journal.pcbi.1002808
Hilton SK, Castro-Nallar E, Pérez-Losada M et al (2016) Metataxonomic and metagenomic approaches vs. culture-based techniques for clinical pathology. Front Microbiol 7:1–12. https://doi.org/10.3389/fmicb.2016.00484
Bromberg JS, Fricke WF, Brinkman CC et al (2015) Microbiota - Implications for immunity and transplantation. Nat Rev Nephrol 11:342–353. https://doi.org/10.1038/nrneph.2015.70
Quince C, Walker AW, Simpson JT et al (2017) Shotgun metagenomics, from sampling to analysis. Nat Biotechnol 35:833–844. https://doi.org/10.1038/nbt.3935
Ghosh A, Mehta A, Khan AM (2018) Metagenomic analysis and its applications. Encycl Bioinforma Comput Biol ABC Bioinforma 1–3:184–193. https://doi.org/10.1016/B978-0-12-809633-8.20178-7
Lemos LN, Pereira RV, Quaggio RB et al (2017) Genome-centric analysis of a thermophilic and cellulolytic bacterial consortium derived from composting. Front Microbiol:8. https://doi.org/10.3389/fmicb.2017.00644
Kanokratana P, Mhuantong W, Laothanachareon T et al (2013) Phylogenetic Analysis and Metabolic Potential of Microbial Communities in an Industrial Bagasse Collection Site. Microb Ecol 66:322–334. https://doi.org/10.1007/s00248-013-0209-0
Mhuantong W, Charoensawan V, Kanokratana P et al (2015) Comparative analysis of sugarcane bagasse metagenome reveals unique and conserved biomass-degrading enzymes among lignocellulolytic microbial communities. Biotechnol Biofuels 8:16. https://doi.org/10.1186/s13068-015-0200-8
Rattanachomsri U, KANOKRATANA P, EURWILAICHITR L et al (2011) Culture-Independent Phylogenetic Analysis of the Microbial Community in Industrial Sugarcane Bagasse Feedstock Piles. Biosci Biotechnol Biochem 75:232–239. https://doi.org/10.1271/bbb.100429
Chandel AK, Antunes FAF, Anjos V et al (2014) Multi-scale structural and chemical analysis of sugarcane bagasse in the process of sequential acid-base pretreatment and ethanol production by Scheffersomyces shehatae and Saccharomyces cerevisiae. Biotechnol Biofuels 7:1–17. https://doi.org/10.1186/1754-6834-7-63
Lim HK, Chung EJ, Kim JC et al (2005) Characterization of a forest soil metagenome clone that confers indirubin and indigo production on Escherichia coli. Appl Environ Microbiol 71:7768–7777. https://doi.org/10.1128/AEM.71.12.7768-7777.2005
Vester JK, Glaring MA, Stougaard P (2015) Improved cultivation and metagenomics as new tools for bioprospecting in cold environments. Extremophiles 19:17–29. https://doi.org/10.1007/s00792-014-0704-3
Mori T, Kamei I, Hirai H, Kondo R (2014) Identification of novel glycosyl hydrolases with cellulolytic activity against crystalline cellulose from metagenomic libraries constructed from bacterial enrichment cultures. Springerplus 3:365. https://doi.org/10.1186/2193-1801-3-365
Venter JC, Remington K, Heidelberg JF et al (2004) Environmental Genome Shotgun Sequencing of the Sargasso Sea. Science (80- ) 304:66–74. https://doi.org/10.1126/science.1093857
Cheng J, Pinnell L, Engel K et al (2014) Versatile broad-host-range cosmids for construction of high quality metagenomic libraries. J Microbiol Methods 99:27–34. https://doi.org/10.1016/j.mimet.2014.01.015
Rondon MR, August PR, Bettermann AD et al (2000) Cloning the soil metagenome: A strategy for accessing the genetic and functional diversity of uncultured microorganisms. Appl Environ Microbiol 66:2541–2547. https://doi.org/10.1128/AEM.66.6.2541-2547.2000
Lopez-Lopez O, Cerdan M, Siso M (2014) New Extremophilic Lipases and Esterases from Metagenomics. Curr Protein Pept Sci 15:445–455. https://doi.org/10.2174/1389203715666140228153801
Lämmle K, Zipper H, Breuer M et al (2007) Identification of novel enzymes with different hydrolytic activities by metagenome expression cloning. J Biotechnol 127:575–592. https://doi.org/10.1016/j.jbiotec.2006.07.036
Yoon MY, Lee KM, Yoon Y et al (2013) Functional screening of a metagenomic library reveals operons responsible for enhanced intestinal colonization by gut commensal microbes. Appl Environ Microbiol 79:3829–3838. https://doi.org/10.1128/AEM.00581-13
Hannig G, Makrides SC (1998) Strategies for optimizing heterologous protein expression in Escherichia coli. Trends Biotechnol 16:54–60. https://doi.org/10.1016/S0167-7799(97)01155-4
Li LL, McCorkle SR, Monchy S et al (2009) Bioprospecting metagenomes: Glycosyl hydrolases for converting biomass. Biotechnol Biofuels 2:1–11. https://doi.org/10.1186/1754-6834-2-10
Flipphi M, Fekete E, Ág N et al (2013) Spliceosome twin introns in fungal nuclear transcripts. Fungal Genet Biol 57:48–57. https://doi.org/10.1016/j.fgb.2013.06.003
Juturu V, Wu JC (2014) Microbial cellulases: Engineering, production and applications. Renew Sustain Energy Rev 33:188–203. https://doi.org/10.1016/j.rser.2014.01.077
Kennedy J, O’Leary ND, Kiran GS et al (2011) Functional metagenomic strategies for the discovery of novel enzymes and biosurfactants with biotechnological applications from marine ecosystems. J Appl Microbiol 111:787–799. https://doi.org/10.1111/j.1365-2672.2011.05106.x
Li Y, Wexler M, Richardson DJ et al (2005) Screening a wide host-range, waste-water metagenomic library in tryptophan auxotrophs of Rhizobium leguminosarum and of Escherichia coli reveals different classes of cloned trp genes. Environ Microbiol 7:1927–1936. https://doi.org/10.1111/j.1462-2920.2005.00853.x
Liebl W, Angelov A, Juergensen J et al (2014) Alternative hosts for functional (meta)genome analysis. Appl Microbiol Biotechnol 98:8099–8109. https://doi.org/10.1007/s00253-014-5961-7
Leis B, Angelov A, Mientus M et al (2015) Identification of novel esterase-active enzymes from hot environments by use of the host bacterium Thermus thermophilus. Front Microbiol 6:1–12. https://doi.org/10.3389/fmicb.2015.00275
Angelov A, Mientus M, Liebl S, Liebl W (2009) A two-host fosmid system for functional screening of (meta)genomic libraries from extreme thermophiles. Syst Appl Microbiol 32:177–185. https://doi.org/10.1016/j.syapm.2008.01.003
Warnecke F, Ivanova N, Ghassemian M et al (2007) Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite.:450. https://doi.org/10.1038/nature06269
Brulc JM, Antonopoulos DA, Berg Miller ME et al (2009) Gene-centric metagenomics of the fiber-adherent bovine rumen microbiome reveals forage specific glycoside hydrolases. Proc Natl Acad Sci 106:1948–1953. https://doi.org/10.1073/pnas.0806191105
Pope PB, Denman SE, Jones M et al (2010) Adaptation to herbivory by the Tammar wallaby includes bacterial and glycoside hydrolase profiles different from other herbivores. Proc Natl Acad Sci 107:14793–14798. https://doi.org/10.1073/pnas.1005297107
Edwards JL, Smith DL, Connolly J et al (2010) Identification of carbohydrate metabolism genes in the metagenome of a marine biofilm community shown to be dominated by Gammaproteobacteria and Bacteroidetes. Genes (Basel) 1:371–384. https://doi.org/10.3390/genes1030371
Cardoso AM, Cavalcante JJV, Cantão ME et al (2012) Metagenomic Analysis of the Microbiota from the Crop of an Invasive Snail Reveals a Rich Reservoir of Novel Genes. PLoS One 7. https://doi.org/10.1371/journal.pone.0048505
Pope PB, Mackenzie AK, Gregor I et al (2012) Metagenomics of the svalbard reindeer rumen microbiome reveals abundance of polysaccharide utilization loci. PLoS One 7:1–10. https://doi.org/10.1371/journal.pone.0038571
Lu HP, Wang Y bin, Huang SW, et al (2012) Metagenomic analysis reveals a functional signature for biomass degradation by cecal microbiota in the leaf-eating flying squirrel (Petaurista alborufus lena). BMC Genomics 13:. https://doi.org/10.1186/1471-2164-13-466
Wongwilaiwalin S, Laothanachareon T, Mhuantong W et al (2013) Comparative metagenomic analysis of microcosm structures and lignocellulolytic enzyme systems of symbiotic biomass-degrading consortia. Appl Microbiol Biotechnol 97:8941–8954. https://doi.org/10.1007/s00253-013-4699-y
Wang L, Hatem A, Catalyurek UV et al (2013) Metagenomic insights into the carbohydrate-active enzymes carried by the microorganisms adhering to solid digesta in the rumen of cows. PLoS One:8. https://doi.org/10.1371/journal.pone.0078507
Do TH, Nguyen TT, Nguyen TN et al (2014) Mining biomass-degrading genes through Illumina-based de novo sequencing and metagenomic analysis of free-living bacteria in the gut of the lower termite Coptotermes gestroi harvested in Vietnam. J Biosci Bioeng 118:665–671. https://doi.org/10.1016/j.jbiosc.2014.05.010
Zhou Y, Pope PB, Li S et al (2014) Omics-based interpretation of synergism in a soil-derived cellulose-degrading microbial community. Sci Rep 4:1–6. https://doi.org/10.1038/srep05288
Ilmberger N, Güllert S, Dannenberg J et al (2014) A comparative metagenome survey of the fecal microbiota of a breast-and a plant-fed asian elephant reveals an unexpectedly high diversity of glycoside hydrolase family enzymes. PLoS One 9:1–12. https://doi.org/10.1371/journal.pone.0106707
Xu B, Xu W, Li J et al (2015) Metagenomic analysis of the Rhinopithecus bieti fecal microbiome reveals a broad diversity of bacterial and glycoside hydrolase profiles related to lignocellulose degradation. BMC Genomics 16:1–11. https://doi.org/10.1186/s12864-015-1378-7
Lopes LD, de Souza Lima AO, Taketani RG et al (2015) Exploring the sheep rumen microbiome for carbohydrate-active enzymes. Antonie van Leeuwenhoek, Int J Gen Mol Microbiol 108:15–30. https://doi.org/10.1007/s10482-015-0459-6
Güllert S, Fischer MA, Turaev D et al (2016) Deep metagenome and metatranscriptome analyses of microbial communities affiliated with an industrial biogas fermenter, a cow rumen, and elephant feces reveal major differences in carbohydrate hydrolysis strategies. Biotechnol Biofuels 9:1–20. https://doi.org/10.1186/s13068-016-0534-x
Jose VL, Appoothy T, More RP, Arun AS (2017) Metagenomic insights into the rumen microbial fibrolytic enzymes in Indian crossbred cattle fed finger millet straw. AMB Express:7. https://doi.org/10.1186/s13568-016-0310-0
Svartström O, Alneberg J, Terrapon N et al (2017) Ninety-nine de novo assembled genomes from the moose (Alces alces) rumen microbiome provide new insights into microbial plant biomass degradation. ISME J 11:2538–2551. https://doi.org/10.1038/ismej.2017.108
Montella S, Ventorino V, Lombard V et al (2017) Discovery of genes coding for carbohydrate-active enzyme by metagenomic analysis of lignocellulosic biomasses. Sci Rep 7:42623. https://doi.org/10.1038/srep42623
Al-Masaudi S, El Kaoutari A, Drula E et al (2017) A metagenomics investigation of carbohydrate-active enzymes along the gastrointestinal tract of Saudi sheep. Front Microbiol 8. https://doi.org/10.3389/fmicb.2017.00666
Gharechahi J, Salekdeh GH (2018) A metagenomic analysis of the camel rumen’s microbiome identifies the major microbes responsible for lignocellulose degradation and fermentation. Biotechnol Biofuels 11:1–19. https://doi.org/10.1186/s13068-018-1214-9
Liu N, Li H, Chevrette MG et al (2019) Functional metagenomics reveals abundant polysaccharide-degrading gene clusters and cellobiose utilization pathways within gut microbiota of a wood-feeding higher termite. ISME J 13:104–117. https://doi.org/10.1038/s41396-018-0255-1
Al-Masaudi S, El Kaoutari A, Drula E et al (2019) A metagenomics investigation of carbohydrate-active enzymes along the goat and camel intestinal tract. Int Microbiol. https://doi.org/10.1007/s10123-019-00068-2
Na OH, Park D, Seong HJ et al (2019) Antarctic tundra soil metagenome as useful natural resources of cold-active lignocelluolytic enzymes. J Microbiol 57:865–873. https://doi.org/10.1007/s12275-019-9217-1
Martinez A, Kolvek SJ, Yip CLT et al (2004) Genetically Modified Bacterial Strains and Novel Bacterial Artificial Chromosome Shuttle Vectors for Constructing Environmental Libraries and Detecting Heterologous Natural Products in Multiple Expression Hosts. Appl Environ Microbiol 70:2452–2463. https://doi.org/10.1128/AEM.70.4.2452-2463.2004
Geng A, Zou G, Yan X et al (2012) Expression and characterization of a novel metagenome-derived cellulase Exo2b and its application to improve cellulase activity in Trichoderma reesei. Appl Microbiol Biotechnol 96:951–962. https://doi.org/10.1007/s00253-012-3873-y
Kanokratana P, Eurwilaichitr L, Pootanakit K, Champreda V (2015) Identification of glycosyl hydrolases from a metagenomic library of microflora in sugarcane bagasse collection site and their cooperative action on cellulose degradation. J Biosci Bioeng 119:384–391. https://doi.org/10.1016/j.jbiosc.2014.09.010
Yeh YF, Chang SC yu, Kuo HW, et al (2013) A metagenomic approach for the identification and cloning of an endoglucanase from rice straw compost. Gene 519:360–366. https://doi.org/10.1016/j.gene.2012.07.076
Pottkämper J, Barthen P, Ilmberger N et al (2009) Applying metagenomics for the identification of bacterial cellulases that are stable in ionic liquids. Green Chem 11:957–996. https://doi.org/10.1039/b820157a
Mai Z, Su H, Yang J et al (2014) Cloning and characterization of a novel GH44 family endoglucanase from mangrove soil metagenomic library. Biotechnol Lett 36:1701–1709. https://doi.org/10.1007/s10529-014-1531-4
Pang H, Zhang P, Duan CJ et al (2009) Identification of cellulase genes from the metagenomes of compost soils and functional characterization of one novel endoglucanase. Curr Microbiol 58:404–408. https://doi.org/10.1007/s00284-008-9346-y
Maruthamuthu M, Jiménez DJ, Stevens P, van Elsas JD (2016) A multi-substrate approach for functional metagenomics-based screening for (hemi)cellulases in two wheat straw-degrading microbial consortia unveilsnovel thermoalkaliphilic enzymes. BMC Genomics 17:1–16. https://doi.org/10.1186/s12864-016-2404-0
Cheng J, Huang S, Jiang H et al (2016) Isolation and characterization of a non-specific endoglucanase from a metagenomic library of goat rumen. World J Microbiol Biotechnol 32:1–8. https://doi.org/10.1007/s11274-015-1957-4
Talamantes D, Biabini N, Dang H et al (2016) Natural diversity of cellulases, xylanases, and chitinases in bacteria. Biotechnol Biofuels 9. https://doi.org/10.1186/s13068-016-0538-6
Feng Y, Duan CJ, Pang H et al (2007) Cloning and identification of novel cellulase genes from uncultured microorganisms in rabbit cecum and characterization of the expressed cellulases. Appl Microbiol Biotechnol 75:319–328. https://doi.org/10.1007/s00253-006-0820-9
Duan C-J, Xian L, Zhao G-C et al (2009) Isolation and partial characterization of novel genes encoding acidic cellulases from metagenomes of buffalo rumens. Appl Microbiol Biotechnol 107:245–256. https://doi.org/10.1007/s00253-006-0820-9
Berlemont R, Delsaute M, Pipers D et al (2009) Insights into bacterial cellulose biosynthesis by functional metagenomics on Antarctic soil samples. ISME J 3:1070–1081. https://doi.org/10.1038/ismej.2009.48
Liu J, Liu WD, Zhao XL et al (2011) Cloning and functional characterization of a novel endo-β-1,4- glucanase gene from a soil-derived metagenomic library. Appl Microbiol Biotechnol 89:1083–1092. https://doi.org/10.1007/s00253-010-2828-4
Gong X, Gruninger RJ, Qi M et al (2012) Cloning and identification of novel hydrolase genes from a dairy cow rumen metagenomic library and characterization of a cellulase gene. BMC Res Notes 5. https://doi.org/10.1271/bbb.110786
Nacke H, Engelhaupt M, Brady S et al (2012) Identification and characterization of novel cellulolytic and hemicellulolytic genes and enzymes derived from German grassland soil metagenomes. Biotechnol Lett 34:663–675. https://doi.org/10.1007/s10529-011-0830-2
Nguyen NH, Maruset L, Uengwetwanit T et al (2012) Identification and characterization of a cellulase-encoding gene from the buffalo rumen metagenomic library. Biosci Biotechnol Biochem 76:1075–1084. https://doi.org/10.1271/bbb.110786
Zhang L, Fan Y, Zheng H et al (2013) Isolation and characterization of a novel endoglucanase from a Bursaphelenchus xylophilus metagenomic library. PLoS One 8:1–8. https://doi.org/10.1371/journal.pone.0082437
Rashamuse KJ, Visser DF, Hennessy F et al (2013) Characterisation of two bifunctional cellulase-Xylanase enzymes isolated from a bovine rumen metagenome library. Curr Microbiol 66:145–151. https://doi.org/10.1007/s00284-012-0251-z
Yan X, Geng A, Zhang J et al (2013) Discovery of (hemi-) cellulase genes in a metagenomic library from a biogas digester using 454 pyrosequencing. Appl Microbiol Biotechnol 97:8173–8182. https://doi.org/10.1007/s00253-013-4927-5
Martin M, Biver S, Steels S et al (2014) Identification and Characterization of a Halotolerant, Cold-Active Marine Endo-β-1,4-Glucanase by Using Functional Metagenomics of Seaweed-Associated Microbiota. Appl Environ Microbiol 80:4958–4967. https://doi.org/10.1128/aem.01194-14
Lee C-M, Lee Y-S, Seo S-H et al (2014) Screening and Characterization of a Novel Cellulase Gene from the Gut Microflora of Hermetia illucens Using Metagenomic Library. J Microbiol Biotechnol 24:1196–1206. https://doi.org/10.1371/journal.pone.0082437
Xiang L, Li A, Tian C et al (2014) Identification and characterization of a new acid-stable endoglucanase from a metagenomic library. Protein Expr Purif 102:20–26. https://doi.org/10.1016/j.pep.2014.07.009
Wang M, Lai GL, Nie Y et al (2015) Synergistic function of four novel thermostable glycoside hydrolases from a long-term enriched thermophilic methanogenic digester. Front Microbiol 6:1–10. https://doi.org/10.3389/fmicb.2015.00509
Zhou Y, Wang X, Wei W et al (2016) A novel efficient β-glucanase from a paddy soil microbial metagenome with versatile activities. Biotechnol Biofuels 9:1–14. https://doi.org/10.1186/s13068-016-0449-6
Yang C, Xia Y, Qu H et al (2016) Discovery of new cellulases from the metagenome by a metagenomics-guided strategy. Biotechnol Biofuels 9:1–12. https://doi.org/10.1186/s13068-016-0557-3
Loaces I, Bottini G, Moyna G et al (2016) EndoG: A novel multifunctional halotolerant glucanase and xylanase isolated from cow rumen. J Mol Catal B Enzym 126:1–9. https://doi.org/10.1016/j.molcatb.2016.01.004
Pandey S, Gulati S, Goyal E et al (2016) Construction and screening of metagenomic library derived from soil for β-1, 4-endoglucanase gene. Biocatal Agric Biotechnol 5:186–192. https://doi.org/10.1016/j.bcab.2016.01.008
Li BF, Zhu YX, Gu ZB et al (2016) Screening and characterization of a novel ruminal cellulase gene (Umcel-1) from a metagenomic library of gayal (Bos frontalis). J Integr Agric 15:855–861. https://doi.org/10.1016/S2095-3119(15)61144-3
Song YH, Lee KT, Baek JY et al (2017) Isolation and characterization of a novel endo-β-1,4-glucanase from a metagenomic library of the black-goat rumen. Brazilian J Microbiol 48:801–808. https://doi.org/10.1016/j.bjm.2017.03.006
Pimentel AC, Ematsu GCG, Liberato MV et al (2017) Biochemical and biophysical properties of a metagenome-derived GH5 endoglucanase displaying an unconventional domain architecture. Int J Biol Macromol 99:384–393. https://doi.org/10.1016/j.ijbiomac.2017.02.075
Gupta P, Mishra AK, Vakhlu J (2017) Cloning and characterization of thermo-alkalistable and surfactant stable endoglucanase from Puga hot spring metagenome of Ladakh (J&K). Int J Biol Macromol 103:870–877. https://doi.org/10.1016/j.ijbiomac.2017.05.113
Shah RK, Patel AK, Davla DM et al (2017) Molecular cloning, heterologous expression, and functional characterization of a cellulolytic enzyme (Cel PRII) from buffalo rumen metagenome. 3 Biotech 7. https://doi.org/10.1007/s13205-017-0895-2
Medina LMP, Ardila DC, Zambrano MM et al (2017) In vitro and in silico characterization of metagenomic soil-derived cellulases capable of hydrolyzing oil palm empty fruit bunch. Biotechnol Reports 15:55–62. https://doi.org/10.1016/j.btre.2017.06.003
Zhao J, Guo C, Zhang L, Tian C (2018) Biochemical and functional characterization of a novel thermoacidophilic, heat and halo-ionic liquids tolerant endo-β-1,4-glucanase from saline-alkaline lake soil microbial metagenomic DNA. Int J Biol Macromol 118:1035–1044. https://doi.org/10.1016/j.ijbiomac.2018.06.141
Lee JP, Lee HW, Na HB et al (2018) Characterization of truncated endo-β-1,4-glucanases from a compost metagenomic library and their saccharification potentials. Int J Biol Macromol 115:554–562. https://doi.org/10.1016/j.ijbiomac.2018.04.102
Jensen MS, Fredriksen L, MacKenzie AK et al (2018) Discovery and characterization of a thermostable two-domain GH6 endoglucanase from a compost metagenome. PLoS One 13. https://doi.org/10.1371/journal.pone.0197862
Kumar N, Sudan SK, Garg R, Sahni G (2019) Enhanced production of novel halostable recombinant endoglucanase derived from the metagenomic library using fed-batch fermentation. Process Biochem 78:1–7. https://doi.org/10.1016/j.procbio.2018.12.033
Wierzbicka-Woś A, Henneberger R, Batista-García RA et al (2019) Biochemical characterization of a novel monospecific endo-β-1,4-glucanase belonging to GH family 5 from a rhizosphere metagenomic library. Front Microbiol 10:1–19. https://doi.org/10.3389/fmicb.2019.01342
Patel M, Patel HM, Dave S (2020) Determination of bioethanol production potential from lignocellulosic biomass using novel Cel-5m isolated from cow rumen metagenome. Int J Biol Macromol 153:1099–1106. https://doi.org/10.1016/j.ijbiomac.2019.10.240
Klippel B, Blank S, Janzer VA et al (2019) Characterization of a thermoactive endoglucanase isolated from a biogas plant metagenome. Extremophiles 23:479–486. https://doi.org/10.1007/s00792-019-01099-3
Wang W, Archbold T, Lam JS et al (2019) A processive endoglucanase with multi-substrate specificity is characterized from porcine gut microbiota. Sci Rep 9:1–13. https://doi.org/10.1038/s41598-019-50050-1
S A, Sheykhabdolahzadeh A, Maleki M, et al (2020) A novel high performance metagenome-derived lignocellulosic biomass hydrolysis in the harsh conditions. Res Sq 1:1–26. 10.21203/rs.2.23802/v1
Chai S, Zhang X, Jia Z et al (2020) Identification and characterization of a novel bifunctional cellulase/hemicellulase from a soil metagenomic library. Appl Microbiol Biotechnol. https://doi.org/10.1007/s00253-020-10766-x
Song YH, Lee KT, Baek JY et al (2016) Isolation and characterization of a novel glycosyl hydrolase family 74 (GH74) cellulase from the black goat rumen metagenomic library. Folia Microbiol (Praha) 62:175–181. https://doi.org/10.1007/s12223-016-0486-3
Beasley DE, Koltz AM, Lambert JE et al (2015) The evolution of stomach acidity and its relevance to the human microbiome. PLoS One 10:1–12. https://doi.org/10.1371/journal.pone.0134116
Pereira J de C, Giese EC, Moretti MM de S, et al (2017) Effect of Metal Ions, Chemical Agents and Organic Effect of Metal Ions, Chemical Agents and Organic Compounds on Lignocellulolytic Enzymes Activities. In: Enzyme Inhibitors and Activators. pp 139–164
Dudev T, Lim C (2014) Competition among metal ions for protein binding sites: Determinants of metal ion selectivity in proteins. Chem Rev 114:538–556. https://doi.org/10.1021/cr4004665
Bostick DL, Brooks CL (2009) Statistical determinants of selective ionic complexation: Ions in solvent, transport proteins, and other “hosts.” Biophys J 96:4470–4492. https://doi.org/10.1016/j.bpj.2009.03.001
Yenenler A, Kurt H, Sezerman OU (2019) Enhancing enzymatic properties of endoglucanase i enzyme from Trichoderma reesei via swapping from cellobiohydrolase I enzyme. Catalysts 9. https://doi.org/10.3390/catal9020130
Tejirian A, Xu F (2010) Inhibition of cellulase-catalyzed lignocellulosic hydrolysis by iron and oxidative metal ions and complexes. Appl Environ Microbiol 76:7673–7682. https://doi.org/10.1128/AEM.01376-10
Seddon AM, Curnow P, Booth PJ (2004) Membrane proteins, lipids and detergents: Not just a soap opera. Biochim Biophys Acta - Biomembr 1666:105–117. https://doi.org/10.1016/j.bbamem.2004.04.011
Hu X, Yu Z, Liu R (2013) Spectroscopic investigations on the interactions between isopropanol and trypsin at molecular level. Spectrochim Acta - Part A Mol Biomol Spectrosc 108:50–54. https://doi.org/10.1016/j.saa.2013.01.072
Tjernberg A, Markova N, Griffiths WJ, Hallén D (2006) DMSO-related effects in protein characterization. J Biomol Screen 11:131–137. https://doi.org/10.1177/1087057105284218
Arakawa T, Kita Y, Timasheff SN (2007) Protein precipitation and denaturation by dimethyl sulfoxide. Biophys Chem 131:62–70. https://doi.org/10.1016/j.bpc.2007.09.004
Nikolaidis A, Andreadis M, Moschakis T (2017) Effect of heat, pH, ultrasonication and ethanol on the denaturation of whey protein isolate using a newly developed approach in the analysis of difference-UV spectra. Food Chem 232:425–433. https://doi.org/10.1016/j.foodchem.2017.04.022
Simpson DM, Beynon RJ (2010) Acetone precipitation of proteins and the modification of peptides. J Proteome Res 9:444–450. https://doi.org/10.1021/pr900806x
Hwang S, Shao Q, Williams H et al (2011) Methanol strengthens hydrogen bonds and weakens hydrophobic interactions in proteins - A combined molecular dynamics and nmr study. J Phys Chem B 115:6653–6660. https://doi.org/10.1021/jp111448a
Green MR, Sambrook J (2017) Isolation of high-molecular-weight DNA using organic solvents. Cold Spring Harb Protoc:356–359. https://doi.org/10.1101/pdb.prot093450
Vagenende V, Yap MGS, Trout BL (2009) Mechanisms of protein stabilization and prevention of protein aggregation by glycerol. Biochemistry 48:11084–11096. https://doi.org/10.1021/bi900649t
Naika GS, Tiku PK (2011) Influence of ethylenediaminetetraacetic acid (EDTA) on the structural stability of endoglucanase from aspergillus aculeatus. J Agric Food Chem 59:7341–7345. https://doi.org/10.1021/jf103889m
Mattos C, Ringe D (2001) Proteins in organic solvents. Curr Opin Struct Biol 11:761–764. https://doi.org/10.1016/S0959-440X(01)00278-0
Alliegro MC (2000) Effects of dithiothreitol on protein activity unrelated to thiol- disulfide exchange: For consideration in the analysis of protein function with cleland’s reagent. Anal Biochem 282:102–106. https://doi.org/10.1006/abio.2000.4557
Wang Y, Annunziata O (2007) Comparison between Protein-polyethylene Glycol (PEG) interactions and the effect of PEG on protein-protein interactions using the liquid-liquid phase transition. J Phys Chem B 111:1222–1230. https://doi.org/10.1021/jp065608u
James GT (1978) Inactivation of the protease inhibitor phenylmethylsulfonyl fluoride in buffers. Anal Biochem 86:574–579. https://doi.org/10.1016/0003-2697(78)90784-4
Hermanson GT (2013) Functional Targets for Bioconjugation
Parisutham V, Kim TH, Lee SK (2014) Feasibilities of consolidated bioprocessing microbes: From pretreatment to biofuel production. Bioresour Technol 161:431–440. https://doi.org/10.1016/j.biortech.2014.03.114
Ribeiro LF, Amarelle V, Alves LDF et al (2019) Genetically engineered proteins to improve biomass conversion: New advances and challenges for tailoring biocatalysts. Molecules:24. https://doi.org/10.3390/molecules24162879
Bashirova A, Pramanik S, Volkov P et al (2019) Disulfide bond engineering of an endoglucanase from Penicillium verruculosum to improve its thermostability. Int J Mol Sci 20. https://doi.org/10.3390/ijms20071602
Patel AK, Singhania RR, Sim SJ, Pandey A (2019) Thermostable cellulases: Current status and perspectives. Bioresour Technol 279:385–392. https://doi.org/10.1016/j.biortech.2019.01.049
Sahoo K, Sahoo RK, Gaur M, Subudhi E (2020) Cellulolytic thermophilic microorganisms in white biotechnology: a review. Folia Microbiol (Praha) 65:25–43. https://doi.org/10.1007/s12223-019-00710-6
Contreras F, Pramanik S, Rozhkova AM et al (2020) Engineering robust cellulases for tailored lignocellulosic degradation cocktails. Int J Mol Sci:21. https://doi.org/10.3390/ijms21051589
Sommer MO, Church GM, Dantas G (2010) A functional metagenomic approach for expanding the synthetic biology toolbox for biomass conversion. Mol Syst Biol 6:1–7. https://doi.org/10.1038/msb.2010.16
Hess M, Sczyrba A, Egan R et al (2011) Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science (80- ) 331:463–467. https://doi.org/10.1126/science.1200387
Graham JE, Clark ME, Nadler DC et al (2011) Identification and characterization of a multidomain hyperthermophilic cellulase from an archaeal enrichment. Nat Commun 2. https://doi.org/10.1038/ncomms1373
Zhu L, Wu Q, Dai J et al (2011) Evidence of cellulose metabolism by the giant panda gut microbiome. Proc Natl Acad Sci 108:17714–17719. https://doi.org/10.1073/pnas.1017956108
van der Lelie D, Taghavi S, McCorkle SM et al (2012) The metagenome of an anaerobic microbial community decomposing poplar wood chips. PLoS One 7. https://doi.org/10.1371/journal.pone.0036740
Lehmann C, Sibilla F, Maugeri Z et al (2012) Reengineering CelA2 cellulase for hydrolysis in aqueous solutions of deep eutectic solvents and concentrated seawater. Green Chem 14:2719–2726. https://doi.org/10.1039/c2gc35790a
Lehmann C, Bocola M, Streit WR et al (2014) Ionic liquid and deep eutectic solvent-activated CelA2 variants generated by directed evolution. Appl Microbiol Biotechnol 98:5775–5785. https://doi.org/10.1007/s00253-014-5771-y
Körfer G, Pitzler C, Vojcic L et al (2016) In vitro flow cytometry-based screening platform for cellulase engineering. Sci Rep 6:1–12. https://doi.org/10.1038/srep26128
Acknowledgments
N.P, A.V., and P.K. are thankful to the National Institute of Technology, Warangal, and M.H.R.D India for the financial research grant for pursuing the Ph.D. work. T.T., S.G., and R.S.R. are thankful to the National Institute of Technology, Warangal.
Funding
This research work was supported by the Department of Science & Technology-Science and Engineering Research Board (DST-SERB), India, for Early career research grant Reference No. ECR/2015/000076.
Author information
Authors and Affiliations
Contributions
Ninian Prem Prashanth P, Aditya Velidandi: conceptualization, writing—original draft preparation, visualization, and editing. Tanvi Tavarna: writing—original draft preparation and reviewing. Ram Sarvesh Raj, Shreyash Gupta: reviewing and editing. Pradeep Kumar Gandam: visualization and editing. Dr. Rama Raju Baadhe: supervision.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Pabbathi, N.P.P., Velidandi, A., Tavarna, T. et al. Role of metagenomics in prospecting novel endoglucanases, accentuating functional metagenomics approach in second-generation biofuel production: a review. Biomass Conv. Bioref. 13, 1371–1398 (2023). https://doi.org/10.1007/s13399-020-01186-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13399-020-01186-y