
Abstract
In the context of the SAMPL7 challenge, we computed, employing a non-equilibrium (NE) alchemical technique, the standard binding free energy of two series of host-guest systems, involving as a host the Isaac’s TrimerTrip, a Cucurbituril-like open cavitand, and the Gilson’s Cyclodextrin derivatives. The adopted NE alchemy combines enhanced sampling molecular dynamics simulations with driven fast out-of-equilibrium alchemical trajectories to recover the free energy via the Jarzynski and Crooks NE theorems. The GAFF2 non-polarizable force field was used for the parametrization. Performances were acceptable and similar in accuracy to those we submitted for Gibb’s Deep Cavity Cavitands in the previous SAMPL6 host-guest challenge, confirming the reliability of the computational approach and exposing, in some cases, some important deficiencies of the GAFF2 non-polarizable force field.





Similar content being viewed by others
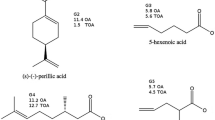
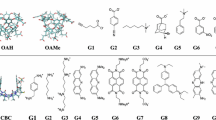
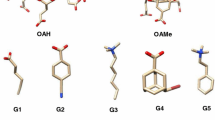
References
Muddana HS, Fenley AT, Mobley DL, Gilson MK (2014) The sampl4 host-guest blind prediction challenge: an overview. J Comput Aided Mol Des 28(4):305–317
Yin J, Henriksen NM, Slochower DR, Shirts MR, Chiu MW, Mobley DL, Gilson MK (2016) Overview of the sampl5 host–guest challenge: are we doing better? J Comput Aided Mol Des, pp 1–19
Rizzi A, Murkli S, McNeill JN, Yao W, Sullivan M, Gilson MK, Chiu MW, Isaacs L, Gibb BC, Mobley DL, Chodera JD (2018) Overview of the sampl6 host-guest binding affinity prediction challenge. J Comput Aided Mol Des 32(10):937–963
Ndendjio SZ, Liu W, Yvanez N, Meng Z, Zavalij PY, Isaacs L (2020) Synthesis and recognition properties Triptycene walled glycoluril trimer. New J Chem 44:338–345
Kellett K, Duggan BM, Gilson MK (2019) Facile synthesis of a diverse library of mono-3-substituted \(\beta\)-cyclodextrin analogues. Supramol Chem 31(4):251–259
Gibb Corinne LD, Gibb Bruce C (2014) Binding of cyclic carboxylates to octa-acid deep-cavity cavitand. J Comput Aided Mol Des 28(4):319–325
Amezcua Martin, Mobley David (2020) SAMPL7 challenge overview: assessing the reliability of polarizable and non-polarizable methods for host-guest binding free energy calculations. ChemrXiv 8 :12768353.v1
https://samplchallenges.github.io/roadmap/submissions/ , Accessed 23 June 2020
Crooks GE (1998) Nonequilibrium measurements of free energy differences for microscopically reversible markovian systems. J Stat Phys 90:1481–1487
Jarzynski C (1997) Nonequilibrium equality for free energy differences. Phys Rev Lett 78:2690–2693
Procacci P, Guarrasi M, Guarnieri G (2018) Sampl6 host-guest blind predictions using a non equilibrium alchemical approach. J Comput Aided Mol Des 32(10):965–982
Procacci P (2016) I. dissociation free energies of drug-receptor systems via non-equilibrium alchemical simulations: a theoretical framework. Phys Chem Chem Phys 18:14991–15004
Nerattini F, Chelli R, Procacci P (2016) Ii. dissociation free energies in drug-receptor systems via nonequilibrium alchemical simulations: application to the fk506-related immunophilin ligands. Phys Chem Chem Phys 18:15005–15018
Procacci P (2018) Myeloid cell leukemia 1 inhibition: An in silico study using non-equilibrium fast double annihilation technology. J Chem Theory Comput 14(7):3890–3902
Procacci P (2016) Hybrid MPI/OpenMP Implementation of the ORAC Molecular Dynamics Program for Generalized Ensemble and Fast Switching Alchemical Simulations. J Chem Inf Model 56(6):1117–1121
Liu P, Kim B, Friesner RA, Berne BJ (2005) Replica exchange with solute tempering: a method for sampling biological systems in explicit water. Proc Acad Sci 102:13749–13754
Marsili S, Signorini GF, Chelli R, Marchi M, Procacci P (2010) Orac: a molecular dynamics simulation program to explore free energy surfaces in biomolecular systems at the atomistic level. J Comput Chem 31:1106–1116
Procacci P (2017) Primadorac: a free web interface for the assignment of partial charges, chemical topology, and bonded parameters in organic or drug molecules. J Chem Inf Model 57(6):1240–1245
O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR (2011) Open babel: an open chemical toolbox. J Cheminf 3(1):33
Izadi S, Onufriev AV (2016) Accuracy limit of rigid 3-point water models. J Chem Phys 145(7):074501
Hasel W, Hendrickson TF, Clark SW (1988) A rapid approximation to the solvent accessible surface areas of atoms. Tetrahedron Comput Methodol 1(2):103–116
Marchi M, Procacci P (1998) Coordinates scaling and multiple time step algorithms for simulation of solvated proteins in the npt ensemble. J Chem Phys 109:5194–5202
Procacci P (2019) Solvation free energies via alchemical simulations: let’s get honest about sampling, once more. Phys. Chem. Chem Phys 25:13826–13834
Procacci P (2019) Accuracy, precision, and efficiency of nonequilibrium alchemical methods for computing free energies of solvation. i. bidirectional approaches. J Chem Phys 151(14):144113
Piero P (2019) Precision and computational efficiency of nonequilibrium alchemical methods for computing free energies of solvation. ii. unidirectional estimates. J Chem Phys 151(14):144115
Beutler TC, Mark AE, van Schaik RC, Gerber PR, van Gunsteren WF (1994) Avoiding singularities and numerical instabilities in free energy calculations based on molecular simulations. Chem Phys Lett 222:5229–539
Anderson TW, Darling DA (1954) A test of goodness of fit. J Am Stat Assoc 49:765–769
Jarque CM, Bera AK (1980) Efficient tests for normality, homoscedasticity and serial independence of regression residuals. Econ Lett 6(3):255–259
Hummer G (2001) Fast-growth thermodynamic integration: error and efficiency analysis. J Chem Phys 114:7330–7337
Procacci P, Marsili S, Barducci A, Signorini GF, Chelli R (2006) Crooks equation for steered molecular dynamics using a nosé-hoover thermostat. J Chem Phys 125:164101
Pohorille A, Jarzynski C, Chipot C (2010) Good practices in free-energy calculations. J Phys Chem B 114(32):10235–10253
Procacci P, Chelli R (2017) Statistical mechanics of ligand-receptor noncovalent association, revisited: binding site and standard state volumes in modern alchemical theories. J Chem Theory Comput 13(5):1924–1933
Zhang C, Chao L, Jing Z, Chuanjie W, Piquemal J-P, Ponder JW, Ren P (2018) Amoeba polarizable atomic multipole force field for nucleic acids. J Chem Theory Comput 14(4):2084–2108
Bannwarth C, Ehlert S, Grimme S (2019) Gfn2-xtb–an accurate and broadly parametrized self-consistent tight-binding quantum chemical method with multipole electrostatics and density-dependent dispersion contributions. J Chem Theory Comput 15(3):1652–1671
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) Autodock4 and autodocktools4: automated docking with selective receptor flexibility. J Comput Chem 30(16):2785–2791
Vassetti D, Pagliai M, Procacci P (2019) Assessment of gaff2 and opls-aa general force fields in combination with the water models tip3p, spce, and opc3 for the solvation free energy of druglike organic molecules. J Chem Theory Comput 15(3):1983–1995
Dewar MJS, Zoebisch EG, Healy EF, Stewart JJP (1985) Am 1: a new general purpose quantum mechanical model. J Am Chem Soc 107:3902–3909
See comments on OVERLAP and INDENT host conformation in the AMOEBA submission file Clip-ponder.txt at https://github.com/samplchallenges/SAMPL7/tree/master/host_guest/Analysis/Submissions/TrimerTrip. Accessed 23 June 2020
Gapsys V, Michielssens S, Peters JH, de Groot BL, Leonov H (2015) Calculation of binding free energies. In: Molecular modeling of protein. Humana Press, pp 73–209
Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA (2004) Development and testing of a general amber force field. J Comp Chem 25:1157–1174
Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, Mackerell AD (2010) Charmm general force field: a force field for drug-like molecules compatible with the charmm all-atom additive biological force fields. J Comput Chem 31(4):671–690
Mobley DL, Bannan CC, Rizzi A, Bayly CI, Chodera JD, Lim VT, Lim NM, Beauchamp KA, Slochower DR, Shirts MR, Gilson MK, Eastman PK (2018) Escaping atom types in force fields using direct chemical perception. J Chem Theory Comput 14(11):6076–6092 PMID: 30351006
Gore J, Ritort F, Bustamante C (2003) Bias and error in estimates of equilibrium free-energy differences from nonequilibrium measurements. Proc Natl Acad Sci USA 100(22):12564–12569
Procacci P (2015) Unbiased free energy estimates in fast nonequilibrium transformations using gaussian mixtures. J Chem Phys 142(15):154117
Procacci P (2020) A remark on the efficiency of the double-system/single-box nonequilibrium approach in the sampl6 sampling challenge. J Comput Aided Mol Des 34(6):635–639
Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E (2015) Gromacs: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2:19–25
Naden Levi N, Shirts Michael R (2015) Linear basis function approach to efficient alchemical free energy calculations. 2. inserting and deleting particles with coulombic interactions. J Chem Theory Comput 11:2536–2549
Sun ZX, Wang XH, Zhang JZH (2017) Bar-based optimum adaptive sampling regime for variance minimization in alchemical transformation. Phys Chem Chem Phys 19:15005–15020
Yildirim A, Wassenaar TA, van der Spoel D (2018) Statistical efficiency of methods for computing free energy of hydration. J Chem Phys 149(14):144111
Khalak Y, Tresadern G, de Groot BL, Gapsys V (2020) Non-equilibrium approach for binding free energies in cyclodextrins in SAMPL7: force fields and software. J Comput Aided Mol Des. https://doi.org/10.1007/s10822-020-00359-1
Bennett CH (1976) Efficient estimation of free energy differences from monte carlo data. J Comput Phys 22:245–268
Shirts MR, Bair E, Hooker G, Pande VS (2003) Equilibrium free energies from nonequilibrium measurements using maximum likelihood methods. Phys Rev Lett 91:140601
Heinzelmann G, Gilson MK (2020) Automated docking refinement and virtual compound screening with absolute binding free energy calculations. bioRxiv. https://doi.org/10.1101/2020.04.15.043240
Boresch S, Tettinger F, Leitgeb M, Karplus M (2003) Absolute binding free energies: a quantitative approach for their calculation. J Phys Chem B 107(35):9535–9551
Tanweer Ul Islam (2017) Stringency-based ranking of normality tests. Commun Stat Simul Comput 46(1):655–668
Gilson MK, Given JA, Bush BL, McCammon JA (1997) The statistical-thermodynamic basis for computation of binding affinities: a critical review. Biophys J 72:1047–1069
Deng Y, Roux B (2006) Calculation of standard binding free energies: aromatic molecules in the t4 lysozyme l99a mutant. J Chem Theory Comput 2(5):1255–1273
Shi Y, Laury ML, Wang Z, Ponder JW (2020) Amoeba binding free energies for the sampl7 trimertrip host-guest challenge. J Comput Aided Mol Des 1–15
Deng Y, Roux B (2009) Computations of standard binding free energies with molecular dynamics simulations. J Phys Chem B 113:2234–2246
Hermans J, Wang L (1997) Inclusion of loss of translational and rotational freedom in theoretical estimates of free energies of binding. Application to a complex of benzene and mutant t4 lysozyme. J Am Chem Soc 119(11):2707–2714
Zhou H-X, Gilson MK (2009) Theory of free energy and entropy in noncovalent binding. Chem Rev 109:4092–4107
Acknowledgements
The computing resources and the related technical support used for this work have been provided by CRESCO/ENEAGRID High Performance Computing infrastructure and its staff. CRESCO/ENEAGRID High Performance Computing infrastructure is funded by ENEA, the Italian National Agency for New Technologies, Energy and Sustainable Economic Development and by Italian and European research programmes (see www.cresco.enea.it for information).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Procacci, P., Guarnieri, G. SAMPL7 blind predictions using nonequilibrium alchemical approaches. J Comput Aided Mol Des 35, 37–47 (2021). https://doi.org/10.1007/s10822-020-00365-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-020-00365-3