
Abstract
In this study, magnetic-dispersive solid-phase extraction coupled with ion pair-dispersive liquid–liquid micro-extraction (MSPE-i-DLLME) was used to the pre-concentration and extraction of five sulfonamides residues (sulfadiazine, sulfathiazole, sulfacetamide, sulfamethazine and sulfamethoxazole) in animal foodstuffs. The sulfonamides are extracted using magnetic carbon nanocomposite and then eluted with acetonitrile. In the DLLME step, the target analytes are collected in 1-octanol containing 10% Aliquat-336 (as extraction solvent). Finally, the compounds are quantified by HPLC with DAD detection. The extraction parameters optimized using the one at the time and central composite design methods. Under the optimized conditions: sample solution volume was 100 mL; initial pH: 12, amounts of MCNs: 30 mg; desorption solvent (ACN) volume, 1 mL; desorption condition, 10 min sonication at two step; extraction solvent (1-octanol + 10% aliquat) volume, 115 µL; pH of DLLME step, 3; salt effect, 24.6%, (w/v) NaCl; centrifuge, 5 min, 4000 rpm. In these extraction conditions, the proposed procedure represented good pre-concentration factors between 130 and 490; detection limits in the range from 0.01 to 5 μg kg−1 (at S/N = 3), and linear response in the 0.1–400 μg kg−1 concentration range. The method is successfully applied to the determination of sulfonamides residues in animal foodstuffs.
Similar content being viewed by others


Introduction
Sulfonamides are one of a series of drugs used to help grow and act as antimicrobial agents in domestic animals in most countries of the world. These compounds, like other chemical agents, are regulated by human health, such as the development of resistance and toxicity, due to its potential side effects. Allergic reactions reported to sulfur-containing compounds such as organosulfur pesticides, antibiotic sulfonamides, non-antibiotic sulfonamides, other sulfur-containing drugs and other sulfur-containing compounds in human foods [1]. Maximum residue limit of sulfonamides in animal foods is set at 100 μg kg−1 by the European Union (EU) [2]. Therefore, there is a need for the development of a simple, rapid, inexpensive, and sensitive method to detect the trace amount of these residues in animal by-product samples. There are numerous literature works about the determination of various animal by-products and water samples using high-performance liquid chromatography (HPLC) and HPLC with mass spectrometry [3,4,5], UV–Vis [6], fluorescence [7] detection and the capillary electrophoresis (CE) [8]. Determination of sulfonamides and other antibacterial residues in animal by-product requires low limits of detection. Therefore, pre-concentration techniques such as liquid–liquid extraction (LLE) [9], solid-phase extraction (SPE) [10], dual-dummy-template molecularly imprinted polymer (DDMIP) [11], and micro-extraction methods such as dispersive liquid–liquid micro-extraction (DLLME [12]) ultrasonic-assisted liquid–liquid micro-extraction (U-LLME) [13], hollow fiber liquid phase [14], three-phase hollow fiber micro-extraction [15] are mandatory. Solid-phase micro-extraction methods are repeatedly developed with the magnetic and non-magnetic multi-walled carbon nanotubes [16] and the invention of new adsorbents such as, spherical mesoporous covalent organic framework [17], tailorable yolk-shell Fe3O4@graphitic carbon submicroboxes [18], thiol-functionalized magnetic carbon nanotubes [19], amino terminated carbon nanotubes with isocyanates [20].
DLLME is a very simple method with very low solvent consumption [12] that can be easily coupled with other methods to obtain higher pre-concentration factor, cleaning and better LOD for detecting analytes. Recently, some developments are applied on DLLME with other micro-extraction methods such as, solid-phase extraction combined with DLLME (SPE-DLLME) [21, 22], supercritical fluid extraction combined with DLLME (SFE-DLLME) [23], and DLLME based on the solidification of floating organic drop (DLLME-SFO) [24].
After our previous works [25, 26], in this project, a combination of MSPE and i-DLLME based on using magnetic carbon nanocomposites (MSPE-i-DLLME) was developed as an extraction method for some sulfonamides (SAs) prior to their determination by HPLC-DAD. Effective parameters for extraction are investigated, and the efficiency of the method for the rapid determination of SAs in animal food samples was identified.
Experimental
Chemical reagents, materials and instrumentation
Activated carbon, ferric nitrate (Fe (NO3)3.9H2O) and HPLC grade water were purchased from Merck (Darmstadt, Germany). Sulfadiazine (SD), sulfathiazole (ST), sulfacetamide (SC), sulfamethazine (SM), and sulfamethoxazole (SX) and Aliquat-336 (Methyl trioctyl ammonium chloride) were purchased from the Sigma–Aldrich (Seelze, Germany). Extra pure and HPLC grade methanol, 1-octanol and acetonitrile were obtained from Samchun (Seoul, Republic of Korea). In addition, other chemicals were analytical and HPLC grade. An ultrasonic bath (Bandelin Sonorex Digital, Germany), an analytical precision scale (Sartorius, Madrid, Spain) and a vortex shaker (IKA, Deutschland, Germany), A Metrohm 781 pH meter (Herisau Switzerland) and a parsia Ind group centrifuge (Tehran, Iran) were used for steps of experiments.
HPLC analysis
Chromatographic analysis is performed on an Agilent HPLC system 1200 series equipped with a diode array detector (DAD). Separations are conducted using an Eclipse-XDB-C18 analytical column (250 mm × 4.6 mm, 5 µm). The mobile phase with the flow rate at 1.0 mL min−1 was 30 mmol L−1 phosphate buffer with pH 4.5 (solvent A) and methanol (solvent B). During the gradient program, from the first 2 to 10 min, the concentration of solvent A was 100% and then decreased to 75% and solvent B increased to 25%, and then from 25 to 50% for solvent B from the 10 to 15 min, the injection volume was 100 µL. The working wavelengths were 210 nm for SD, SM, and SX and 265 nm for SC and ST.
Mass spectra were obtained using an electrospray ionization (ESI) source on an ion trap mass spectrometer detector (Agilent, Wilmington, USA). The following optimal MS parameters were set up; the capillary exit voltage was set at 109.8 V, the voltage of skimmer 1 and skimmer 2 was set at 40 and − 5 V, respectively.
Preparation of magnetic carbon nanocomposites (MCNs)
The magnetic carbon nanocomposites as adsorbent for pre-concentration of SAs from aqueous solution were prepared and characterized based on the combustion method that we described in our previous research [25]. First, activated carbon was added to concentrated nitric acid (65%) and heated at 80 °C for 3–4 h, to make hydrophilic. Then, 1 g of the modified carbon was dispersed in 40 mL aqueous solution containing 8 g of Fe (NO3)3·9H2O and sonicated for 45 min to adsorb iron salt. After filtration and drying, the active carbon that impregnated with iron salt was placed under a N2 atmosphere at 750° C for 3 h, forming Fe3O4 magnetic nanoparticles. These prepared MCNs were stable for several months under environmental conditions.
Micro-extraction procedure
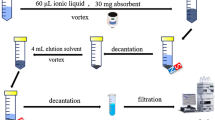
100 mL of SAs sample solution (pH 12) was transferred to 100 mL glass beaker. Then, 30 mg of magnetic carbon nanocomposites was added to it and shaked for 9 min. After about 3 min, an adsorbent precipitated by the adsorbent, an Nd–Fe–B strong magnet was placed at the bottom of the glass to separate the adsorbent particles from the sample solution. Adsorbed SAs eluted from the sorbent in two stages using 0.5 mL of ACN for each stage. Next, 115 µL extraction solvent (1-octanol with 10% aliquat) was added to the 1 mL collected ACN as disperser and was rapidly injected into an aliquot of 5 mL of 24.6 %w/v NaCl solution. Immediately after adding this mixture, a cloudy solution was formed in the test tube. The extraction solvent is lighter than water, so it floats on the surface of the water. To completely separate this solvent, we centrifuged the mixture for 5 min at 4000 rpm. The extracted solvent was collected, and finally, 100 µL of it was injected into the HPLC-DAD using a micro-syringe (Hamilton, Reno, NV, USA).
Preparation of real samples
Several samples of animal by-products such as honey, milk, fish, parts of lamb and chicken (tissue and liver, purchased from local supermarkets of Tehran, Iran) are stored at 4 °C. For pretreatment of lamb, chicken, and fish samples, 5 g of sodium sulfate and 15 mL of ACN were added to 20 g of homogenized sample and the samples were kept in ultrasonic bath for 5 min, and then the solvent was separated by centrifugation. This process was repeated twice to complete the extraction. The extraction solvent is evaporated under N2 bubbling to reduce its volume to 1 mL. The condensed specimen was diluted to 100.0 mL with aqueous solution (pH 12.0). For preparation of honey samples, 20 g of sample was dissolved in 10 mL of 2 mol L−1 HCl, and then it is dissolved in distilled water after filtering to a volume of 100 mL (pH 12). [14]
Preparation of milk samples is different, because of the presence of fat and protein; for the precipitation of proteins, 10 mL of 0.5 mol L−1 K3[Fe(CN)6].3H2O solution and 10 mL of 2 mol L−1 Zn(CH3COO)2·2H2O [6] were added to a 20 mL milk sample. The sample was mixed for 2 min, until the protein is completely precipitated, the samples are centrifuged and filtered through a PTFE filter and diluted to 100 mL prior to the MSPE-i-DLLME procedure.
Calculations
The pre-concentration factors (PF) and extraction recoveries (ER) are calculated based on the following equations:
Va and Vd are volumes of the extractant and donor solutions, respectively, Cex, final is the final concentration of the analyte in the extractant solution, and Cd, initial is the initial analyte concentration in the donor phase.
Relative recovery (RR) is acquired from the following equation:
Cfound, Creal, and Cadded are the analyte concentration after addition of a known amount of standard into the real sample, the analyte concentration in the real sample, and the concentration of a known amount of the standard, which spiked into the real sample, respectively.
The pre-concentrated SAs are confirmed by the comparison of their retention time and UV spectra with pure standard samples. Procedure for determination of the limit of detection (LOD) is to set the LOD at three times the noise level of the baseline in the chromatogram, and the LOQ is set three times that of the LOD.
Results and discussion
Optimization of the developed method
MSPE parameters such as pH of initial sample solution, type and volume of solvents, extraction time, amount of magnetic sorbent, breakthrough volume, and DLLME parameters such as the ionic strength, extraction time and pH of sample solution were investigated and optimized. We used to evaluate the effect of type of elution solvent and desorption conditions for MSPE-MCNs, extraction solvent in DLLME, and breakthrough volume using the one-factor-at-a-time approach, and the effect of other parameters on extraction efficiency was investigated through experimental design simultaneously using by “Design-Expert 7.1.3” software.
Effect of type of elution solvent for MSPE
In the MSPE stage, the analytes were adsorbed onto the nanoparticles and rinsed with a suitable solvent, in addition, this solvent should play the role of dispersing solvent in the DLLME step. Therefore, it should be a good solvent for the SAs and should be incompatible with both organic and aqueous phases. Methanol, ethanol, acetone, acetonitrile (ACN), and ACN with 10 mmol L−1 ammonium acetate were investigated for this purpose. Finally, ACN was chosen for the solvent extraction due to its best efficiency. Figure 1a compares the performance of different solvents.

Investigation of desorption solvent (a) and condition (b) in MSPE; extraction conditions: sample solution volume, 100 mL; initial pH: 12, amounts of MCNs: 30 mg; desorption solvent volume, 1 mL; extraction solvent (1-octanol + 10% aliquat), 115 µL; pH of DLLME step, 3; salt effect, 24.6%, (w/v) NaCl; centrifuge, 5 min, 4000 rpm
Desorption condition for MSPE
The time and desorption conditions by ACN were optimized. The sorbent was eluted by different volume of ACN in various condition (vortex or sonication at different time) (Fig. 1b). Twice eluting of sorbent by 0.5 mL ACN along 5 min sonication at each step produced better extraction efficiency than sonication or vortex at longer times (> 10 min) at one step. Longer times of sonication can bring out iron oxide nanoparticles from the pores of activated carbon. Therefore, it is observed that the sorbent is not separated completely by the external magnet from the elution solvent and the extraction efficiency is decreased. Therefore, two-step elution of sorbent in total by 1 mL ACN in 10 min sonication is selected for elution condition.
The effect of initial sample volume in MSPE
The main purpose of the micro-extraction method is to extract the concentration of sample and collect analytes in a small volume of extraction solvent. Therefore, higher sample size is better. Fortunately, the carbon nanocomposites used in this research are magnetic and, therefore, being well dispersed in the solution, and then it is possible to quickly and fully collect it from larger volumes of the sample solution. In order to obtain the best sample volume, different volumes of the water samples 25–200 mL are studied, all of these solutions contain 20 µg of SAs. The best pre-concentration factors are obtained when the sample volume is 100 mL.
Extraction solvent in DLLME
The organic solvent for pre-concentration of target analytes was selected between the higher and lower density solvents rather than water and the ability to extract the desired compounds and the proper behavior of chromatography [27, 28]. Several solvents with a density lower and more than water such as chloroform (density: 1.49 g mL−1), 1-undecanol (density: 0.83 g mL−1), 1-octanol (density: 0.82 g mL−1), 1-octanol with 10% Aliquat-336 and 1-octanol with 1 mmol L−1 CTAB were examined. The highest extraction efficiencies were obtained by using 1-octanol with 10% Aliquat-336. Aliquat-336 acts as an ion pairing agent for extraction of hydrophilic drug [29, 30]. Therefore, 1-octanol was added to various concentrations of Aliquat-336 as the ion pair agent in the range of 5–20% (v/v) as investigated. The highest extraction rate was observed in 10% Aliquat-336 in 1-octanol, and then this solvent is chosen. If Aliquat-336 is added more than this amount, the viscosity of the solvent is increased and the analyte is not well extracted.
Due to hydrophilic property of SAs, using of polar solvent with a carrier is useful for extraction of them from aqueous solution to organic solvent.
Optimization of other parameters by experimental design
Other parameters are optimized by central composite design (CCD). It is a combination of full or fractional factorial points (Nf = 2f), axial points (Na = 2f), and a set of center points (N0) [31, 32]. In rotatable designs, the predicted error is the same for all of the investigated points. The value needed to ensure rotatability for half fraction CCD (Nf = 2f−1) was ± 2.378 which was calculated by using Eq. (4) [33].
Regarding Eq. (5), for half fraction CCD with 6-factor and 3-center points, totally 47 experiments had to be run.
The main factors and levels tested in accordance with the design matrix were mg of sorbent in the range of 2–30 mg, extraction time in MSPE 5–65 min, initial pH, and DLLME pH from 2 to 12, organic solvent volume 20–120 µL and amount of salt in DLLME solution from 0 to 30 percent.
Figure 2a shows the simultaneous effect of extraction time (A) and the amount of sorbent (B) on the response. By increasing the amount of sorbent, the extraction efficiency is increased, because the surface of sorbent for adsorbing the analyte is increased, and after this, with the increase in sorbent amount, the extraction efficiency decreased due to insufficient elution of sorbent with the solvent [26]. The pre-concentration factor decreased gently on increasing the extraction time after 9 min, because the mass transfer of components needs specified time to reach the equilibrium, and after among longer times, analyte may be extracted back to solution [34]. The effect of DLLME pH (D) and the % salt (F) on the response is shown in Fig. 2b. Increasing salt concentration reduces the solubility of SAs in the aqueous phase, thus extraction efficiency is increased. In addition, pKa value of SAs is in the range of 2.2–7.4, in lower pH, it is converted to neutral form and extracted better to organic solvent. Figure 2c indicates the response surface plot of initial pH (C) and volume of organic solvent (E) on the extraction efficiency. PH is one of the main factors affecting the adsorption behavior of the nanoparticle mixture system due to the different charge densities of nanoparticles at different pHs, electrostatic reactions, etc. [35]. The extraction efficiency increases with increasing initial pH, because at basic pH, SAs have ionic form and adsorbed slightly better on carboxylic site on magnetic activated carbon. With the increase in volume of organic solvent, solubility and transferring of SAs in extraction phase increase. The optimal conditions were calculated as pH 12 for initial pH, 9 min for extraction time and 30 mg for magnetic sorbent in SPE and pH 3 for DLLME solution, 115 µL for organic solvent (1-octanol + 10% aliquat), and 24.6% (w/v) salt (NaCl) concentration in DLLME. Finally, five experiments were carried out in optimal conditions to verify the accuracy of the optimization results by the response surface method.

Response surface and contour plot of SAs using the CCD, which illustrates the relationship among extraction time and amount of sorbent (a), % salt and pH of DLLME (b) and volume of extraction solvent of DLLME and Initial pH (c) and their counter plots, d Pareto charts of the main and interaction effects obtained from the central composite design
The quality of fit of the polynomial model equation is expressed by the coefficients of determination (R2). R2 is a measure of the amount of variation around the mean explained by the model and was equal to 0.8535. Figure 2d shows the Pareto charts of the main and interaction effects obtained from the central composite design: AA, BB, CC, DD, EE and FF are the quadratic effects of the amount of sorbent, extraction solvent volume, extraction time, initial pH, DLLME pH, and % salt in DLLME, respectively. AB, BC and AC are the interaction effects between them.
Analytical performance and validation of MSPE-i-DLLME
Analytical method performances, i.e., pre-concentration factors (PFs), limit of detections and quantifications (LODs and LOQs, respectively), linear dynamic ranges (LDRs), extraction recoveries (ER), intra- and inter-day precision (RSDs) were calculated in different samples, after determining the optimal conditions and shown in Table 1. Twelve spiking level of five SAs in the range of 0.01–500 μg kg−1 were used. Linearity dynamic ranges of 0.1–400-μg kg−1 were obtained for water sample. The homoscedasticity of the data was examined by F-test of the variances at the lower and high limit of range. The difference between the observed and the critical value of F was not significant (α = 0.05).
The LODs for each SAs were obtained in the range of 0.01–0.1 μg kg−1 practically based on signal-to-noise ratio of 3. RSDs for intra- and inter-day precision were obtained in the range of 5.6–12% and 8.2–13%, respectively. The PFs are obtained using Eq. 1 between 130 and 490. Analytical performances for other matrixes (honey, milk, lamb, chicken and fish) are displayed in Table 1.
Comparison of MSPE-i-DLLME with other methods
Analytical performances of other reported literature works such as LODs, and LOQs for extraction and determination of SAs in different matrixes are summarized in Table 2 for comparison [4, 17,18,19,20, 36, 37]. LODs, LOQs, and extraction times of the presented method are more appropriate and comparable with the previous methods that reported for determination of SAs. Furthermore, in this work, variety of investigated real samples is more than other methods.
Analysis of real samples
Match matrix method is investigated in the study to achieve results with maximum accuracy. The application possibility of match matrix method was calculated by comparing the peak area ratios of SAs from the real samples to those obtained in working curve solutions at the concentrations of 1–200 µg kg−1. The obtained relative recovery (RR%) values are indicated in Table 3. The RR% of the SAs from honey, milk, chicken, lamb, and fish was in the range of 70.5–90%, 72.5–92%, 70–89.6%, 75.2–91.4 and 75.1–89.7, respectively.
Figure 3 is the chromatogram of MSPE-DLLME/HPLC-DAD analysis of lamb (Fig. 3a1) and fish (Fig. 3b1) samples spiked with 100 µg kg−1 of SAs. In the case of contaminated real samples, LC-MS technique is used to confirm the results. Mass spectra of liver lamb (Fig. 3a2) and fish (Fig. 3b2) analysis are displayed in this figure. Table 3 shows the results obtained from the analysis of real samples by MSPE-i-DLLME/HPLC-DAD method.

The chromatograms of MSPE-DLLME/HPLC-DAD of infect lamb (a1) and fish (b1) samples (down chromatograms) and spiked samples with 100 µg kg−1 of SAs (up chromatograms) with mass spectra of liver lamb (a2) and fish (b2) analysis. Extraction conditions: sample solution volume, 100 mL; initial pH: 12, amounts of MCNs: 30 mg; desorption solvent (ACN) volume, 1 mL; desorption condition, 10 min sonication at two step; extraction solvent (1-octanol + 10% aliquat) volume, 115 µL; pH of DLLME step, 3; salt effect, 24.6%, (w/v) NaCl; centrifuge, 5 min, 4000 rpm
Conclusion
In this work, for the first time, we applied MSPE-i-DLLME for extraction and pre-concentration of SAs from various animal foodstuff samples. Two major purposes followed by this method: obtaining the validated method with high accuracy and precision for determination of SAs, and the powerful method for determination of SAs in complex and various real samples, that all of these purposes achieved. A low limit of detection, up to 0.01–5 μg kg−1, and pre-concentration factors of 130–490 are the main advantages of this analytical procedure in comparison with other extraction methods. The presented method was fast and two extraction steps were done during 15 min; therefore, the method is very suitable for the rapid extraction of SAs from various samples at ppb levels. Accordingly, the presented method can be a guideline for the extraction and determination of trace amounts of antibiotics in animal by-products.
References
S.J. Stohs, M.J. Miller, Food Chem. Toxicol. 63, 240–243 (2014)
S.Y. Won, C.H. Lee, H.S. Chang, S.O. Kim, S.H. Lee, D.S. Kim, Food Control 22(7), 1101–1107 (2011)
J. Premarathne, D. Satharasinghe, A. Gunasena, D. Munasinghe, P. Abeynayake, Food Control 72, 276–282 (2017)
K.H. Lu, C.Y. Chen, M.R. Lee, Talanta 72(3), 1082–1087 (2007)
Y. Li, J. Han, Y. Yan, B. Chen, G. Zhang, Y. Liu, C. Sheng, J. Iran. Chem. Soc. 10(2), 339–346 (2013)
G.Z. Fang, J.X. He, S. Wang, J. Chromatogr. A 1127(1–2), 12–17 (2006)
J. Raich-Montiu, J. Folch, R. Compañó, M. Granados, M.D. Prat, J. Chromatogr. A 1172(2), 186–193 (2007)
Y.M. Park, H. Pyo, S.J. Park, S.K. Park, Anal. Chim. Acta 548(1–2), 109–115 (2005)
A.R. Ghiasvand, S. Shadabi, E. Mohagheghzadeh, P. Hashemi, Talanta 66(4), 912–916 (2005)
U. Koesukwiwat, S. Jayanta, N. Leepipatpiboon, J. Chromatogr. A 1149(1), 102–111 (2007)
Y.P. Song, L. Zhang, G.N. Wang, J.X. Liu, Food Control 82, 233–242 (2017)
S. Berijani, A. Yaghoub, A. Mansoor, M.R. Milani Hosseini, E. Aghaee, J. Chromatogr. A 1123(1), 1–9 (2006)
Y. Ji, Z. Meng, J. Zhao, H. Zhao, L. Zhao, J. Chromatogr. A 1609, 460520 (2020)
F. Tajabadi, M. Ghambarian, Y. Yamini, N. Yazdanfar, Talanta 160, 400–408 (2016)
F. Tajabadi, M. Ghambarian, Y. Yamini, Anal. Methods 7(7), 2959–2967 (2015)
A.V. Herrera-Herrera, J. Hernández-Borges, M.M. Afonso, J.A. Palenzuela, M.A. Rodríguez-Delgado, Talanta 116, 695–703 (2013)
L. Wen, L. Lu, W. Xia, M.-L. Wang, J.-M. Lin, R.-S. Zhao, J. Chromatogr. A 1625, 461275 (2020)
X. Liu, Y. Tong, L. Zhang, Food Chem. 303, 125369 (2020)
A. Nasir, N. Yahya, N. Zain, B. Lim, S. Kamaruzaman, B. Saad, N. Nishiyama, N. Yoshida, Y. Hirota, Food Chem. 276, 458–466 (2019)
L. Fu, H. Zhou, E. Miao, S. Lu, S. Jing, Y. Hu, L. Wei, J. Zhan, M. Wu, Food Chem. 289, 701–707 (2019)
X. Yuan, D. Wu, C. Liu, X. Li, Z. Xiong, L. Zhao, New J. Chem. 42(24), 19578–19590 (2018)
M. Shamsipur, N. Yazdanfar, M. Ghambarian, Food Chem. 204, 289–297 (2016)
M. Jowkarderis, F. Raofie, Talanta 88, 1360–1367 (2010)
M. Shamsipur, N. Fattahi, Y. Assadi, M. Sadeghi, K. Sharafi, Talanta 130, 26–32 (2014)
N. Yazdanfar, M. Shamsipur, M. Ghambarian, A. Esrafili, Chromatographia 81(3), 487–499 (2018)
N. Yazdanfar, M. Shamsipur, M. Ghambarian, Microchim. Acta 186(3), 178–188 (2019)
H. Sereshti, A. Rohanifar, S. Bakhtiari, S. Samadi, J. Chromatogr. A 1238, 46–53 (2012)
H. Sereshti, S. Samadi, M. Jalali-Heravi, J. Chromatogr. A 1280, 1–8 (2013)
L. Adlnasab, H. Ebrahimzadeh, Y. Yamini, Microchim. Acta 179(1–2), 179–184 (2012)
S. Bahar, Iran. J. Anal. Chem. 2(1), 1–6 (2015)
M. Zeaiter, J.M. Roger, V. Bellon-Maurel, N. Rutledge, Trends Anal. Chem. 23(2), 157–170 (2004)
M. Mousavi, E. Noroozian, M. Jalali-Heravi, A. Mollahosseini, Anal. Chim. Acta 581(1), 71–77 (2007)
H. Sereshti, M. Karimi, S. Samadi, J. Chromatogr. A 1216(2), 198–204 (2009)
L. Zhao, H.K. Lee, J. Chromatogr. A 931(1–2), 95–105 (2001)
S.A. Mousavi, H. Janjani, Toxin Rev. 39, 87–98 (2018)
T. Chatzimitakos, C. Stalikas, Microchem. J. 155, 104670 (2020)
H. Yu, H. Mu, Y.M. Hu, J. Pharm. Anal. 2(1), 76–81 (2012)
Acknowledgements
The authors thank the research council of Razi University of Kermanshah (Iran) and Iranian Research, and Development Center for Chemical Industries, Tehran, Iran (ACECR) for the financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yazdanfar, N., Shamsipur, M. & Ghambarian, M. Determination of sulfonamide residues in animal foodstuffs by magnetic dispersive solid-phase extraction using magnetic carbon nanocomposites coupled with ion pair-dispersive liquid–liquid micro-extraction combined with HPLC-DAD. J IRAN CHEM SOC 18, 1433–1442 (2021). https://doi.org/10.1007/s13738-020-02127-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-020-02127-2