
Abstract
Virus-like particles (VLPs) are high-priority antigens with highly ordered repetitive structures, which are similar to natural viral particles. We have developed a competitive enzyme-linked immunosorbent assay (cELISA) for detecting antibodies directed against Senecavirus A (SVA). Our assay utilizes SVA VLPs that were expressed and assembled in an E. coli expression system as the coating antigens. VLPs have better safety and immunogenicity than intact viral particles or peptides. The VLPs-based cELISA was used to test 342 serum samples collected from different pig farms, and the results showed that its specificity and sensitivity were 100% and 94%, respectively. The consistency rates of cELISA with the BIOSTONE AsurDx™ Senecavirus A (SVA) Antibody Test Kit and an indirect immunofluorescent assay were 90.0% and 94.2%, respectively. Therefore, this VLPs-based cELISA can be effectively and reliably used for the detection and discrimination of SVA infection in serum samples.
Similar content being viewed by others


Keypoints
-
1.
We successfully obtained SVA VLPs by using the prokaryotic expression system.
-
2.
We successfully established a SVA antibody-specific competitive ELISA test method by using SVA VLPs as antigens.
Introduction
SVA, also known as Seneca Valley virus (SVV), is a single-stranded, positive-sense RNA virus. It is the only member of the genus Senecavirus in the family Picornaviridae (Adams et al. 2015; Hales et al. 2008). SVA has a nonenveloped capsid, approximately 25–30 nm in diameter, with icosahedral symmetry. Its genome consists of a 5′ untranslated region (5′-UTR), an open reading frame (ORF), and a 3′-UTR. The ORF encodes a polyprotein precursor that is cleaved into a leader protein (L) and three proteins (P1, P2, and P3). P1 is processed into VP1, VP3, and VP0, and VP0 is further cleaved into VP2 and VP4, which compose the viral capsid. P2 and P3 are the nonstructural proteins of the virus (Hales et al. 2008; Leme et al. 2016; Liu et al. 2018; Venkataraman et al. 2008).
SVA was initially identified as a contaminant in a culture of adenovirus in human embryonic retinal cells (PER.C6) (Fallaux et al. 1998). It causes vesicular disease in pigs, and its clinical features are very similar to those of foot-and-mouth disease (Canning et al. 2016; Leme et al. 2016; Vannucci et al. 2015). SVA can cause death in piglets (up to 4 days of age), with a mortality rate of 30–70% (Vannucci et al. 2015). In recent years, cases of SVA infection in pigs have been detected in several provinces of China, including Hubei, Fujian, Henan, and Guangdong province (Liu et al. 2018; Qian et al. 2016; Wu et al. 2016). This virus may cause huge economic losses in the pig industry (Knight-Jones and Rushton 2013; Porphyre et al. 2018). Therefore, a rapid, safe, and highly specific diagnostic method is required to prevent and control the spread of SVA.
At present, the serological diagnosis methods for SVA mainly include virus neutralization tests (VNT), indirect immunofluorescent assays (IFAs), competitive enzyme-linked immunosorbent assays (cELISA) and indirect enzyme-linked immunosorbent assays (Dvorak et al. 2017; Leme et al. 2015; Rudin et al. 2011; Yang et al. 2012). VNT is the gold standard for the detection of antibodies in animal sera, and is recommended by the World Organisation for Animal Health (OIE). However, compared with other methods, VNT and IFA are too time-consuming and complex to be suitable for clinical field testing. ELISA is widely used because it is simple, inexpensive, and easy to perform. Inactivated viruses and monomeric proteins are currently used as the coating antigens in ELISA systems for the detection of serum antibodies. However, using complete viral particles as these antigens poses a security risk, and the immunogenicity of monomeric proteins or peptides is relatively poor (Brocchi et al. 1995). Therefore, a recombinant protein that is both safe and highly immunogenic as a diagnostic antigen is critical to the development of new diagnostic techniques (Ko et al. 2009).
VLPs are composed of one or more structural proteins of virus, and do not contain viral genetic material. That structure is similar to native virus particles (Wu et al. 2008). Therefore, VLPs as coating antigens for ELISA systems is safer than that of whole virus particles. Moreover, VLPs exhibit better sensitivity, specificity, and immunogenicity than monomeric proteins or peptides given their highly organized and repetitive surface structures. VLPs have been used to develop safe candidates for immunological detection methods (Michel and Tiollais 2010). In this study, we established a cELISA using SVA VLPs as the coating antigens for the detection of SVA antibody in swine serum samples.
Materials and methods
Serum samples and cells
A total of 342 serum samples were harvested from pigs infected with SVA. Antibodies directed against SVA in the serum samples were detected with the commercial BIOSTONE AsurDx™ Senecavirus A (SVA) Antibody Test Kit (South Lake Tahoe, State of California, CA, USA) and an indirect IFA. Serum samples and porcine kidney epithelial cells (IBRS-2) were obtained from the Key Laboratory of the Lanzhou Veterinary Research Institute of the Chinese Academy of Agricultural Sciences, Lanzhou, Gansu Province, China.
Plasmid construction
pSMK, pSMA and SUMO fusion protein expression vectors, were constructed as described previously (Xiao et al. 2016). The SUMO-tagging recombinant protein expression vectors containing the SVA VP0, VP1 and VP3 gene were constructed as described previously (Mo et al. 2019). In brief, the recombinant plasmids pSMA-VP0, pSMK-VP1 and pSMK-VP3 were constructed using pSMK and pSMA as the expression vector.
Expression of recombinant proteins
The recombinant plasmids were transferred into E. coli BL21 (DE3). The E. coli cells were cultured in LB medium containing ampicillin (50 µg/ml) and kanamycin (10 µg/ml) at 37 ℃. When the OD600 was 0.7–0.9, isopropylthio-β-d-galactoside (0.05 mM) was added to LB medium at 16 ℃ to induce coexpression of the recombinant proteins for 16 h.
Purification of recombinant proteins and quantification of VLPs
The recombinant protein was purified and analyzed with SDS-PAGE and western blotting, as previously described (Mo et al. 2019). The His-SUMO-tag of recombinant protein was cleaved by SUMO protease and the SVA capsid proteins VP0, VP1 and VP3 can self-assemble into VLPs in the assembly buffer (400 mM Tris-HCl, 250 mM NaCl, 1 mM CaCl2, and 1 mM DTT, pH = 8.0) for 24 h at 4 °C (Yin et al. 2010). SVA VLPs were purifed by sucrose density gradient centrifugation, the samples were centrifuged at 35,000 rpm for 3 h using an Optima L-100 XP ultracentrifuge (Beckman Coulter, Fullerton, CA, USA). Then the fractions with the highest OD280 value was detected using dynamic light scattering instrument (DLS) and transmission electron microscope (TEM; CM 100, JEOL Ltd., Tokyo, Japan) to determine the molecular size and shape of the SVA VLPs, respectively (Guo et al. 2013).
Preparation of rabbit serum
Each of three adult rabbits was subcutaneously injected with VLPs (200 µg) and an equal volume of Freund’s complete adjuvant to induce antibody production against the antigen. Two booster immunizations with the same dose of VLPs plus Freund’s incomplete adjuvant were administered at 2-weekly intervals. Two weeks after the final booster injection, the blood of rabbits was collected, and the sera were prepared and stored at − 80 ℃ before analysis.
Preparation of competitive antibody
The immunoglobulin G (IgG) in the rabbit sera obtained in the previous step was separated with the saturated ammonium sulfate method. The rabbit IgG was further purified with Protein A sepharose affinity column chromatography and then labeled with horseradish peroxidase (HRP) with the improved NaIO4 method (Minaeian et al. 2012). The HRP-conjugated rabbit IgG was stored at − 80 ℃ before use.
Establishment of cELISA method
A 96-well ELISA microplate was coated with various concentrations (0.5–1.0 µg/ml) of SVA VLPs in carbonate buffer solution (0.05 M, pH 9.6) and incubated at 4 ℃ overnight. After the microplate was washed three times with phosphate-buffered saline (PBS) containing 0.1% Tween (PBST), it was blocked with 1% bovine serum albumin (BSA) in distilled water for 1 h at 37 ℃, washed three times with PBST, and patted dry. In the antibody-coated wells of the plate, 50 µl of SVA-positive or SVA-negative serum and 50 µl of serially diluted HRP–IgG were mixed to ensure that the competitive reaction was as efficient as possible. The plates were incubated for 60 min at 37 ℃ and washed 3–4 times with 300 µl of PBST.
Tetramethylbenzidine (TMB, 50 µl) was added and the samples incubated for 15 min at 37 ℃. The color reaction was stopped with 50 µl of 2 M H2SO4. To optimize the reaction conditions of the SVA cELISA, the best results under each set of condition were determined on the basis of the OD450 values and the percentage inhibition (PI), which was calculated with the following formula: PI = (OD450 of standard negative serum – OD450 of measured individual sample)/(OD450 of standard negative serum – OD450 of standard positive serum) × 100%.
Determination of the cELISA cut-off value (PI)
In this procedure, 50 SVA-positive seras with different antibody titers and 50 SVA-negative sera were tested by the established cELISA to determine the PI. The cut-off value was determined based on a receiver operating characteristic (ROC) curve.
Analysis of the sensitivity and specificity of the cELISA
The sensitivity of the cELISA was assessed with SVA-positive sera with different antibody titers. To determine whether the SVA cELISA system reacted positively with antibodies directed against viruses other than SVA, six randomly selected pig sera positive for serotype O FMDV, PCV2, PPV, CSFV, Actinobacillus pleuropheumoniae, or Haemophilus parasuis were tested with the SVA cELISA.
cELISA repeatability test
A repeatability test was conducted to determine the stability of the SVA cELISA. The intra-assay repeatability of the SVA cELISA was tested with 10 randomly selected serum samples, with three replicates of each, in the same microplate. The inter-assay repeatability of the SVA cELISA was used to test the serum samples in triplicate in microplates in different production batches. The coefficients of variation (CVs) of the three replicates of each of the 10 serum samples were calculated.
Comparison of cELISA and commercial ELISA
To validate the SVA cELISA, a total of 342 swine sera samples were tested with both the SVA cELISA and the BIOSTONE AsurDx™ Senecavirus A (SVA) Antibody Test Kit.
Comparison of cELISA and IFA
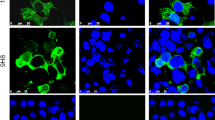
When cells dense were about 60% in cell culture flasks, they were infected with the virus, and the virus solution was discarded at the appropriate time. The cells were then washed three times with PBS, fixed with 4% paraformaldehyde for 10–15 min, and washed three times with PBS. The cells were treated with 0.1% Triton X-100 for 15 min and washed three times with PBS. An anti-SVA polyclonal antibody was added, and the cells were incubated at 37 ℃ for 2 h. The cells were washed three times with PBS, FITC-labeled anti-porcine IgG was added, and the cells were incubated for a further 1 h at 37 ℃ in an incubator. The cells were washed three times with PBS and observed under an inverted fluorescence microscope.
Results
Expression and identification of recombinant VP0, VP1 and VP3 proteins
The recombinant fusion proteins (VP0, VP1, and VP3) were purified with nickel-chelating affinity chromatography. The results of SDS-PAGE indicated that the recombinant fusion proteins were successfully expressed (Fig. 1a), and the results of a western blotting analysis indicated that the recombinant fusion proteins were recognized by standard SVA-positive serum (Fig. 1b). The yields of the soluble recombinant proteins were greater when the expression was induced at 20 ℃ than when it was incubated at 37 ℃ (Fig. 1c), which is consistent with previous findings (Mo et al. 2019).

Analysis of three SUMO-tagged recombinant proteins using SDS-PAGE and western blotting (a). SDS-PAGE: M, protein molecular marker; lane 1, prior to induction; lane 2, post induction. The SUMO-tagged recombinant proteins were induced as described in "Materials and methods". Identification of recombinant proteins with varied preparations using western blot (b). Lanes 1, recombinant proteins generated in varied preparations. Expression of recombinant proteins induced at 20 °C and 37 °C (c). Protein molecular marker (M), supernatant (lane 1) and pellet (lane 2) of lysated recombinant E. coli incubated at 20 °C, supernatant (lane 3) and pellet (lane 4) of lysated recombinant E. coli incubated at 37 °C; and supernatant (lane 5) and pellet (lane 6) of lysated recombinant E. coli prior to induction
Assembly of SVA VLPs
After the assembly of the SVA VLPs in assembly buffer, the product was filtered, and the immunogenicity of the SVA VLPs was confirmed with SDS-PAGE (Fig. 2a) and western blotting (Fig. 2b). The molecular size and shape of the SVA VLPs was determined with TEM (Fig. 2c) and DLS (Fig. 2d), the results show that the diameter of SVA VLPs was ~ 23 nm, which was similar to natural virions of SVA.

Expression and assembly of purified SVA-VLPs (a). Analysis of SVA virus-like particles using SDS-PAGE. M, protein molecular marker; lane1, SUMO Protease; lane 2–4, digested recombinant proteins with SUMO protease; lane 5, nondigested recombinant proteins. Analysis of SVA virus-like particles using western blot (b). Lanes 1–2, purified SVA-VLPs generated in varied preparations. Visualization of SVA virus-like particles using TEM (c). The bar indicates 200 nm. DLS diagram (d)
Preparation of rabbit HRP-conjugated hyperimmune serum
A direct ELISA was used to determine the titer of HRP–IgG, and showed it to be 1:512,000 (Table 1).
Development of cELISA based on SVA VLPs
The optimal reaction conditions for the SVA cELISA were confirmed with OD450 and P/N (positive/negative) values: 0.9 µg/ml of VLPs in 100 µl volume (carbonate solution) were used to coat each well of the plate at 4 ℃ overnight; the plate was blocked with 1% BSA for 75 min at 37 ℃; the competitive reaction was performed for 30 min at 37 ℃; a 1:3 dilution of the test serum sample and a 1:7000 dilution of HRP–IgG were used; reaction with TMB substrate for 15 min at 37 ℃ to visualize the products; the reaction was terminated with 50 µl of 2 M H2SO4.
Determination of the cELISA cut-off value (PI)
A total of 50 positive sera and 50 negative sera were tested with the SVA VLP cELISA. The PI cut-off point was confirmed that minimized the false negative and false positive rates of the SVA cELISA system (Fig. 3a, b). A ROC curve analysis showed that the specificity and sensitivity of the cELISA were optimal when the cut-off value was PI = 45%. Therefore, serum samples were defined as positive when the PI of the serum tested was ≥ 45%, and negative when PI < 45%.

Validation of cELISA by ROC curve (a). The PI values of 104 confirmed sera calculated by cELISA were compared with the virus neutralization test results via ROC analysis. AUC stands for area under curve. The value of sensitivity or specificity of the cELISA calculated at varied cutoff value (b)
Evaluation of the cELISA
A total of 342 swine serum samples were tested using the SVA VLP cELISA, the BIOSTONE AsurDx™ Senecavirus A (SVA) Antibody Test Kit, and IFA. The calculated sensitivity and specificity of the SVA cELISA were 94% and 100%, respectively. The coincidence rates when the cELISA was compared with the commercial AsurDx™ Senecavirus A (SVA) Antibody Test Kit Manual and IFA were 90% and 94.2%, respectively (Table 2). These results confirm that the SVA cELISA is highly accurate. The repeatability of the assay was evaluated by determining the OD450 of 10 serum samples processed in triplicate in the same plate (intra-assay repeatability) or different plates at different times (inter-assay reproducibility). The standard deviations and CVs were then calculated. The low values of CV achieved in the repeatability (CV < 10%) and reproducibility (CV < 15%) assays demonstrate the reliability of this new approach (Tables 3 and 4).
Discussion
Since 2015, SVA has spread increasingly widely, and the symptoms of SVA infection are similar to those of foot-and-mouth disease, which has caused great economic losses in the pig industry. Therefore, the rapid diagnosis of SVA infection is crucial to its prevention and control (Montiel et al. 2016). We have developed a sensitive and specific SVA ELISA for the detection and diagnosis of SVA-specific antibodies. Previous studies have shown that the immunization of animals with FMDV VLPs produced in E. coli was sufficient to induce antibodies that protected guinea pigs, pigs, and cattle from virulent infection, indicating the potential utility of VLPs as candidate vaccines (Guo et al. 2013). The E. coli expression system can be easily used to manufacture VLPs and does not require cell culture or bio-protection facilities. Our previous studies and those of others have confirmed the immunogenicity of VLPs (Guo et al. 2013; Li et al. 2016; Li et al. 2011). However, the reactivity of VLPs as coating antigens for cELISA remained unclear. Therefore, we developed a cELISA based on SVA VLPs produced in an E. coli expression system. Therefore, we developed a cELISA method based on the SVA VLP generated in the E. coli expression system, and tested the serum samples. The result demonstrated that it has high specificity, sensitivity, and repeatability. In this experiment, the SVA capsid protein genes were inserted into the pSMK or pSMA plasmids with His and SUMO (small ubiquitin-modified system) tags, to generate pSMA-VP0, pSMK-VP1, and pSMK-VP3. Competent E. coli cells were cotransformed with the plasmids and the coexpression of the capsid proteins was induced. In this experiment, the animals were immunized with the recombinant proteins to obtain highly immune serum. Therefore, the basic components of the diagnostic test, including the diagnostic antigens and related reference sera, can be generated independently. At present, the development of diagnostic reagents based on inactivated SVA requires complex biosafety equipment, increasing its cost. For example, in the BIOSTONE AsurDx™ Senecavirus A (SVA) Antibody Test Kit (BIOSTONE, USA), inactivated SVA and a monoclonal antibody are used to detect serum antibodies, and the kit can test 450 sera samples. However, it costs $1500, and is consequently too expensive for developing countries. Therefore, we have established a bacterial expression system for SVA VLPs, which can be prepared inexpensively in any laboratory with a device for bacterial culture. In this assay, SVA VLPs replace inactivated SVA in the ELISA, so that the cELISA can be used to assess clinical herd immunity, particularly in developing countries.
In some cases, different monoclonal antibodies (mAbs) should be used in combination to achieve the optimal performance of an ELISA and to overcome the weak affinity of serum antibodies for the coating antigen. Although mAbs may show high specificity, high sensitivity, and consistent performance, their use will increase the complexity and uncertainty of the test method (Yang et al. 2015). In the present study, polyclonal antibodies were used as the competitive antibodies to evaluate the level of clinical herd immunity. Polyclonal antibodies have greater affinity for the antigen because they recognize more epitopes on the coating antigen than mAbs, which represented high specificity and good performance.
In summary, this cELISA based on SVA VLPs expressed in an E. coli system has high specificity, sensitivity, and reproducibility. The SVA VLPs cELISA does not require high-level biosafety facilities, is easy to produce, and inexpensive, so it is especially suitable for use in underdeveloped countries. And it can be used to assess herd immunity induced with a variety of inactivated SVA vaccines, promoting the prevention and control of SVA.
Availability of data and materials
Accession number, DQ641257.1 identified the nucleic acid sequence of the SVA. All materials are available upon request.
References
Adams MJ, Lefkowitz EJ, King AM, Bamford DH, Breitbart M, Davison AJ, Ghabrial SA, Gorbalenya AE, Knowles NJ, Krell P, Lavigne R, Prangishvili D, Sanfacon H, Siddell SG, Simmonds P, Carstens EB (2015) Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2015). Arch Virol 160(7):1837–1850 https://doi.org/10.1007/s00705-015-2425-z
Brocchi E, Berlinzani A, Gamba D, De Simone F (1995) Development of two novel monoclonal antibody-based ELISAs for the detection of antibodies and the identification of swine isotypes against swine vesicular disease virus. J Virol Methods 52(1–2):155–167 https://doi.org/10.1016/0166-0934(94)00158-d
Canning P, Canon A, Bates JL, Gerardy K, Linhares DC, Pineyro PE, Schwartz KJ, Yoon KJ, Rademacher CJ, Holtkamp D, Karriker L (2016) Neonatal mortality, vesicular lesions and lameness associated with Senecavirus A in a U.S. sow farm. Transbound Emerg Dis 63(4):373–378 https://doi.org/10.1111/tbed.12516
Dvorak CM, Akkutay-Yoldar Z, Stone SR, Tousignant SJ, Vannucci FA, Murtaugh MP (2017) An indirect enzyme-linked immunosorbent assay for the identification of antibodies to Senecavirus A in swine. BMC Vet Res 13(1):50 https://doi.org/10.1186/s12917-017-0967-x
Fallaux FJ, Bout A, van der Velde I, van den Wollenberg DJ, Hehir KM, Keegan J, Auger C, Cramer SJ, van Ormondt H, van der Eb AJ, Valerio D, Hoeben RC (1998) New helper cells and matched early region 1-deleted adenovirus vectors prevent generation of replication-competent adenoviruses. Hum Gene Ther 9(13): 1909–1917 https://doi.org/10.1089/hum.1998.9.13-1909
Guo HC, Sun SQ, Jin Y, Yang SL, Wei YQ, Sun DH, Yin SH, Ma JW, Liu ZX, Guo JH, Luo JX, Yin H, Liu XT, Liu DX (2013) Foot-and-mouth disease virus-like particles produced by a SUMO fusion protein system in Escherichia coli induce potent protective immune responses in guinea pigs, swine and cattle. Vet Res 44:48. https://doi.org/10.1186/1297-9716-44-48
Hales LM, Knowles NJ, Reddy PS, Xu L, Hay C, Hallenbeck PL (2008) Complete genome sequence analysis of Seneca Valley virus-001, a novel oncolytic picornavirus. J Gen Virol 89(Pt5):1265–1275 https://doi.org/10.1099/vir.0.83570-0
Knight-Jones TJD, Rushton J (2013) The economic impacts of foot and mouth disease-what are they, how big are they and where do they occur? Prev Vet Med 112(3–4):161–173 https://doi.org/10.1016/j.prevetmed.2013.07.013
Ko YJ, Jeoung HY, Lee HS, Chang BS, Hong SM, Heo EJ, Lee KN, Joo HD, Kim SM, Park JH, Kweon CH (2009) A recombinant protein-based ELISA for detecting antibodies to foot-and-mouth disease virus serotype Asia 1. J Virol Methods 159(1):112–118 https://doi.org/10.1016/j.jviromet.2009.03.011
Leme RA, Zotti E, Alcantara BK, Oliveira MV, Freitas LA, Alfieri AF, Alfieri AA (2015) Senecavirus A: an emerging vesicular infection in Brazilian pig herds. Transbound Emerg Dis 62(6):603–611 https://doi.org/10.1111/tbed.12430
Leme RA, Oliveira TE, Alcantara BK, Headley SA, Alfieri AF, Yang M, Alfieri AA (2016) Clinical manifestations of Senecavirus A infection in neonatal pigs, Brazil, 2015. Emerg Infect Dis 22(7):1238–1241 https://doi.org/10.3201/eid2207.151583
Li ZY, Yin XP, Yi YZ, Li XR, Li BY, Lan X, Zhang ZF, Liu JX (2011) FMD subunit vaccine produced using a silkworm-baculovirus expression system: Protective efficacy against two type Asia1 isolates in cattle. Vet Microbiol 149(1–2):99–103 https://doi.org/10.1016/j.vetmic.2010.10.022
Li HT, Li ZY, Xie YL, Qin XD, Qi XC, Sun P, Bai XW, Ma YJ, Zhang ZD (2016) Novel chimeric foot-and-mouth disease virus-like particles harboring serotype O VP1 protect guinea pigs against challenge. Vet Microbiol 83:92–96 https://doi.org/10.1016/j.vetmic.2015.12.004
Liu J, Ren X, Li Z, Xu G, Lu R, Zhang K, Ning Z (2018) Genetic and phylogenetic analysis of reemerged novel Seneca Valley virus strains in Guangdong province, 2017. Transbound Emerg Dis 65(3):614–617 https://doi.org/10.1111/tbed.12839
Michel ML, Tiollais P (2010) Hepatitis B vaccines: Protective efficacy and therapeutic potential. Pathol Biol, 58, 288–295 https://doi.org/10.1016/j.patbio.2010.01.006
Minaeian S, Rahbarizadeh F, Zarkesh Esfahani SH, Ahmadvand D (2012) Characterization and enzyme-conjugation of a specific anti-L1 nanobody. J Immunoass Immunochem 33(4):422–434 https://doi.org/10.1080/15321819.2012.665407
Mo Y, Song X, Mu P, Dong S, Cao H, Guo S, Yao H, Sun X S (2019) Prokaryotic expression of capsid protein of Seneca virus type A and assembly of its pentamer. Chin Vet Sci 08:12 https://doi.org/10.16656/j.issn.1673-4696.2019.0098
Montiel N, Buckley A, Guo B, Kulshreshtha V, VanGeelen A, Hoang H, Rademacher C, Yoon KJ, Lager K (2016) Vesicular disease in 9-week-old pigs experimentally infected with Senecavirus A. Emerg Infect Dis 22(7):1246–1248 https://doi.org/10.3201/eid2207.151863
Porphyre T, Rich KM, Auty HK (2018) Assessing the economic impact of vaccine availability when controlling Foot and Mouth Disease outbreaks. Front Vet Sci 5:47 https://doi.org/10.3389/fvets.2018.00047
Qian S, Fan W, Qian P, Chen H, Li X (2016) Isolation and full-genome sequencing of Seneca Valley virus in piglets from China, 2016. Virol J 13(1):173 https://doi.org/10.1186/s12985-016-0631-2
Rudin CM, Poirier JT, Senzer NN, Stephenson J, Loesch D, Burroughs KD, Reddy PS, Hann CL, Hallenbeck PL (2011) Phase I clinical study of Seneca Valley Virus (SVV-001), a replication-competent picornavirus, in advanced solid tumors with neuroendocrine features. Clin Cancer Res 17(4):888–895 https://doi.org/10.1158/1078-0432.Ccr-10-1706
Vannucci FA, Linhares DC, Barcellos DE, Lam HC, Collins J, Marthaler D (2015) Identification and complete genome of Seneca Valley Virus in vesicular fluid and sera of pigs affected with idiopathic vesicular disease, Brazil. Transbound Emerg Dis 62(6):589–593. https://doi.org/10.1111/tbed.12410
Venkataraman S, Reddy SP, Loo J, Idamakanti N, Hallenbeck PL, Reddy VS (2008) Structure of Seneca Valley Virus-001: an oncolytic picornavirus representing a new genus. Structure 16(10):1555–1561 https://doi.org/10.1016/j.str.2008.07.013
Wu PC, Chien MS, Tseng YY, Lin J, Lin WL, Yang CY, Huang C (2008) Expression of the porcine circovirus type 2 capsid protein subunits and application to an indirect ELISA. J Biotechnol, 133, 58–64 https://doi.org/10.1016/j.jbiotec.2007.09.015
Wu Q, Zhao X, Chen Y, He X, Zhang G, Ma J (2016) Complete genome sequence of Seneca Valley Virus CH-01-2015 identified in China. Genome Announc. https://doi.org/10.1128/genomeA.01509-15
Xiao Y, Chen HY, Wang Y, Yin B, Lv C, Mo X, Yan H, Xuan Y, Huang Y, Pang W, Li X, Yuan YA, Tian K (2016) Large-scale production of foot-and-mouth disease virus (serotype Asia1) VLP vaccine in Escherichia coli and protection potency evaluation in cattle. BMC Biotechnol 16(1):56. https://doi.org/10.1186/s12896-016-0285-6
Yang M, van Bruggen R, Xu W (2012) Generation and diagnostic application of monoclonal antibodies against Seneca Valley virus. J Vet Diagn Invest 24(1):42–50 https://doi.org/10.1177/1040638711426323
Yang M, Parida S, Salo T, Hole K, Velazquez-Salinas L, Clavijo A (2015) Development of a competitive enzyme-linked immunosorbent assay for detection of antibodies against the 3B protein of foot-and-mouth disease virus. Clin Vaccine Immunol 22(4):389–397 https://doi.org/10.1128/CVI.00594-14
Yin SH, Sun SQ, Yang SL, Shang YJ, Cai XP, Liu XT (2010) Self-assembly of virus-like particles of porcine circovirus type 2 capsid protein expressed from Escherichia coli. Virol J 7:166. https://doi.org/10.1186/1743-422X-7-166
Acknowledgements
Not applicable.
Funding
This study was funded by the National Natural Science Foundation of China (31672592), Development and validation of a competitive ELISA, and Central Public-interest Scientific Institution Basal Research Fund (1610312016002, 1610312017010), The National Key Research and Development Program (2016YFD0500700, 2017YFD0500906, 2017YFD0501100), Gansu Province Youth Science and Technology Fund Project (17JR5RA324), Elite Youth program of Chinese Academy of Agricultural Sciences.
Author information
Authors and Affiliations
Contributions
MB and RW contributed to the preparation of the manuscript. MB, RW and YZ conducted the experiment. SS and HG designed this analysis. HD analyzed the data. All authors reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All of the animal experiments were conducted in accordance with the regulations for the administration of affairs concerning experimental animals approved by the State Science and Technology Commission of the People’s Republic of China.
Consent for publication
Not applicable.
Competing interests
Authors declared no potential competing interests with respect to the research, authorship, and/or publication of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bai, M., Wang, R., Sun, S. et al. Development and validation of a competitive ELISA based on virus-like particles of serotype Senecavirus A to detect serum antibodies. AMB Expr 11, 7 (2021). https://doi.org/10.1186/s13568-020-01167-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13568-020-01167-4