
Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a recently emanating human infectious coronavirus that causes COVID-19 disease. On 11th March 2020, it has been announced as a pandemic by the World Health Organization (WHO). Recently, several repositioned drugs have been subjected to clinical investigations as anti-COVID-19 drugs. Here, in silico drug discovery tools were utilized to evaluate the binding affinities and features of eighteen anti-COVID-19 drug candidates against SARS-CoV-2 main protease (Mpro). Molecular docking calculations using Autodock Vina showed considerable binding affinities of the investigated drugs with docking scores ranging from − 5.3 to − 8.3 kcal/mol, with higher binding affinities for HIV drugs compared to the other antiviral drugs. Molecular dynamics (MD) simulations were performed for the predicted drug-Mpro complexes for 50 ns, followed by binding energy calculations utilizing molecular mechanics-generalized Born surface area (MM-GBSA) approach. MM-GBSA calculations demonstrated promising binding affinities of TMC-310911 and ritonavir towards SARS-CoV-2 Mpro, with binding energy values of − 52.8 and − 49.4 kcal/mol, respectively. Surpass potentialities of TMC-310911 and ritonavir are returned to their capabilities of forming multiple hydrogen bonds with the proximal amino acids inside Mpro's binding site. Structural and energetic analyses involving root-mean-square deviation, binding energy per-frame, center-of-mass distance, and hydrogen bond length demonstrated the stability of TMC-310911 and ritonavir inside the Mpro's active site over the 50 ns MD simulation. This study sheds light on HIV protease drugs as prospective SARS-CoV-2 Mpro inhibitors.
Graphic Abstract

Similar content being viewed by others

1 Introduction
The coronavirus 2019 (COVID-19) pandemic that has emerged from severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a universal crisis [1, 2]. According to the World Health Organization (WHO), there were > 1,500,000 accumulative cases universally, with a ~ 5.9% case mortality reported in April 2020 [3]. One of the most significant challenges is the absence of a specific vaccine and treatment for COVID-19 [4]. Repositioning of approved drugs is therefore required as the fast track in the fight against COVID-19 infection [5,6,7]. Since January 2020, several clinical trials have tested antiviral, anti-inflammatory, and anti-malarial drugs for treating COVID-19 [8,9,10]. Among these drugs, remdesivir (GS-5734) is the most promising drug candidate to combat COVID-19 [11, 12]. Consequently, remdesivir has been recently declared to have a positive influence in a clinical trial for the treatment of COVID-19 [13]. Remdesivir, a nucleotide analog antiviral drug, targets the RNA-dependent RNA polymerase (RdRp) enzyme of SARS-CoV-2 and, in turn, prevents viral replication [11]. Prevention of the SARS-CoV-2 replication could also be achieved by targeting the viral main protease (Mpro) (also called 3-chymotrypsin-like protease (3CLpro)) and papain-like protease (PLpro). Because of the essential role of Mpro in the viral life cycle, numerous experimental and in silico studies have attempted to identify small molecules, natural products, and repurposed drugs as potential SARS-CoV-2 Mpro inhibitors [14,15,16,17,18,19,20,21,22,23]. Until now, the outcomes of the initiated clinical trials for protease inhibitors have not yet been released [18, 24, 25]. Therefore, the present study was set to investigate eighteen drugs in clinical development as potential Mpro inhibitors using in silico drug discovery techniques. The investigated drugs included anti-influenza drugs, anti-malarial drugs, anti-HIV drugs, anti-inflammatory drugs, anti-HCV drugs, anti-Ebola drugs, and anti-genitourinary infections drugs. Molecular docking calculations were first performed to predict the binding poses of the investigated drugs against SARS-CoV-2 Mpro. The binding affinities and features were highlighted. Molecular dynamics (MD) simulations for 50 ns were executed on the predicted docked drug-Mpro complexes, and a molecular mechanical-generalized Born surface area (MM-GBSA) approach was utilized to evaluate the binding energies (∆Gbinding). Post-dynamics analyses, including structural and energetic aspects, were executed to obtain insight into the stability and affinity of the drugs as potential Mpro inhibitors. The presented results are promising and proposing prospect inhibition for obtainable therapeutics against COVID-19.
2 Computational Methodology
2.1 Drug Preparation
Eighteen repurposed drugs for COVID-19 treatment were retrieved in SDF format from the DrugBank database before molecular docking calculations [26, 27]. Omega software was utilized to generate the 3D structures of the drugs [28, 29], and their geometrical structures were then minimized with the MMFF94S force field utilizing SZYBKI software [30]. The 2D chemical structures of the investigated anti-COVID-19 drug candidates are depicted in Table 1.
2.2 Main Protease Preparation
For molecular docking and molecular dynamics calculations, the crystal structure of SARS-CoV-2 main protease (Mpro; PDB code: 6LU7 [18]) was taken as a template. Ions and water molecules were removed. H++ server was employed to investigate the protonation state of Mpro. As well, all missing hydrogen atoms were added [31]. For molecular docking calculations, the Autodock protocol was utilized to prepare the pdbqt file of Mpro [32]. The pKa for SARS-CoV-2 Mpro amino acid residues was estimated under physical conditions of salinity = 0.15, pH 6.5, external dielectric = 80, and internal dielectric = 10.
2.3 Molecular Docking
Autodock Vina software was used to perform all molecular docking calculations [33]. All docking parameters were conserved to the default, except the exhaustiveness parameter was set to 200. The binding site was realized by a docking box around the active site with XYZ dimensions of 25 Å × 25 Å × 25 Å and a spacing value of 1.00 Å. The center of the grid was positioned at − 13.069, 9.74, 68.49 (XYZ coordinates) for SARS-CoV-2 Mpro.
2.4 Molecular Dynamics
AMBER16 software was utilized to perform molecular dynamics (MD) simulations on the docked structures of the investigated drugs inside the active site of Mpro [34]. The minutiae description of the applied MD simulations was demonstrated in Ref. [7, 14]. Concisely, Mpro was described using AMBER force field FF14SB [35], while the general AMBER force field (GAFF2) was utilized to describe the drugs [36]. The restrained electrostatic potential (RESP) approach with the assistance of Gaussian09 software was applied to assign the atomic partial charges of the investigated drugs [37, 38]. The drug-Mpro complexes were solvated in a cubic water box with 15 Å distances between the edges of the box and any atom of drug or drug-Mpro complexes. The solvated drug-Mpro complexes were minimized for 5000 steps, gradually heated from 0 to 300 K over 50 ps, and equilibrated for 1 ns. The NPT ensembles were adopted, and drug-Mpro complexes were simulated for 50 ns. Pmemd.cuda implemented in AMBER16 was utilized to perform all molecular dynamics simulations. CompChem GPU/CPU cluster (hpc.compchem.net) was applied to carry out molecular docking calculations and molecular dynamics simulations. 2D and 3D of the drug-Mpro interactions were visualized using the Discovery studio module of Biovia software (Dassault Systemes of France).
2.5 MM-GBSA Binding Energy
The binding energies of the investigated anti-COVID-19 drug candidates with SARS-CoV-2 Mpro were estimated using molecular mechanics-generalized Born surface area (MM-GBSA) approach with modified GB model (igb = 2) implemented in AMBER16 software [39]. For the MM-GBSA calculations, binding energy was calculated according to uncorrelated snapshots collected every 10 ps over the production run. The binding energy (ΔGbinding) was evaluated as follows:
where the energy term (G) is estimated as:
Evdw and Eele are van der Waals and electrostatic energies, respectively. GGB is the electrostatic solvation free energy calculated from the generalized Born equation and GSA is the nonpolar contribution to the solvation free energy from the solvent-accessible surface area (SASA). A single-trajectory approach was used, in which the coordinates of each drug-receptor, receptor and drug were extracted from a single trajectory. For all investigated drugs, entropy contributions were neglected.
3 Results and Discussion
Several repurposed drugs alone or in combinations have been subjected to clinical trials to treat COVID-19 [8, 40]. The mechanism of action of most of these drugs is to target the viral replication process or block viral entry into the host cell. Among these drugs, sixteen approved and investigational drugs were originally developed to treat influenza, HIV, HCV, and other respiratory infections. Besides, two approved anti-malarial drugs were proposed for clinical investigation as prospective anti-COVID-19 drugs. The chemical structure, original usage, and mechanism of action of these eighteen anti-COVID-19 drug candidates are summarized in Table 1.
The efficacy and safety of these drugs as promising anti-COVID-19 drugs are still under evaluation. In the present study, binding affinities, features, and stabilities of these anti-COVID-19 drug candidates were evaluated against SARS-CoV-2 main protease (Mpro) using in silico drug discovery techniques.
3.1 Molecular Docking Calculations
Molecular docking is used as a fundamental tool in the drug discovery pipeline [41]. In the present study, Autodock Vina software was applied to perform all molecular docking calculations and predict the binding modes of the repurposed drugs with SARS-CoV-2 Mpro. The predicted binding affinities and features of the investigated drugs towards Mpro are listed in Table 2. The 2D and 3D representations of interactions of the inspected drugs with the key amino acid residues of SARS-CoV-2 Mpro are illustrated in Fig. S1.
As can be seen from data in Table 2, the docking scores of the investigated drugs with SARS-CoV-2 Mpro ranged from − 5.8 to − 8.3 kcal/mol. Inspecting the drug-Mpro interactions revealed that most of the drugs share a similar binding pose inside the active site of Mpro, exhibiting a fundamental hydrogen bond with GLU166. Further interactions, including hydrogen bonds, hydrophobic interactions, and pi-based interactions, were also noticed between the drugs and the proximal amino acids inside the active site of SARS-CoV-2 Mpro (Fig. S1). It is also worth noting that a small group of the examined drugs could not form such a fundamental hydrogen bond with GLU166. This is returned to the preferability of these drugs to form a parallel hydrogen bond with HIS164 or HIS163, as in the case of chloroquine and triazavirin (Table 2). Several studies have indicated the vital role of the catalytic CYS145 residue in suppressing the protease activity [42, 43]. As shown in Table 2, seven out of the eighteen examined drugs exhibited a potential hydrogen bond with the catalytic CYS145 residue with bond lengths in the range of 2.06 − 2.99 Å.
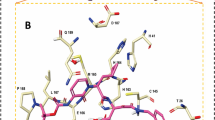
Among the examined drugs, HIV protease inhibitors demonstrated higher binding affinities against SARS-CoV-2 Mpro, compared to the other antiviral and anti-malarial drugs. For instance, TMC-310911 showed the highest binding affinity against Mpro with a docking score of − 8.3 kcal/mol. Structural insights into the binding mode of the TMC-310911 with the Mpro demonstrated that the sulfone moiety forms a hydrogen bond with the backbone NH of GLU166 residue with a bond length of 1.89 Å (Fig. 1 and Table 2). While, the carbamate moiety of TMC-310911 interacts with the backbone atoms of SER144, and CYS145 via hydrogen bonds of lengths 3.02 and 2.34 Å, respectively (Fig. 1 and Table 2). The strong interaction of TMC-310911 with the conserved residue GLU166 of Mpro has been recently reported with a docking score of − 7.1 kcal/mol [25].

2D representations of the predicted binding poses of the scrutinized HIV protease drugs inside the active site of SARS-CoV-2 main protease (Mpro)
The other four examined HIV protease drugs ─namely lopinavir, ritonavir, darunavir, and tenofovir alafenamide─ demonstrated promising binding affinities against Mpro with docking scores of − 8.0, − 7.7, − 7.5, and − 7.5 kcal/mol, respectively, forming two, four, five, and four hydrogen bonds with the key amino acids of Mpro, respectively (Fig. 1). Inspecting the binding mode of ritonavir with the Mpro revealed that (i) the C=O of the carbamoyl moiety demonstrates a hydrogen bond with the backbone NH of GLU166, with a bond length of 2.38 Å, (ii) the thiazole ring interacts with the imidazole ring of HIS163 via a hydrogen bond of length 2.01 Å, (iii) the C=O of the urea group forms a hydrogen bond with the backbone NH of GLY143 with a bond length of 1.83 Å, and (iv) the OH group participates in a hydrogen bond with GLN189 with a bond length of 2.63 Å (Fig. 1 and Table 2).
In agreement with the current results, the higher potency of ritonavir, compared to lopinavir, has been recently reported with SARS-CoV-2 Mpro [44].
Interestingly, remdesivir, an RNA-dependent RNA polymerase (RdRp) inhibitor developed for Ebola-virus, demonstrated the second-highest binding affinity towards SARS-CoV-2 Mpro with a docking score of − 8.2 kcal/mol. The other examined anti-Ebola drug ─namely galidesivir─ showed a satisfactory binding affinity with a docking score of − 7.1 kcal/mol with Mpro.
For anti-influenza drugs, relatively weak binding affinities were observed with SARS-CoV-2 Mpro, with docking scores of − 6.2, − 6.1, − 5.8, − 5.8, and − 5.3 kcal/mol for umifenovir, baloxavir marboxil, triazavirin, oseltamivir, and favipiravir, respectively. Besides, the anti-malarial, HCV, and anti-informatory drugs showed relatively weak potency towards Mpro (docking scores of − 6.2, − 5.8, − 6.7, − 6.3, and − 6.4 kcal/mol for hydroxychloroquine, chloroquine, methylprednisolone, ribavirin, and azithromycin, respectively).
In summary, the results of molecular docking calculations suggested HIV protease drugs and remdesivir as promising SARS-CoV-2 Mpro inhibitors.
3.2 Molecular Dynamics Simulations
It has been demonstrated that solvent effects, conformational flexibilities of drug-receptor complexes, and dynamics must be applied to improve the reliability of the predicted ligand–protein binding energies. Therefore, molecular dynamics (MD) simulations combined with MM-GBSA binding energy calculations were carried out for the repurposed drugs complexed with SARS-CoV-2 Mpro.
3.2.1 10 ns MD simulations
Eighteen repurposed drugs in clinical development to treat COVID-19 were further inspected using MD techniques over a simulation time of 10 ns, followed by MM-GBSA binding energy calculations. The evaluated MM-GBSA binding energies for the investigated drugs with Mpro are listed in Fig. 2.

Evaluated MM-GBSA binding energies for the investigated COVID-19 drug candidates as SARS-CoV-2 main protease (Mpro) inhibitors
What is interesting about the data in Fig. 2 that five out of the studied drugs demonstrated considerable binding energies (ΔGbinding < − 40.0 kcal/mol), while the rest of the drugs were noticed with relatively weak binding energies ranged from − 10.5 to − 36.0 kcal/mol. The single most striking observation to emerge from the data comparison was the significant-high binding affinities of TMC-310911 and ritonavir towards SARS-CoV-2 Mpro with binding energies (ΔGbinding) of − 59.6 and − 54.4 kcal/mol, respectively.
Overall, the MM-GBSA//10 ns MD binding energies demonstrated the promising potentiality of TMC-310911, ritonavir, remdesivir, lopinavir, and darunavir to inhibit SARS-CoV-2 Mpro.
3.2.2 50 ns MD simulations
To increase the reliability of the predicted binding affinities, the top potent drugs (ΔGbinding < − 40.0 kcal/mol with Mpro) were further subjected to longer MDs of 50 ns, and the corresponding MM-GBSA binding energies were consequently estimated (Fig. 2).
According to the calculated MM-GBSA//50 ns MD binding energies, TMC-310911 and ritonavir showed promising binding affinities against SARS-CoV-2 Mpro with ΔGbinding of − 52.8 and − 49.4 kcal/mol, respectively. The calculated values of Gdrug, Greceptor, and Gcomplex for the TMC-310911 and ritonavir in complex with Mpro are listed in Table S1. Compared to TMC-310911 and ritonavir, remdesivir showed a lower binding affinity with Mpro (ΔGbinding of − 42.7 kcal/mol). The estimated ΔGbinding over the 50 ns MD for lopinavir and darunavir with Mpro were observed to be higher than − 40.0 kcal/mol (ΔGbinding of − 35.6 and − 34.8 kcal/mol, respectively).
To reveal the nature of interactions of TMC-310911 and ritonavir with Mpro, MM-GBSA binding energy decomposition was performed over the 50 ns MD simulations. The evaluated energy components are listed in Table 3.
It is apparent from data in Table 3 that the Evwd interactions were predominant forces in the TMC-310911-Mpro and ritonavir-Mpro complexes with average values of − 65.8 and − 67.5 kcal/mol, respectively. As well, the Eele interactions were favorable in both TMC-310911-Mpro and ritonavir-Mpro complexes with average values of − 23.1 and − 10.5 kcal/mol, respectively.
Considering the promising binding affinities of TMC-310911 and ritonavir with Mpro, further investigations were carried out to inspect the drug-Mpro stability over the 50 ns MD simulation.
3.3 Post-Dynamics Analyses
Post-dynamics analyses were performed on the collected trajectories over the 50 ns MD simulations for TMC-310911-Mpro and ritonavir-Mpro complexes. The post-dynamics analyses involved root-mean-square deviation (RMSD), binding energy per-frame, center-of-mass (CoM) distance, and hydrogen bond length.
3.4 Binding Energy Per-Frame
To get a more in-depth insight into the stability of the drug inside the active site of Mpro, the correlation between MM-GBSA binding energy and time was investigated. The MM-GBSA binding energy per-frame for TMC-310911-Mpro and ritonavir-Mpro complexes is plotted versus time in Fig. 3.

MM-GBSA binding energies vs. time for TMC-310911 (in black) and ritonavir (in red) towards SARS-CoV-2 main protease (Mpro)
As shown in Fig. 3, there was outright stability for TMC-310911-Mpro and ritonavir-Mpro over the generated MD trajectories with average values of − 52.8 and − 49.4 kcal/mol, respectively.
3.5 Hydrogen Bond Length and Center-of-Mass Distance
The stabilities of TMC-310911-Mpro and ritonavir-Mpro complexes were further investigated by measuring the hydrogen bond lengths and center-of-mass (CoM) distances between the drug and the proximal amino acid GLU166 residue over the 50 ns MD simulations (Fig. 4).

a Hydrogen bond lengths and b center-of-mass (CoM) distances between TMC-310911 (in black) and ritonavir (in red) and the essential residue amino acid GLU166 of SARS-CoV-2 main protease (Mpro) over the 50 ns MD simulations
As can be concluded from data in Fig. 4a, TMC-310911 and ritonavir demonstrated high stabilities inside the active site of SARS-CoV-2 Mpro with average hydrogen bond lengths of 2.03 and 2.07 Å, respectively. Besides, the measured CoM distances were approximately constant around 7.2 and 10.1 Å over the 50 ns MD simulations for TMC-31091-Mpro and ritonavir-Mpro complexes, respectively (Fig. 4b).
3.6 Root-Mean-Square Deviation
The root-mean-square deviations (RMSD) throughout the 50 ns MD simulations were estimated to delineate the structural changes of drug-Mpro complexes. The RMSD's of backbone as a function of time with respect to the starting structure of TMC-310911- and ritonavir-Mpro complexes are plotted in Fig. 5.

Root-mean-square-deviation (RMSD) of SARS-CoV-2 main protease (Mpro) backbone atoms from the initial structure bound with TMC-310911 (in black) and ritonavir (in red) over the 50 ns MD simulations
It can be seen from data in Fig. 5 that the RMSD values of TMC-310911-Mpro and ritonavir-Mpro stayed beneath 0.35 nm for the course of the MD simulations. The RMSD results indicated that TMC-310911 and ritonavir are tightly bonded in the active site of the SARS-CoV-2 Mpro, and they do not impact the overall topology of Mpro.
Overall, the post-dynamics analyses provided evidence on the high stability of TMC-310911-Mpro and ritonavir-Mpro complexes, demonstrating their potentiality as prospective anti-COVID-19 drugs.
4 Conclusion
Several clinical trials have recently been launched to evaluate the efficacy and safety of repurposed drugs to treat COVID-19. In the current study, the potencies of eighteen repurposed drugs in clinical development were evaluated against SARS-CoV-2 Mpro using combined molecular docking and molecular dynamics (MD) techniques. Molecular docking calculations revealed the high binding affinities of TMC-310911 and ritonavir towards Mpro with docking scores of − 8.3 and − 7.7 kcal/mol, respectively. The calculated MM-GBSA binding energies for TMC-310911 and ritonavir with Mpro over the 50 ns MD were − 52.8 and − 49.4 kcal/mol, respectively, demonstrating their high potencies as Mpro inhibitors. Post-dynamics analyses over the 50 ns MD confirmed the promising binding affinities and stabilities of TMC-310911 and ritonavir with SARS-CoV-2 Mpro. The present results shed light on TMC-310911 and ritonavir as prospective repurposed drugs for the treatment of COVID-19.
Data Availability
All data generated or analysed during this study are included in this published article and its supplementary information files.
References
Zhou P et al (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579:270–273. https://doi.org/10.1038/s41586-020-2012-7
Wu F et al (2020) A new coronavirus associated with human respiratory disease in China. Nature 579:265–269. https://doi.org/10.1038/s41586-020-2008-3
Gorbalenya AE et al (2020) The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol 5:536–544. https://doi.org/10.1038/s41564-020-0695-z
Shereen MA, Khan S, Kazmi A, Bashir N, Siddique R (2020) COVID-19 infection: Origin, transmission, and characteristics of human coronaviruses. J Adv Res 24:91–98. https://doi.org/10.1016/j.jare.2020.03.005
Khan SA, Zia K, Ashraf S, Uddin R, Ul-Haq Z (2020) Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1751298
Tazikeh-Lemeski E, Moradi S, Raoufi R, Shahlaei M, Janlou MAM, Zolghadri S (2020) Targeting SARS-COV-2 non-structural protein 16: a virtual drug repurposing study. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1779133
Ibrahim MAA, Abdelrahman AHM, Hegazy MF (2020) In-silico drug repurposing and molecular dynamics puzzled out potential SARS-CoV-2 main protease inhibitors. J Biomol Struct Dyn 1:12. https://doi.org/10.1080/07391102.2020.1791958
Harrison C (2020) Coronavirus puts drug repurposing on the fast track. Nat Biotechnol 38:379–381. https://doi.org/10.1038/d41587-020-00003-1
Kupferschmidt K, Cohen J (2020) Race to find COVID-19 treatments accelerates. Science 367:1412–1413. https://doi.org/10.1126/science.367.6485.1412
Rosa SGV, Santos WC (2020) Clinical trials on drug repositioning for COVID-19 treatment. Rev Panam Salud Publ 44:e40. https://doi.org/10.26633/RPSP.2020.40
Yin W et al (2020) Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 368:1499–1504. https://doi.org/10.1126/science.abc1560
Beigel JH et al (2020) Remdesivir for the treatment of covid-19 - preliminary report. N Engl J Med. https://doi.org/10.1056/NEJMoa2007764
Administration FD (2020) Coronavirus (COVID-19) Update: FDA issues emergency use authorization for potential COVID-19 treatment. vol. 1.
Ibrahim MAA, Abdeljawaad KAA, Abdelrahman AHM, Hegazy MF (2020) Natural-like products as potential SARS-CoV-2 M(pro) inhibitors: in-silico drug discovery. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1790037
Kandeel M, Abdelrahman AHM, Oh-Hashi K, Ibrahim A, Venugopala KN, Morsy MA, Ibrahim MAA (2020) Repurposing of FDA-approved antivirals, antibiotics, anthelmintics, antioxidants, and cell protectives against SARS-CoV-2 papain-like protease. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1784291
Liu Z et al (2020) Composition and divergence of coronavirus spike proteins and host ACE2 receptors predict potential intermediate hosts of SARS-CoV-2. J Med Virol 92:595–601. https://doi.org/10.1002/jmv.25726
Cimolai N (2020) Potentially repurposing adamantanes for COVID-19. J Med Virol 92:531–532. https://doi.org/10.1002/jmv.25752
Jin Z et al (2020) Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature 582:289–293. https://doi.org/10.1038/s41586-020-2223-y
Riva L et al (2020) Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature. https://doi.org/10.1038/s41586-020-2577-1
Cavasotto CN, Di Filippo JI (2020) In silico drug repurposing for COVID-19: targeting SARS-CoV-2 proteins through docking and consensus ranking. Mol Inform. https://doi.org/10.1002/minf.202000115
Alves VM et al (2020) QSAR modeling of SARS-CoV Mpro inhibitors identifies sufugolix, cenicriviroc, proglumetacin, and other drugs as candidates for repurposing against SARS-CoV-2. Mol Inform. https://doi.org/10.1002/minf.202000113
Battisti V, Wieder O, Garon A, Seidel T, Urban E, Langer T (2020) A computational approach to identify potential novel inhibitors against the coronavirus SARS-CoV-2. Mol Inform. https://doi.org/10.1002/minf.202000090
Ibrahim MAA et al (2020) In silico drug discovery of major metabolites from spices as SARS-CoV-2 main protease inhibitors. Comput Biol Med 126:104046–104058. https://doi.org/10.1016/j.compbiomed.2020.104046
Muralidharan N, Sakthivel R, Velmurugan D, Gromiha MM (2020) Computational studies of drug repurposing and synergism of lopinavir, oseltamivir and ritonavir binding with SARS-CoV-2 protease against COVID-19. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1752802
Ancy I, Sivanandam M, Kumaradhas P (2020) Possibility of HIV-1 protease inhibitors-clinical trial drugs as repurposed drugs for SARS-CoV-2 main protease: a molecular docking, molecular dynamics and binding free energy simulation study. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1786459
Wishart DS et al (2006) DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res 34:D668–672. https://doi.org/10.1093/nar/gkj067
Wishart DS et al (2018) DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 46:D1074–D1082. https://doi.org/10.1093/nar/gkx1037
Hawkins PC, Skillman AG, Warren GL, Ellingson BA, Stahl MT (2010) Conformer generation with OMEGA: algorithm and validation using high quality structures from the protein databank and cambridge structural database. J Chem Inf Model 50:572–584. https://doi.org/10.1021/ci100031x
OMEGA (2013), 2.5.1.4 edn. OpenEye Scientific Software, Santa Fe, NM, USA
SZYBKI OpenEye Scientific Software: Santa Fe, NM, USA.
Gordon JC, Myers JB, Folta T, Shoja V, Heath LS, Onufriev A (2005) H++: a server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res 33:W368–W371. https://doi.org/10.1093/nar/gki464
Forli S, Huey R, Pique ME, Sanner MF, Goodsell DS, Olson AJ (2016) Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat Protoc 11:905–919. https://doi.org/10.1038/nprot.2016.051
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31:455–461. https://doi.org/10.1002/jcc.21334
Case DA et al (2016) AMBER 2016. University of California, San Francisco
Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C (2015) ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput 11:3696–3713. https://doi.org/10.1021/acs.jctc.5b00255
Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA (2004) Development and testing of a general amber force field. J Comput Chem 25:1157–1174. https://doi.org/10.1002/jcc.20035
Bayly CI, Cieplak P, Cornell W, Kollman PA (1993) A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J Phys Chem 97:10269–10280. https://doi.org/10.1021/j100142a004
Frisch MJ et al (2009) Gaussian 09, Revision, E01 edn. Gaussian Inc., Wallingford CT, USA
Massova I, Kollman PA (2000) Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect Drug Discov Des 18:113–135. https://doi.org/10.1023/A:1008763014207
McKee DL, Sternberg A, Stange U, Laufer S, Naujokat C (2020) Candidate drugs against SARS-CoV-2 and COVID-19. Pharmacol Res 157:104859. https://doi.org/10.1016/j.phrs.2020.104859
de Ruyck J, Brysbaert G, Blossey R, Lensink MF (2016) Molecular docking as a popular tool in drug design, an in silico travel. Adv Appl Bioinform Chem 9:1–11. https://doi.org/10.2147/AABC.S105289
Yoshino R, Yasuo N, Sekijima M (2020) Identification of key interactions between SARS-CoV-2 main protease and inhibitor drug candidates. Sci Rep 10:12493. https://doi.org/10.1038/s41598-020-69337-9
Ghahremanpour MM et al (2020) Identification of 14 known drugs as inhibitors of the main protease of SARS-CoV-2. ACS Med Chem Lett. https://doi.org/10.1021/acsmedchemlett.0c00521
Nutho B et al (2020) Why are lopinavir and ritonavir effective against the newly emerged coronavirus 2019? atomistic insights into the inhibitory. Mech Biochem 59:1769–1779. https://doi.org/10.1021/acs.biochem.0c00160
Acknowledgements
Dr. Mahmoud F. Moustafa extends his appreciation to the Deanship of Scientific Research at King Khalid University for funding this work under Grant No. (R.G.P 2/90/41). The computational work was completed with resources supported by the Science and Technology Development Fund, STDF, Egypt, Grants Nos. 5480 & 7972 (Granted to Dr. Mahmoud A. A. Ibrahim).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ibrahim, M.A.A., Abdelrahman, A.H.M., Allemailem, K.S. et al. In Silico Evaluation of Prospective Anti-COVID-19 Drug Candidates as Potential SARS-CoV-2 Main Protease Inhibitors. Protein J 40, 296–309 (2021). https://doi.org/10.1007/s10930-020-09945-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10930-020-09945-6