Genomic Variations in Drug Resistant Mycobacterium tuberculosis Strains Collected from Patients with Different Localization of Infection

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
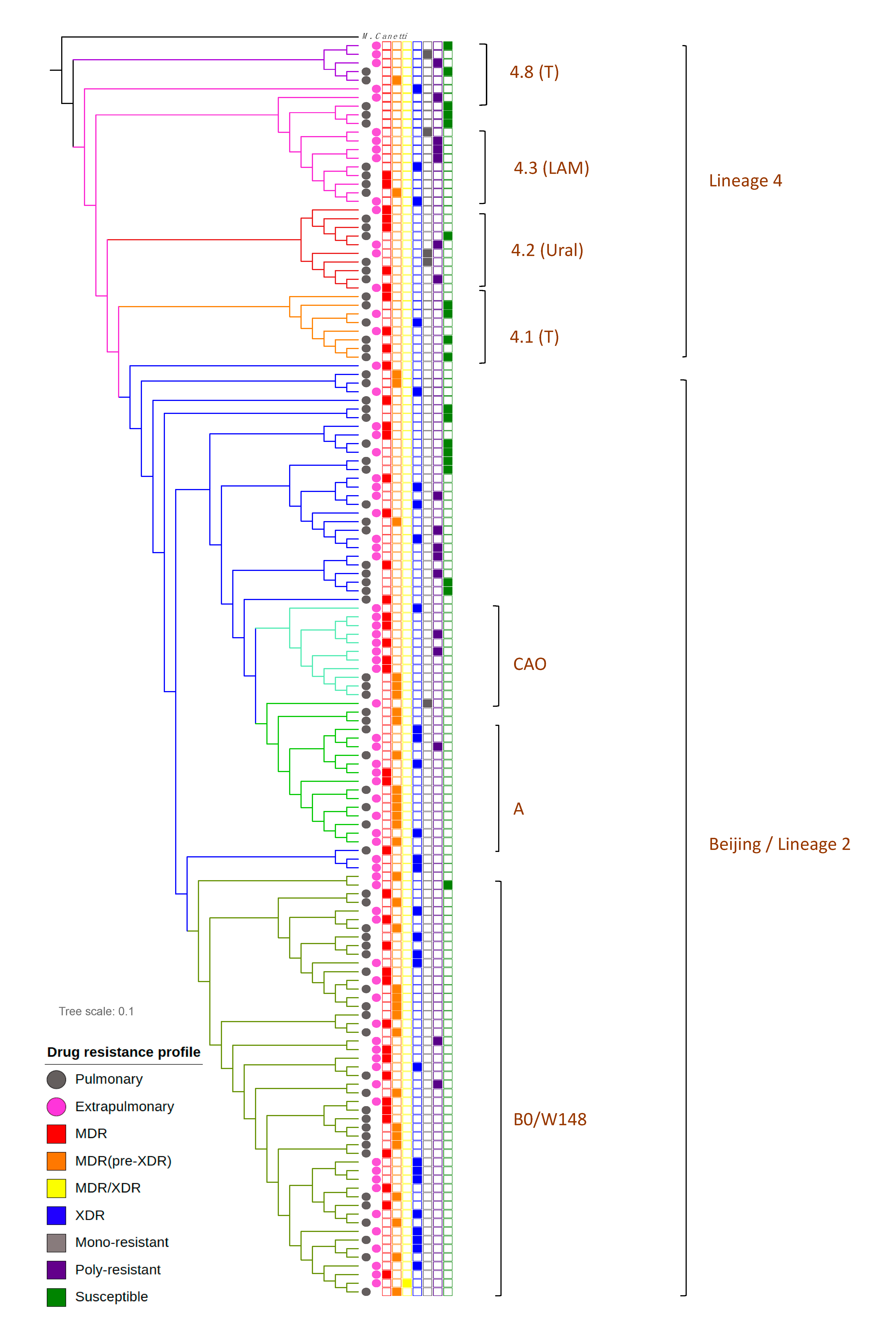
2.1. M. tuberculosis Phylogenetic Analysis
2.2. Drug Resistance of PTB and XPTB Strains
2.3. Mutations Associated with Drug Resistance
2.4. TB/HIV Coinfection
3. Discussion
4. Materials and Methods
- INH—1.0 (LJ), 0.1 (MIGIT)
- RIF—40.0(LJ), 1.0 (MIGIT)
- SM—10.0 (LJ), 1.0 (MIGIT)
- EMB—2.0 (LJ), 5.0 (MIGIT)
- PZA—100.0 (MIGIT), LJ was not used
- ETH—40.0 (LJ), 5.0 (MIGIT)
- OFL—2.0 (LJ), 2.0 (MIGIT)
- KM—30.0 (LJ), 2.5 (MIGIT)
- AM—1.0 (MIGIT), LJ was not used
- CS—30.0 (LJ), MIGIT was not used
- CAP—30.0 (LJ), 2.5 (MIGIT)
- PAS—1.0 (LJ), MIGIT was not used
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report 2019; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Yablonsky, P.; Mushkin, A.; Belilovsky, E.; Galkin, V. Extrapulmonary tuberculosis. Indian J. Med Res. 2004, 120, 316–353. [Google Scholar]
- Chernyaeva, E.; Rotkevich, M.; Krasheninnikova, K.; Yurchenko, A.; Vyazovaya, A.; Mokrousov, I.; Solovieva, N.; Zhuravlev, V.; Yablonsky, P.; O’Brien, S.J. Whole-Genome Analysis ofMycobacterium tuberculosisfrom Patients with Tuberculous Spondylitis, Russia. Emerg. Infect. Dis. 2018, 24, 579–583. [Google Scholar] [CrossRef] [Green Version]
- Benavente, E.D.; Coll, F.; Furnham, N.; McNerney, R.; Glynn, J.R.; Campino, S.; Pain, A.; Mohareb, F.; Clark, T.G. PhyTB: Phylogenetic tree visualisation and sample positioning for M. tuberculosis. BMC Bioinform. 2015, 16, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Q.-Q.; Liu, H.-C.; Jiao, W.-W.; Li, Q.-J.; Han, R.; Tian, J.-L.; Liu, Z.-G.; Zhao, X.-Q.; Li, Y.-J.; Wan, K.-L.; et al. Evolutionary History and Ongoing Transmission of Phylogenetic Sublineages of Mycobacterium tuberculosis Beijing Genotype in China. Sci. Rep. 2016, 6, 34353. [Google Scholar] [CrossRef] [PubMed]
- Mokrousov, I. Insights into the Origin, Emergence, and Current Spread of a Successful Russian Clone of Mycobacterium tuberculosis. Clin. Microbiol. Rev. 2013, 26, 342–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shitikov, E.; Kolchenko, S.; Mokrousov, I.; Bespyatykh, J.; Ischenko, D.; Ilina, E.; Govorun, V. Evolutionary pathway analysis and unified classification of East Asian lineage of Mycobacterium tuberculosis. Sci. Rep. 2017, 7, 9227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. What Is Multidrug-Resistant Tuberculosis (MDR-TB) and How Do We Control It; World Health Organization: Geneva, Switzerland, 2018; Available online: https://www.who.int/news-room/q-a-detail/what-is-multidrug-resistant-tuberculosis-(mdr-tb)-and-how-do-we-control-it (accessed on 11 October 2020).
- WHO. Drug-Resistant TB: XDR-TB FAQ. Available online: https://www.who.int/tb/areas-of-work/drug-resistant-tb/xdr-tb-faq/en/ (accessed on 11 October 2020).
- Zhang, Y.; Yew, W.-W. Mechanisms of drug resistance in Mycobacterium tuberculosis: Update 2015. Int. J. Tuberc. Lung Dis. 2015, 19, 1276–1289. [Google Scholar] [CrossRef]
- Reeves, A.Z.; Campbell, P.J.; Sultana, R.; Malik, S.; Murray, M.; Plikaytis, B.B.; Shinnick, T.M.; Posey, J.E. Aminoglycoside Cross-Resistance in Mycobacterium tuberculosis Due to Mutations in the 5′ Untranslated Region ofwhiB7. Antimicrob. Agents Chemother. 2013, 57, 1857–1865. [Google Scholar] [CrossRef] [Green Version]
- Yakrus, M.A.; Driscoll, J.; McAlister, A.; Sikes, D.; Hartline, D.; Metchock, B.; Starks, A.M. Molecular and Growth-Based Drug Susceptibility Testing ofMycobacterium tuberculosisComplex for Ethambutol Resistance in the United States. Tuberc. Res. Treat. 2016, 2016, 3404860. [Google Scholar] [CrossRef] [Green Version]
- Yadon, A.N.; Maharaj, K.; Adamson, J.H.; Lai, Y.-P.; Sacchettini, J.C.; Ioerger, T.R.; Rubin, E.J.; Pym, A.S. A comprehensive characterization of PncA polymorphisms that confer resistance to pyrazinamide. Nat. Commun. 2017, 8, 588. [Google Scholar] [CrossRef]
- Vyazovaya, A.A.; Mokrousov, I.; Solovieva, N.; Mushkin, A.; Manicheva, O.; Vishnevsky, B.; Zhuravlev, V.; Narvskaya, O. Tuberculous Spondylitis in Russia and Prominent Role of Multidrug-Resistant Clone Mycobacterium tuberculosis Beijing B0/W148. Antimicrob. Agents Chemother. 2015, 59, 2349–2357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chernyaeva, E.; Fedorova, E.; Zhemkova, G.; Korneev, Y.; Kozlov, A. Characterization of multiple and extensively drug resistant Mycobacterium tuberculosis isolates with different ofloxacin-resistance levels. Tuberculosis 2013, 93, 291–295. [Google Scholar] [CrossRef] [PubMed]
- De Steenwinkel, J.E.; Kate, M.T.T.; De Knegt, G.J.; Kremer, K.; Aarnoutse, R.E.; Boeree, M.J.; Verbrugh, H.A.; Van Soolingen, D.; Bakker-Woudenberg, I.A. Drug Susceptibility ofMycobacterium tuberculosisBeijing Genotype and Association with MDR TB. Emerg. Infect. Dis. 2012, 18, 660–663. [Google Scholar] [CrossRef]
- Mokrousov, I. Mycobacterium tuberculosis phylogeography in the context of human migration and pathogen’s pathobiology: Insights from Beijing and Ural families. Tuberculosis 2015, 95, S167–S176. [Google Scholar] [CrossRef] [PubMed]
- Stucki, D.; Brites, D.; Jeljeli, L.; Coscolla, M.; Liu, Q.; Trauner, A.; Fenner, L.; Rutaihwa, L.; Borrell, S.; Luo, T.; et al. Mycobacterium tuberculosis lineage 4 comprises globally distributed and geographically restricted sublineages. Nat. Genet. 2016, 48, 1535–1543. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Chen, J.; Shen, Y.; Jin, J.; Wu, J.; Sun, F.; Wu, Y.; Xie, L.; Zhang, Y.; Zhang, W. Phenotypic and genotypic characterization of pyrazinamide resistance among multidrug-resistant Mycobacterium tuberculosis clinical isolates in Hangzhou, China. Clin. Microbiol. Infect. 2018, 24, 1016.e1–1016.e5. [Google Scholar] [CrossRef] [Green Version]
- Slutsker, L.; Castro, K.G.; Ward, J.W.; Dooley, J.S.W. Epidemiology of Extrapulmonary Tuberculosis among Persons with AIDS in the United States. Clin. Infect. Dis. 1993, 16, 513–518. [Google Scholar] [CrossRef]
- Naing, C.; Mak, J.W.; Maung, M.; Wong, S.F.; Kassim, A.I.B.M. Meta-Analysis: The Association BETWEEN HIV Infection and Extrapulmonary Tuberculosis. Lung 2013, 191, 27–34. [Google Scholar] [CrossRef]
- Qian, X.; Nguyen, D.T.; Lyu, J.; Albers, A.E.; Bi, X.; Graviss, E.A. Graviss, Risk factors for extrapulmonary dis-semination of tuberculosis and associated mortality during treatment for extrapulmonary tuberculosis. Emerg. Microbes Infect. 2018, 7, 102. [Google Scholar] [CrossRef]
- World Health Organization WHO. Guidelines for Surveillance of Drug Resistance in Tuberculosis; WHO: Geneva, Switzerland, 2009. [Google Scholar]
- Van Embden, J.D.; Cave, M.D.; Crawford, J.T.; Dale, J.W.; Eisenach, K.D.; Gicquel, B.; Hermans, P.; Martin, C.; McAdam, R.; Shinnick, T.M. Strain identification of Mycobacterium tuberculosis by DNA fingerprinting: Recommendations for a standardized methodology. J. Clin. Microbiol. 1993, 31, 406–409. [Google Scholar] [CrossRef] [Green Version]
- FastQC. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 16 January 2018).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- BOWTIE2. Available online: http://bowtie-bio.sourceforge.net/bowtie2/index.shtml (accessed on 16 January 2018).
- SAMtools Homepage. Available online: http://samtools.sourceforge.net/ (accessed on 13 March 2012).
- VCFtools. Available online: https://vcftools.github.io/index.html (accessed on 16 January 2018).
- FreeBayes. Available online: https://github.com/ekg/freebayes (accessed on 16 January 2018).
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Xia, E.; Teo, Y.-Y.; Ong, R.T.-H. SpoTyping: Fast and accurate in silico Mycobacterium spoligotyping from sequence reads. Genome Med. 2016, 8, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Commander. Available online: https://socialsciences.mcmaster.ca/jfox/Misc/Rcmdr/ (accessed on 16 January 2018).


{kind=link}
| PTB | XPTB | Total | ||||
|---|---|---|---|---|---|---|
| Genotype | N | % | N | % | N | % |
| Beijing: | 48 | 66.67 | 60 | 82.19 | 108 | 74.48 |
| Beijing unclustered | 16 | 22.22 | 15 | 20.55 | 31 | 21.38 |
| Beijing B0/W148 | 26 | 36.11 | 23 | 31.51 | 49 | 33.79 |
| Beijing Clade A | 3 | 4.17 | 14 | 19.18 | 17 | 11.72 |
| Beijing CAO | 3 | 4.17 | 8 | 10.96 | 11 | 7.59 |
| 4.1 | 5 | 6.94 | 3 | 4.11 | 8 | 5.52 |
| 4.2 | 6 | 8.33 | 4 | 5.48 | 10 | 6.9 |
| 4.3 | 11 | 15.27 | 2 | 2.74 | 13 | 8.97 |
| 4.4 | 1 | 1.39 | 0 | 0 | 1 | 0.69 |
| 4.8 | 1 | 1.39 | 4 | 5.48 | 5 | 3.45 |
| Diagnosis | Susceptible | Mono-Resistant | Poly-Resistant | MDR | XDR | Total |
|---|---|---|---|---|---|---|
| Pulmonary | 12 (16.44) | 3 (4.11) | 9 (12.33) | 44 (60.27) | 5 (6.85) | 73 |
| Extrapulmonary | 7 (9.72) | 2 (2.78) | 9 (12.5) | 30 (41.67) | 24 (33.33) | 72 |
| Total number | 19 (13.10) | 5 (3.45) | 18 (12.41) | 74 (51.03) | 29 (20.00) | 145 |
| Genotype | Susceptible | Mono- Resistant | Poly- Resistant | MDR | XDR | Total Number |
|---|---|---|---|---|---|---|
| Beijing: | 9 | 1 | 11 | 62 | 25 | 108 |
| Beijing unclustered | 8 | 0 | 5 | 12 | 6 | 31 |
| Beijing B0/W148 | 1 | 0 | 3 | 31 | 14 | 49 |
| Beijing A | 0 | 1 | 1 | 11 | 4 | 17 |
| Beijing CAO | 0 | 0 | 2 | 8 | 1 | 11 |
| 4.1 | 4 | 0 | 0 | 3 | 1 | 8 |
| 4.2 | 1 | 2 | 2 | 5 | 0 | 10 |
| 4.3 | 3 | 1 | 4 | 3 | 2 | 13 |
| 4.4 | 0 | 0 | 0 | 0 | 1 | 1 |
| 4.8 | 2 | 1 | 1 | 1 | 0 | 5 |
| Diagnosis | HIV+ | HIV− | N |
|---|---|---|---|
| PTB | 3 | 69 | 72 |
| XPTB | 14 | 41 | 55 |
| Generalized TB | 8 | 10 | 18 |
| Geneinc Clade | HIV Coinfection | Diagnosis | |
|---|---|---|---|
| PTB | XPTB | ||
| 4.8 | + | 0 | 1 |
| − | 1 | 3 | |
| 4.3 | + | 1 | 1 |
| − | 10 | 1 | |
| 4.2 | + | 1 | 0 |
| − | 5 | 4 | |
| 4.1 | + | 0 | 0 |
| − | 5 | 3 | |
| 4.4 | + | 0 | 0 |
| − | 1 | 0 | |
| Beijing unclustered | + | 0 | 6 |
| − | 16 | 9 | |
| Beijing B0/W148 | + | 1 | 10 |
| − | 25 | 13 | |
| Beijing CAO | + | 0 | 1 |
| − | 4 | 7 | |
| Beijing A | + | 0 | 3 |
| − | 2 | 11 | |
| Total | 72 | 73 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chernyaeva, E.; Rotkevich, M.; Krasheninnikova, K.; Lapidus, A.; Polev, D.E.; Solovieva, N.; Zhuravlev, V.; Yablonsky, P.; O’Brien, S.J. Genomic Variations in Drug Resistant Mycobacterium tuberculosis Strains Collected from Patients with Different Localization of Infection. Antibiotics 2021, 10, 27. https://doi.org/10.3390/antibiotics10010027
Chernyaeva E, Rotkevich M, Krasheninnikova K, Lapidus A, Polev DE, Solovieva N, Zhuravlev V, Yablonsky P, O’Brien SJ. Genomic Variations in Drug Resistant Mycobacterium tuberculosis Strains Collected from Patients with Different Localization of Infection. Antibiotics. 2021; 10(1):27. https://doi.org/10.3390/antibiotics10010027
Chicago/Turabian StyleChernyaeva, Ekaterina, Mikhail Rotkevich, Ksenia Krasheninnikova, Alla Lapidus, Dmitrii E. Polev, Natalia Solovieva, Viacheslav Zhuravlev, Piotr Yablonsky, and Stephen J. O’Brien. 2021. "Genomic Variations in Drug Resistant Mycobacterium tuberculosis Strains Collected from Patients with Different Localization of Infection" Antibiotics 10, no. 1: 27. https://doi.org/10.3390/antibiotics10010027