
Phosphorylation and Dephosphorylation of Tau Protein by the Catalytic Subunit of PKA, as Probed by Electrophoretic Mobility Retard
Abstract
Background:
Tau is a microtubule associated protein that regulates the stability of microtubules and the microtubule-dependent axonal transport. Its hyperphosphorylated form is one of the hallmarks of Alzheimer’s disease and other tauopathies and the major component of the paired helical filaments that form the abnormal proteinaceous tangles found in these neurodegenerative diseases. It is generally accepted that the phosphorylation extent of tau is the result of an equilibrium in the activity of protein kinases and phosphatases. Disruption of the balance between both types of enzyme activities has been assumed to be at the origin of tau hyperphosphorylation and the subsequent toxicity and progress of the disease.
Objective:
We explore the possibility that, beside the phosphatase action on phosphorylated tau, the catalytic subunit of PKA catalyzes both tau phosphorylation and also tau dephosphorylation, depending on the ATP/ADP ratio.
Methods:
We use the shift in the relative electrophoretic mobility suffered by different phosphorylated forms of tau, as a sensor of the catalytic action of the enzyme.
Results:
The results are in agreement with the long-known thermodynamic reversibility of the phosphorylation reaction (ATP + Protein = ADP+Phospho-Protein) catalyzed by PKA and many other protein kinases.
Conclusion:
The results contribute to put the compartmentalized energy state of the neuron and the mitochondrial-functions disruption upstream of tau-related pathologies.
INTRODUCTION
Tau is one of the proteins that, associated with microtubules, participates in regulating cellular morphogenesis, axonal transport, and cytoskeleton functionality. Together with the extracellular senile plaques, composed of amyloid-β, tangles of filaments composed mainly of hyperphosphorylated tau protein are the second type of aberrant structures found associated with Alzheimer’s disease and other tauopathies [1–8]. Tau protein contains 85 sites susceptible of phosphorylation on the largest modified CNS tau isoform (441 residues). This number is surprisingly large, especially considering that the number of phosphorylated sites is not as large as would be expected. The cytosolic tau contains about 2 moles of phosphate per mol of protein and, although hyperphosphorylation can be considered as one of the hallmarks of tauopathies, only about 9 to 10 phosphorylated sites per mol of tau are found in the disease protein [2, 9–13]. Different phosphorylation sites can be found at different stages of the disease. A survey of literature about tau phosphorylation leads one to think that hyperphosphorylation and aggregation (probably in this order) must have an important role in neurodegenerative disorders [6, 14, 15].
It is generally accepted that the phosphorylation extent of tau protein is the result of the opposite actions of kinases and phosphatases, and that disruption of this balance is at the origin of the abnormal tau phosphorylation [16–18]. Herein we show the results of exploring the possibility that a protein kinase may also participate in the regulation of the tau phosphorylation by catalyzing the transference of phosphate between ATP and tau, and also the reverse reaction of phosphate transference from phospho-tau (Tau-P) to ADP:
(1)
The first description of the reversibility of a phosphorylation reaction was that of phosvitin, published in 1960 by Rabinowitz and Lipmann [20]. These authors found values of 22 and 58 for the equilibrium constant of the phosphate transference from ATPMg2+ to phosvitin, showing how phosphate was transferred from phosphoprotein to ADP to form ATP. In 1975, the Krebs’s group reported the reversible phosphorylation of lysozyme by the cyclic AMP-dependent Protein Kinase [21]. The average result of several determinations in the forward and the reverse reaction, at pH 6.9 and 30°C, was K = 23.8 for the equilibrium constant expressed as the transfer of phosphate from ATP to the protein. A similar value was reported by El Maghrabi et al. for the reversible phosphorylation of pyruvate kinase by the catalytic subunit of PKA [22]. The phosphorylation of glycogen phosphorylase b by glycogen phosphorylase kinase was also shown to be reversible under experimental conditions preventing phosphorylase from tetramerization [23]. The transfer of phosphate can also occur to an OH group of tyrosine. Phosphorylation of a tyrosine in the Rous-sarcoma-specific immunoglobulin is catalyzed by the pp60src kinase with an equilibrium constant K = 2.6 at pH 6.5 and 30°C [24]. The enzyme associated to membranes phosphatidylinositol kinase catalyzes the transfer of phosphate from ATPMg2+ to yield the phosphorylated form of phosphatidylinositol. This reaction was shown to be reversible with an equilibrium constant the value of which indicated that the reverse reaction of transfer of phosphate to ADP was favored in vitro [25]. Autophosphorylation of tyrosine kinase has been shown to be reversible with an equilibrium constant 3.31 [26]. The insulin receptor kinase contains two classes of tyrosine residues susceptible of being phosphorylated: one of them can be reversibly phosphorylated by the receptor itself, depending on the [ADP]/[ATP] ratio, the other one is irreversible; the equilibrium constant for the reversible phosphorylation of one site has been reported to be 8.7 [27].
The change in electrophoretic mobility suffered by tau protein when it is phosphorylated by C-PKA has been chosen as a sensor for monitoring changes in the phosphorylation state of the protein. The upward shift of electrophoretic mobility of tau, induced by phosphorylation, is known from long ago [28, 29] and has been reported by several groups [18, 30–33]. This phosphorylation-induced change in electrophoretic mobility is not exclusive of tau but is exhibited by many proteins after being phosphorylated by different kinases [33]. The membrane protein phospholamban changes its electrophoretic mobility after phosphorylation by PKA and also by the Ca2+-calmodulin dependent protein kinase [34]. The regulatory subunit of PKA [35], the calmodulin-dependent protein kinase [36] and the glycogen synthase kinase 3 [37] suffer the same kind of upward shift of electrophoretic mobility upon phosphorylation. This phosphorylation-dependent mobility shift has been recently claimed to be common to most eukaryotic proteins [38]. Herein we show how, in the presence of ATPMg2+, the catalytic subunit of PKA catalyzes the upward mobility shift of tau protein and also, in the presence of ADPMg2+, the reverse process of downward mobility shift of that protein, therefore emphasizing the plausible role that the ATP/ADP ratio may have in regulating protein phosphorylation.
MATERIALS AND METHODS
Chemical/reagents
The catalytic subunit of the cAMP-dependent protein kinase (C-PKA) from bovine heart was obtained from Sigma-Aldrich and the purified catalytic subunit from recombinant E. coli was from Promega. P1, P5-Di(adenosine- 5’)pentaphosphate, ATP, ADP and AMP were from Sigma Aldrich. Antibody 7.51, which recognizes tau independently of the extent of phosphorylation, was a gift from Dr Wischik [39]. Antibody AT8 was from Innogenetics. It recognizes tau phosphorylated at serine 202 and threonine 205 [40]. 12E8 was a gift from Dr P. Seubert, Elan Pharmaceuticals. It recognizes tau phosphorylated at both serine 262 and 356 [41].
Tau purification and characterization
The longest human tau isoform, htau 42 clone, was kindly provided by M. Goedert (MRC, Cambridge, UK). Tau purification protocol is based on the procedure described by Lindwall [42] with some modifications. Htau 42 was expressed in E. coli BL21 cells and was induced by adding IPTG to a final concentration of 0.4 mM for 2 h. After centrifugation, the pellet was suspended in 0.1 M Mes, 2 mM EGTA, 0.5 mM MgCl2, pH 6.7, containing 1 mM Phenylmethylsulfonyl fluoride, 0.5 M NaCl and 5 mM of 2-mercaptoethanol and then sonicated in ice. Bacterial lysates were centrifuged at 13,000 rpm for 10 min using a Beckman JA rotor. The supernatant obtained was boiled for 10 min and, after keeping it on ice for an additional 5 min, centrifuged again at 13,000 rpm for 10 min, using the Beckman JA rotor. This supernatant was then adjusted to pH 11 by adding Tris 1 M. A saturated solution of (NH4)2SO4 was added to the supernatant to a final concentration of 50% and kept at 4° C from 1 h to O/N. After centrifugation for 1 h at 13,000 rpm using the Beckman JA rotor, the ammonium sulphate pellet was suspended in 0.1 M Mes, 2 mM EGTA, 0.5 mM MgCl2, pH 6.7 or PBS. Purified tau was incubated with RNAsa (1 μl of RNAsa per ml of the purified tau) for 30 min at 37 °C with stirring at 300 rpm. After boiling for 1 min, the tau solution was centrifuged at 14,000 rpm and 4°C. The supernatant containing the tau solution was then stored at –20°C.
In some experiments, a C-PKA-Tau mixture was prepared by diluting 20 μl of C-PKA from PROMEGA and 1 ml of Ampicillin 100 mg/ml into 1 ml of a tau solution at 0.03 mg/ml. This mixture was then dialyzed O/N against two successive liters of a buffer composed of potassium phosphate 250 mM, 2-mercaptoethanol 1.4 mM at pH 7 in the presence of protease inhibitors. This dialyzed solution was then aliquoted and stored at –20°C.
The phosphorylated form of tau used in dephosphorylation experiments was prepared by incubation at 30–35°C of a reaction mixture composed of 400 μl of tau, 400 μl of MgCl2 50 mM, 200 μl of EGTA 2.5 mM, 20 μl of ATP 100 mM, 775 μl of Hepes buffer (10 mM Hepes, 0.1 M NaCl, pH 7), and 205 μl of a PKA-C solution composed of 5 μl of recombinant C-PKA from PROMEGA and 200 μl of Hepes-ME buffer (20 μl of 2.5 μM 2-mercaptoethanol plus 180 μl of Hepes-buffer). After 6–8 h, the mixture was boiled for 15 min and dialyzed O/N against 2 liters of Hepes buffer containing proteases inhibitors. The dialyzed solution was then stored at –20°C. In some cases, 0.1 mg/ml of Ampicillin was included in the dialysis buffer. Tau protein concentration was estimated by densitometry of the silver stained bands, using the ImageJ software [43], and taking bovine serum albumin as a reference.
Electrophoresis
The different forms of tau were analyzed by electrophoresis on 8% of sodium dodecyl sulphate–polyacrylamide gel (SDS-PAGE). The electrophoretic bands were visualized by Coomassie blue or silver stained and quantified by using the software ImageJ [43]. Scanning of the electrophoresis bands was carried out after subtracting background with a rolling ball radius of 50 pixels (except where indicated). The height of the scanning rectangle was the same in all cases, so that all curves are comparable on the x-axis. The y-axis was normalized to 100%. Except where indicated, original gel photos were used, without photographic treatment. Identification of phosphorylated residues was made by western blots developed with antibodies 7.51, AT8, and 12E8, after electrophoresis and transference to nitrocellulose membranes. Incubation was made O/N at 4°C in PBS containing 0.1% Tween and 5% of non-fat milk. Incubation with secondary antibodies (mouse 800) was for 1 h at room temperature. Odyssey was used for visualizing tau bands.
Reversion of the phosphorylated form of tau, prepared as described in the previous section, was observed in seven experiments out of a total number of nine incubations for long periods of time, in which the ADP conversion into ATP and the bacterial degradation was avoided. Reversion of the tau phosphorylation, using the C-PKA-Tau mixture prepared as described in the previous section, was observed in four experiments out of a total of five in which the absence of ADP conversion into ATP could be avoided. The experiments aimed to obtain the equilibrium constants were done twice (Figs. 7 and 8) at two different concentrations of ATP.
RESULTS
Electrophoretic mobility (EM) retard of tau protein upon phosphorylation
Figure 1A shows the Coomassie blue stained of tau protein separated by SDS-PAGE, after 4 h of incubation at 37°C in the absence (lane 1) and the presence of ATP and C-PKA (lane 2). As can be observed, the incubation with ATPMg2+ and C-PKA causes a delay in the electrophoresis mobility. Figure 1B, C, and D show the corresponding western blots using antibodies to total tau (7.51), Ser 202 and Thr 205 (AT8), and Ser 262 and 356 (12E8) respectively, therefore confirming that the electrophoretic retard caused by incubation with ATPMg2+ and C-PKA is related to serine phosphorylation catalyzed by C-PKA. Figure 1E shows the silver stained of differently phosphorylated forms of tau protein. Lanes 2 and 5 correspond to the unphosphorylated and phosphorylated forms of tau protein, respectively. As can be observed, phosphorylation induces an upward mobility shift of tau. Lanes 3 and 4 show a time-dependent generation of bands showing a retarded mobility. Three bands can be distinguished as an intermediate (lanes 3 and 4) step between the initial unphosphorylated tau (lane 2) and the final state of phosphorylated tau protein (lane 5). As can be observed in Fig. 2, tau phosphorylation, as probed by the retained electrophoretic mobility, was dependent on the incubation time with ATP, and also on the ATP concentration.
Fig. 1
SDS-PAGE (8% of polyacrylamide) of different forms of tau protein. A) Lane 1, tau protein (0.05 mg/ml) incubated for 4 h at 37°C in the presence of MgCl2 (10 mM), EGTA (0.25 mM), 2-mercaptoethanol (0.25 mM), Hepes (5 mM), and NaCl (50 mM), pH 7. Lane 2, the same tau protein incubation as Lane 1, but in the presence of ATP (2 mM) and C-PKA (4 units). B-D) Tau protein incubated as in (A) showing the western blots using antibody 7.51 to total tau (B), AT8 to Ser 202 and Thr 205 (C), and 12E8 to Ser 262 and Ser 356 (D). E) Silver stained of different forms of tau protein. Lane 1, Albumin used as a control. Lane 2, tau protein. Lanes 3 and 4, tau protein (3.6 ng/μl) incubated with ATP (5 mM) and C-PKA for 2.5 h (lane 3) and 17 h (lane 4). Lane 5 shows the phosphorylated tau protein prepared as described in the Materials and Methods. The amounts of tau and albumin loaded in lanes 1 and 2 were approximately 70 and 60 ng respectively. F) shows tau protein (0.05 mg/ml) incubated for 1 h with ATP 0.1 mM in the absence (lane 2) or the presence of C-PKA (lane 1). Lane 3 shows a mixture of bovine serum albumin and ovalbumin used as controls.
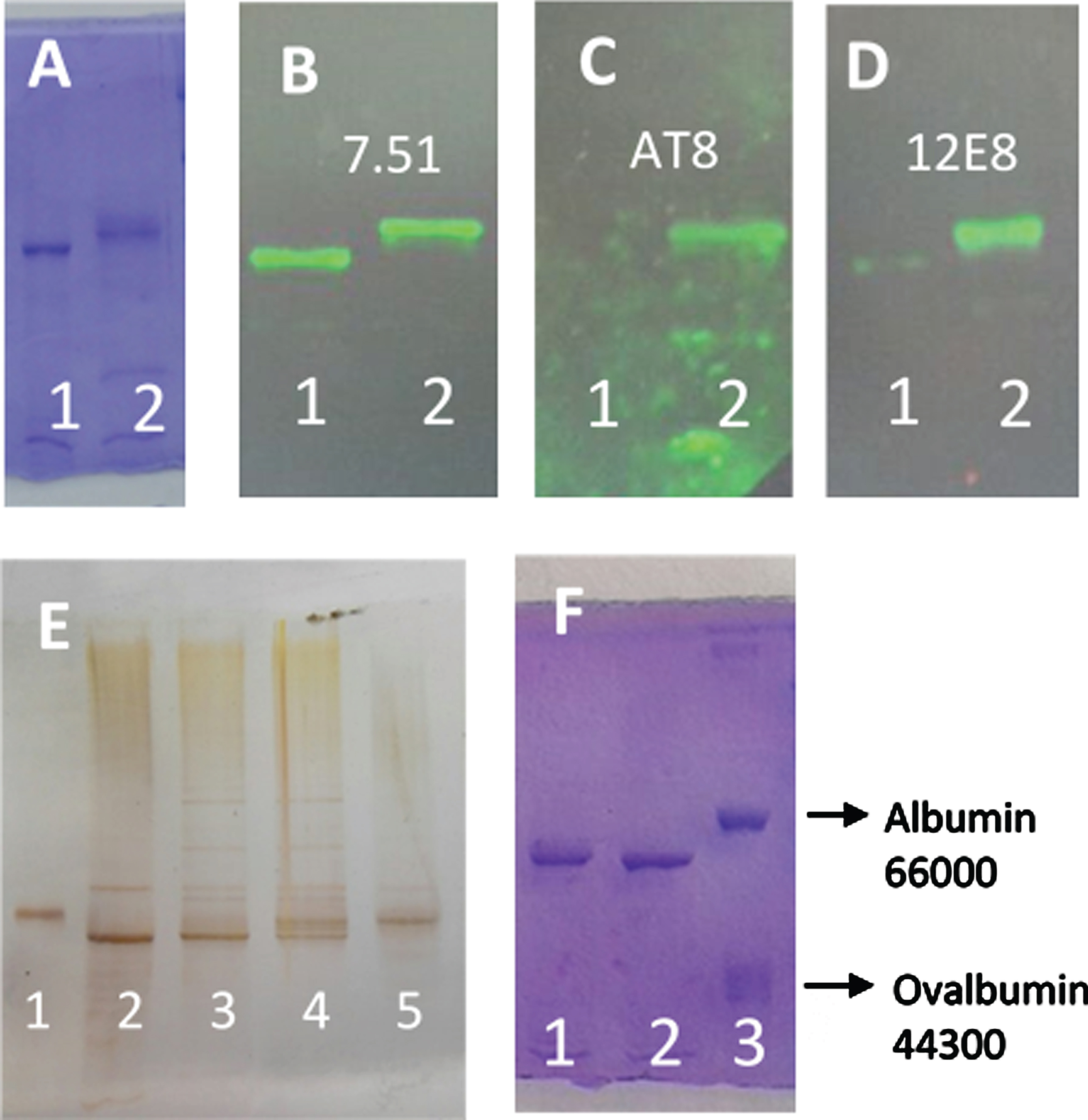
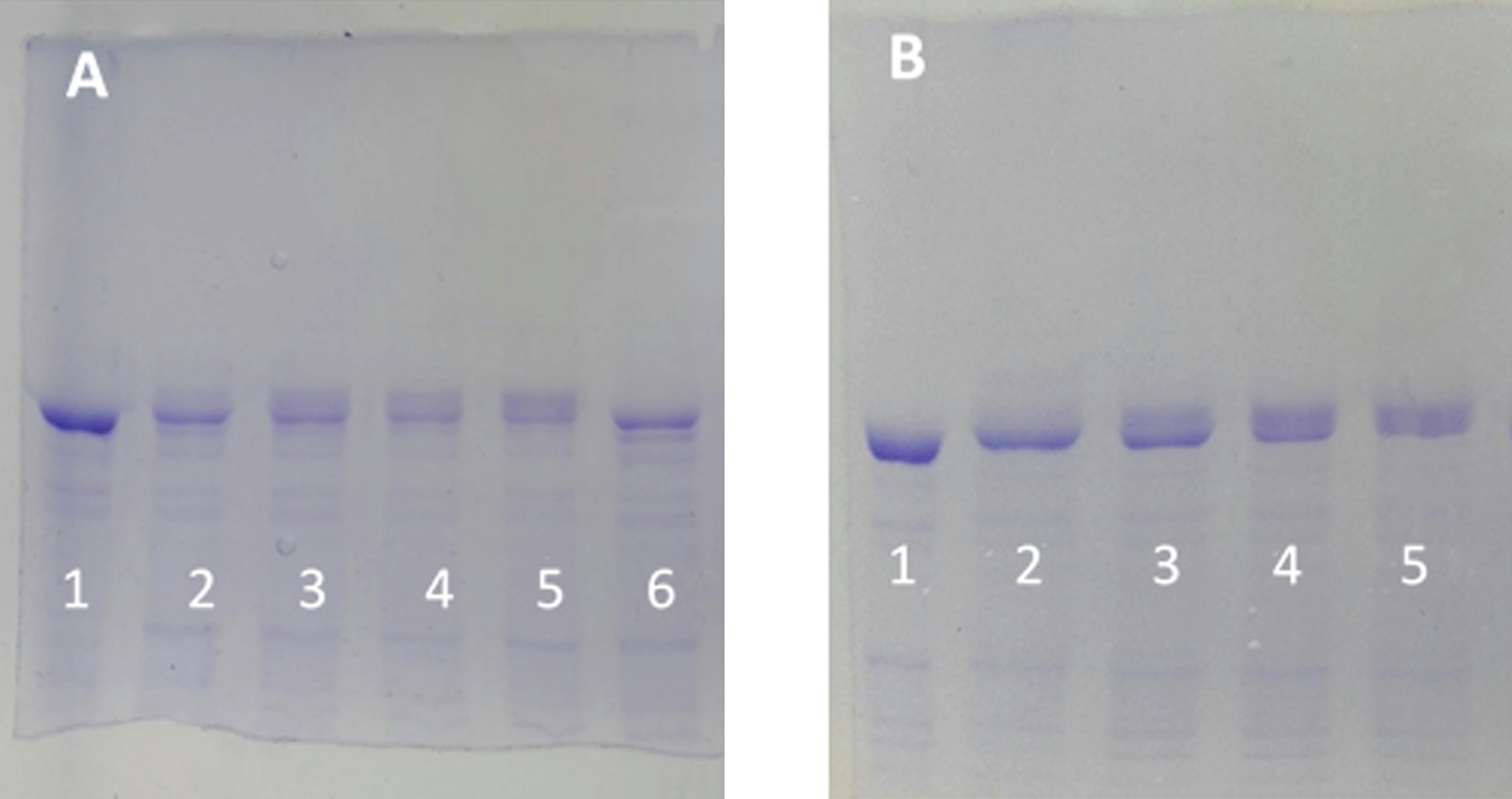
Fig. 2
Electrophoretic mobility retard of tau protein as a function of the ATP concentration and as a function of time. A) SDS-PAGE (8% of polyacrylamide) of tau protein (0.05 mg/ml, in a buffer composed of 10 mM Hepes, 0.1 M NaCl, 10 mM MgCl2, 0.25 mM EGTA, and 0.25 mM 2-mercaptoethanol 10 mM at pH 7) in the absence of ATP and C-PKA (Lane 1). The rest of the lanes correspond to the same tau protein at 0.05 mg/ml incubated at 37°C for 2 h in the presence of the same buffer as in lane 1 but in the presence of 3.2 units of C-PKA and ATP 0.01 mM (lane 2), ATP 0.05 mM (lane 3), ATP 0.1 mM (lane 4), ATP 0.5 mM (lane 5), and no ATP (lane 6). B) Time dependence of the mobility retard. Tau protein as in (A), incubated with 0.1 mM of ATP at time 0 (lane 1), 30 min (lane 2), 1.5 h (lane 3), 3 h (lane 4), and 4 h (lane 5).
Reversibility of the EM retard of phosphorylated tau protein. Experiments with phosphorylated tau, as described under Materials and Methods
The catalytic subunit of PKA catalyzes the reversion of the EM retard of tau protein induced by phosphorylation. As can be observed in Fig. 3, phosphorylated tau (TauP) suffers an electrophoretic retard (Lane 1, TauPo) as compared with the electrophoretic mobility of tau (Lane 6, Tauo); incubation of TauP with C-PKA and ADPMg2+ for 47 and 67 h at 20°C (lanes 4 and 3) produces the partial reversion of the electrophoretic mobility of phosphorylated tau (compare scanning of bands in lanes 1, 4 and 3). Scanning of lanes 4 and 3 show bands with the EM corresponding to unphosphorylated tau. The incubation of TauP with the same nucleotide solution in the absence of C-PKA (lane 2, TauP67) does not alter the EM of TauP, which shows essentially the same EM than TauP in the presence of the same mixture of nucleotides and enzyme, but without incubation (time zero) (lane 1). These results strongly suggest that C-PKA catalyzes the TauP dephosphorylation by ADPMg2+.
Fig. 3
Silver stained of tau and phosphorylated tau separated by SDS-PAGE (8% acrylamide). Lanes 5–8: Tau protein (2.4 μg/ml final concentration) was diluted in a buffer composed of 31 μl of 10 mM Hepes, 0.15 M KCl, 0.25 mM 2-mercaptoethanol, 15 mM MgCl2, and 0.1 mg/ml Ampicillin at pH 6 adjusted with KOH; plus 10 μl of 10 mM Hepes, 0.15 M NaCl at pH 7; plus 5 μl of a solution of 250 mM potassium phosphate 10 mM and 0.1 mg/ml of Ampicillin at pH 7, containing 10 mM of P1, P5-Di(adenosine-5’) pentaphosphate. This solution was incubated at 20°C in the presence of 25 mM of AMP and 2.5 mM ATP for 47 h (lane 8) and 67 h (lane 7) or 2.5 mM ADP for 67 h (lane 5) and 4 units of C-PKA (Promega); lane 6 is a control in the absence of C-PKA, without incubation. Lanes 1–4: Phosphorylated tau (TauP) (1.8 μg/ml, final concentration) was diluted similarly to tau and incubated at 20°C with 2.5 mM ADP and 4 units of C-PKA for 47 h (lane 4) and 67 h (lane 3); dephosphorylation percentages of TauP are indicated. Lane 1 is a control in the absence of C-PKA, without incubation. Lane 2 is a control of TauP incubated for 67 h in the presence of ADP but in the absence of C-PKA. Tau and TauP samples were boiled before use.
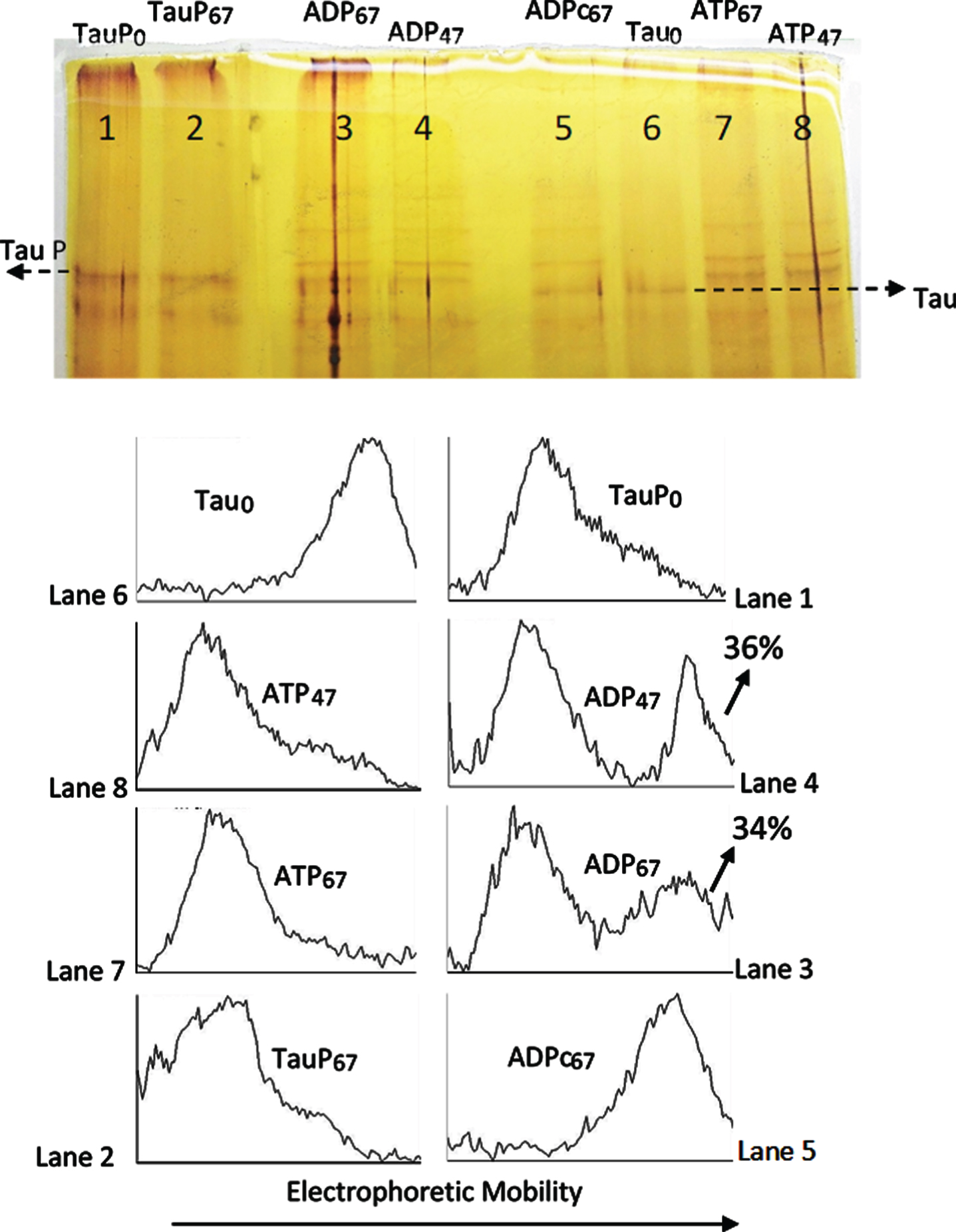
The reversion of the electrophoretic mobility seems to be much slower than the forward reaction. Lanes 8 and 7 of Fig. 3 show the upward mobility shift of tau after incubation of the protein with ATPMg2+ and C-PKA, under the same conditions as those use for the reverse process, except for the presence of ATP instead of ADP. As can be observed, the EM retard after 47 h of incubation is the same as after 67 h, and in both cases the conversion of tau into TauP seems to be complete, therefore suggesting that tau phosphorylation, as observed by the EM retard, has reached the observable maximum. However, the reverse reaction hardly takes about 50% of the original TauP, after 67 h of incubation with ADPMg2+ and C-PKA. It seems that if C-PKA catalyzes the dephosphorylation reaction of Tau-P, the equilibrium constant in the forward (phosphorylation) direction must be very large. It would mean that only under conditions of a high excess of ADP over the ATP concentration could the TauP dephosphorylation by C-PKA be relevant.
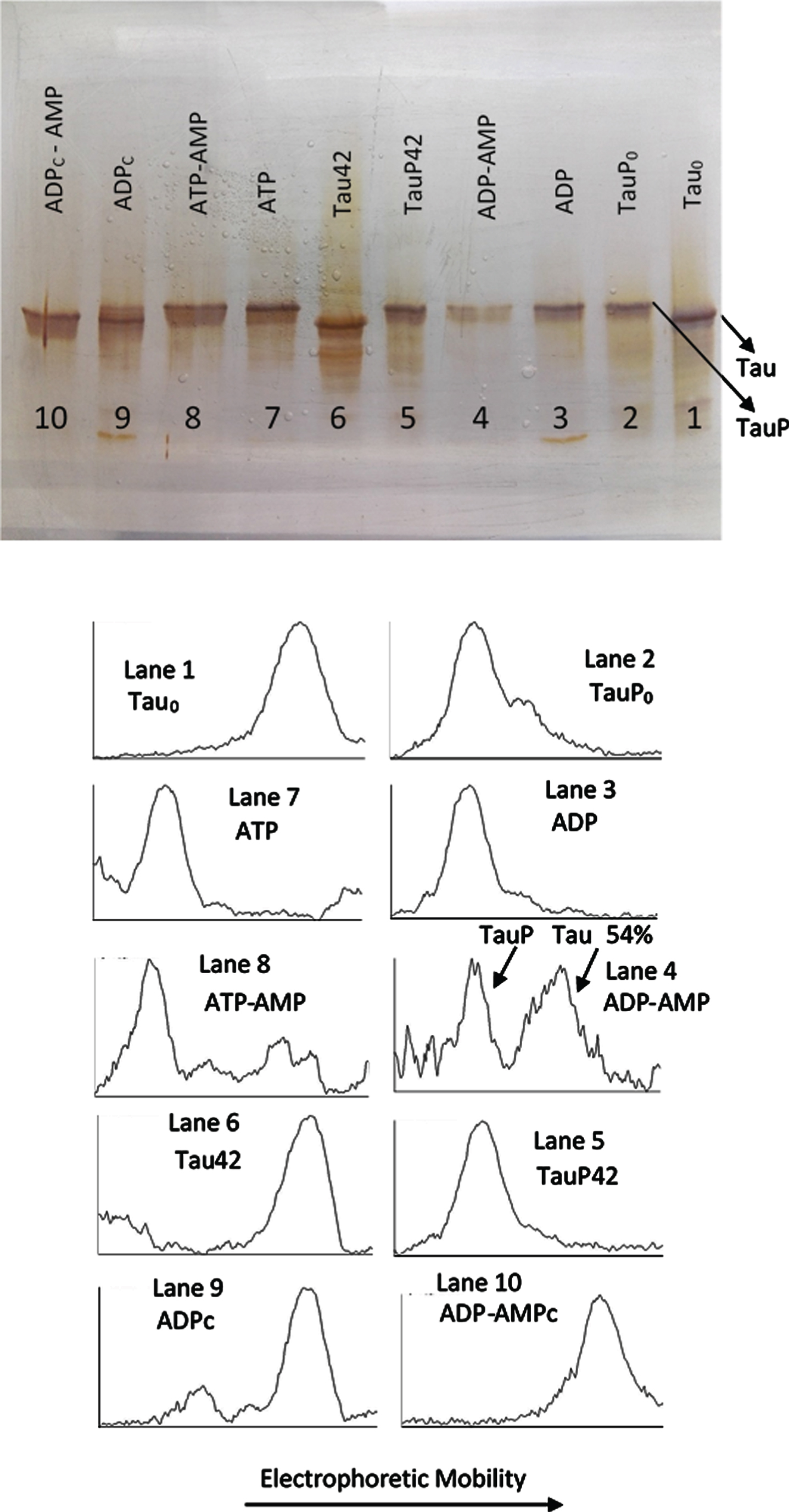
The experiment described under Fig. 3 required from the presence of AMP so that the C-PKA catalyzed reversion of the EM retard could be observed. In a similar experiment showed in Fig. 4, it can be seen how, in the absence of AMP (Lane 3), incubation with ADPMg2 + and C-PKA does not affect the EM of TauP. Lane 4 shows how the EM of TauP is, however, reversed in the presence of AMP, in a similar extent to that shown in Fig. 3. Although some inhibition of the C-PKA activity is observed in the presence of AMP, under the conditions of these experiments, the presence of AMP does not seem to significantly affect the activity of the enzyme, as can be deduced from the results showed in lanes 7 and 8: the EM retard of tau induced by phosphorylation with ATPMg2 + and C-PKA is essentially the same, independently of the presence of AMP. These results suggest the possibility that C-PKA preparations could contain adenylate kinase as an impurity. This enzyme catalyzes the conversion of two moles of ADP into one mole of ATP and one mole of AMP with an equilibrium constant close to unity [44, 45]. The specific activity of adenylate kinase is at least two orders of magnitude greater than that of C-PKA [21, 44, 46]; therefore, undetectable traces of adenylate kinase would be more than enough to transform ADP into ATP, thus making it difficult to observe the dephosphorylation reaction. The presence of a high concentration of AMP could push the reaction of ATP production back to the ADP synthesis, therefore preventing the formation of ATP from ADP. In agreement with this suggestion, lane 9 shows how the incubation with ADPMg2+ produces the same upward mobility shift of tau as that produced by ATPMg2+. In the presence of AMP this mobility shift is not observed (lane 10). This ATP-like behavior of ADP was repeatedly observed in all experiments carried out in the absence of AMP and/or the adenylate kinase inhibitor, P1, P5-Di(adenosine-5’) pentaphosphate.
Fig. 4
Silver stained of tau and phosphorylated tau separated by SDS-PAGE (8% acrylamide). Lanes 1, 6–10: Tau protein (3 μg/ml final concentration) was diluted in a buffer composed of 22.5 μl of 10 mM Hepes, 0.15 M KCl, 0.25 mM 2-mercaptoethanol, 15 mM MgCl2, and 0.1 mg/ml Ampicillin at pH 7 plus 2.5 μl of a solution of 250 mM potassium phosphate 10 mM and 0.1 mg/ml of Ampicillin at pH 7, containing 10 mM of P1, P5-Di(adenosine-5’) pentaphosphate. This solution was incubated with C-PKA, at 20°C for 42 h, in the presence of 2.5 mM ATP plus the presence (Lane 8) or absence (Lane 7) of 25 mM of AMP and in presence of 2.5 mM ADP plus the presence (Lane 10) or absence (Lane 9) of 25 mM of AMP. Lanes 1 and 6 are controls of tau in the absence of C-PKA and nucleotides, incubated for 0 h (Lane 1) and 42 h (Lane 2). Lanes 2-5: Phosphorylated tau (TauP) (2.7 μg/ml, final concentration) was diluted in the same buffer solution as described for tau and incubated with C-PKA, at 20°C for 42 h, in the presence of 2.5 mM ADP plus the presence (Lane 4) or absence (Lane 3) of 25 mM of AMP; dephosphorylation percentage of TauP is indicated. Lanes 2 and 5 are controls of TauP in the presence of 2.5 mM ADP but in the absence of C-PKA, incubated for 0 h (Lane 2) and 42 h (Lane 5). 5 units of C-PKA from Promega were used, when added. Both tau and TauP solutions were boiled before used.
Reversibility of the EM retard of phosphorylated tau protein. Experiments with freshly preparations of partially phosphorylated tau
The experiments of EM retard reversibility of phosphorylated tau, shown in Figs. 3 and 4, were carried out with tau protein that had been exhaustively phosphorylated at high concentration, at 30°C for 6 h, dialyzed and stored at –20°C. Freshly preparations of partially phosphorylated tau were also used to assay the reversibility of the EM retard (Figs. 5 and 6). Figure 5 shows the ADP- induced reversion of the EM retard of a tau protein solution which had been previously phosphorylated by incubation for 2 h with ATPMg2+. Scanning of lane 4 with ImageJ shows the band of tau at time zero of the incubation period. Scanning of lane 3 allowed to observe the appearance of two bands with less mobility after two hours of incubation of tau with ATPMg2+. When the partially phosphorylated tau corresponding to lane 3 was incubated with ADP for two additional hours, the TauP band had decreased its intensity (lane 2), therefore suggesting the dephosphorylation of tau. Under these experimental conditions, both processes of phosphorylation and the corresponding reversion seem to occur approximately with a similar rate. A similar result is shown in Fig. 6. Lane 1 shows a partially phosphorylated tau after 2 h of incubation with ATP. When this mixture of tau and phosphorylated tau was incubated with ADP for 30 min, the bands corresponding to the retarded phosphorylated tau, TauP and TauPα had decreased their intensity while the band corresponding to the unphosphorylated tau increased it (lane 2).
Fig. 5
Successive incubation of tau protein with ATP and ADP. The mixture of tau protein and C-PKA prepared as described in Materials and Method (Tau-C-PKA) was diluted 1/10 in a buffer composed of 75 μl of buffer Hepes containing 10 mM Hepes and 0.1 M NaCl, at pH 7 and 50 μl of phosphate buffer containing 250 mM phosphate and 0.1 mg/ml Ampicillin at pH 7. This reaction mixture contained 15 mM MgCl2, 0.25 mM EGTA, 50 mM P1, P5-Di(adenosine-5’)pentaphosphate, 0.25 mM 2-mercaptoethanol and ATP 0.1 mM. Lane 4 corresponds to tau protein in this reaction mixture at time 0 of incubation (ATP0). After 2 h of incubation at 30°C, in the presence of ATPMg2+, the reaction mixture was divided in three parts: the first one (ATP1) was loaded in the gel (lane 3); the second one was incubated at 30°C for two additional hours in the presence of 1.1 mM ADP(ATP/ADP) (lane 2); the third one was incubated for the same time, at 30°C, in the presence of Hepes buffer, (ATP/Hepes) (lane 1). Scanning of the electrophoresis bands was carried out after applying a filter to improve the photo quality. The table at the bottom of the figure shows the results of peak percentages resulting from the scanning. TauP represent the band displaying the lowest mobility, and TauPα represents the intermediate band usually observed at short times of incubation. Scanning was done after applying a filter to improve the photo quality.
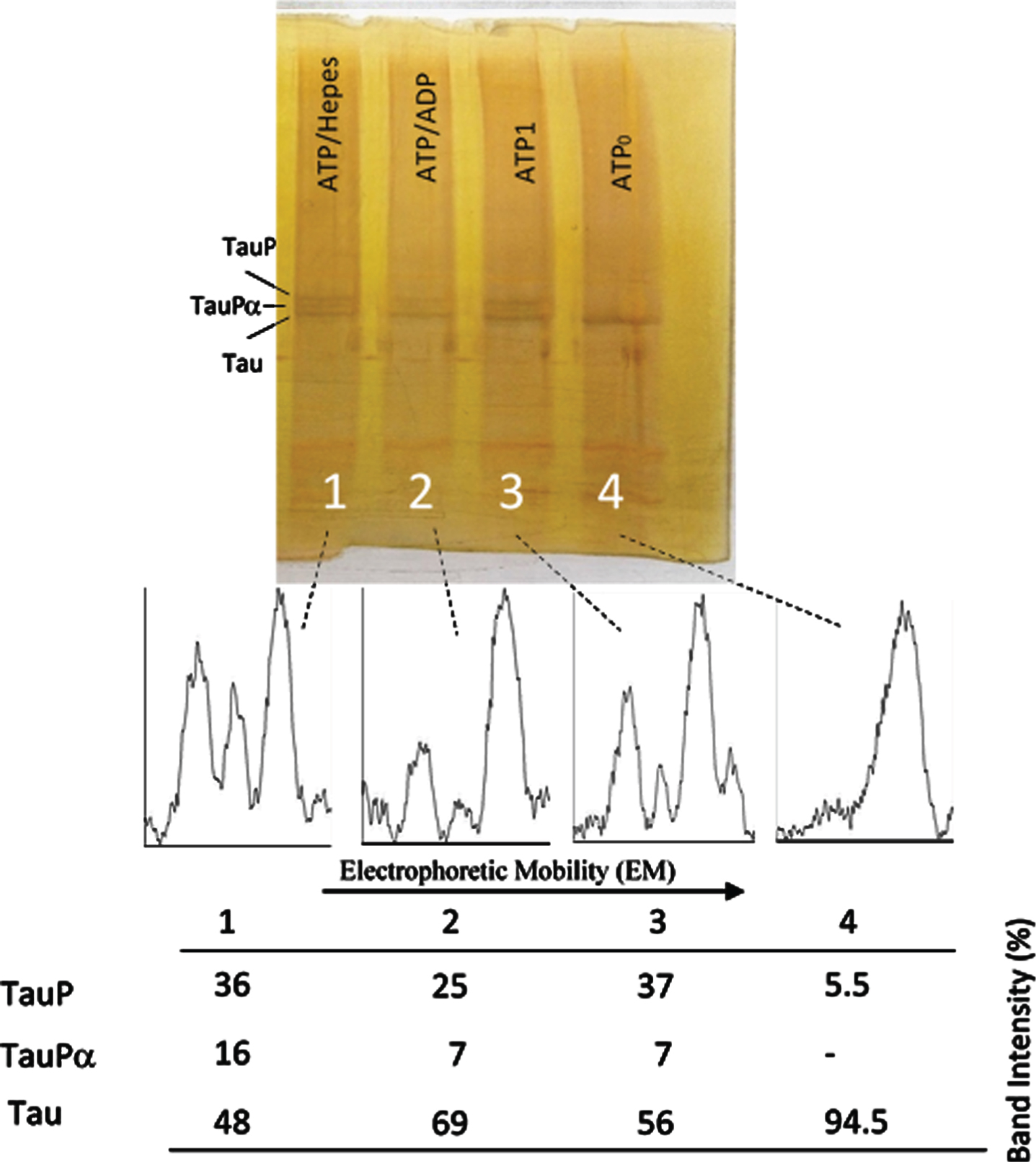
Fig. 6
Successive incubation of tau protein with ATP and ADP. The mixture of tau protein and C-PKA prepared as described in Materials and Method (Tau-C-PKA) was diluted 1/10 in a buffer composed of 75 μl of Hepes buffer containing 10 mM Hepes and 0.1 M NaCl, at pH 7 and 50 μl of phosphate buffer containing 250 mM phosphate and 0.1 mg/ml Ampicillin at pH 7. This reaction mixture contained 15 mM MgCl2, 0.25 mM EGTA, 50 mM P1, P5-Di(adenosine- 5’)pentaphosphate, 0.25 mM 2-mercaptoethanol, 0.05 mg/ml DNA, and ATP 0.1 mM. After 2 h of incubation at 30°C, in the presence of ATPMg2+, the reaction mixture was divided in three parts: the first one (25 μl) (ATP1) was loaded in the gel (lane 1); the second one (22.5 μl) was incubated at 30°C for 30 min after adding 2.5 ml of 10 mM ADP (ATP/ADP) (lane 2); the third one (22.5 μl) was incubated for the same time, at 30°C, after adding 2.5 μl of Hepes buffer containing 10 mM Hepes, 0.1 M NaCl, 0.1 mg/ml DNA, and 100 μM P1, P5-Di(adenosine- 5’)pentaphosphate at pH 7 (lane 3). The Table at the bottom of the figure shows the results of peak percentages after the scanning of the bands. TauP represent the band displaying the lowest mobility, and TauPα represents the intermediate band usually observed at short times of incubation. The rolling ball radius used for subtraction of background was of 5 pixels.
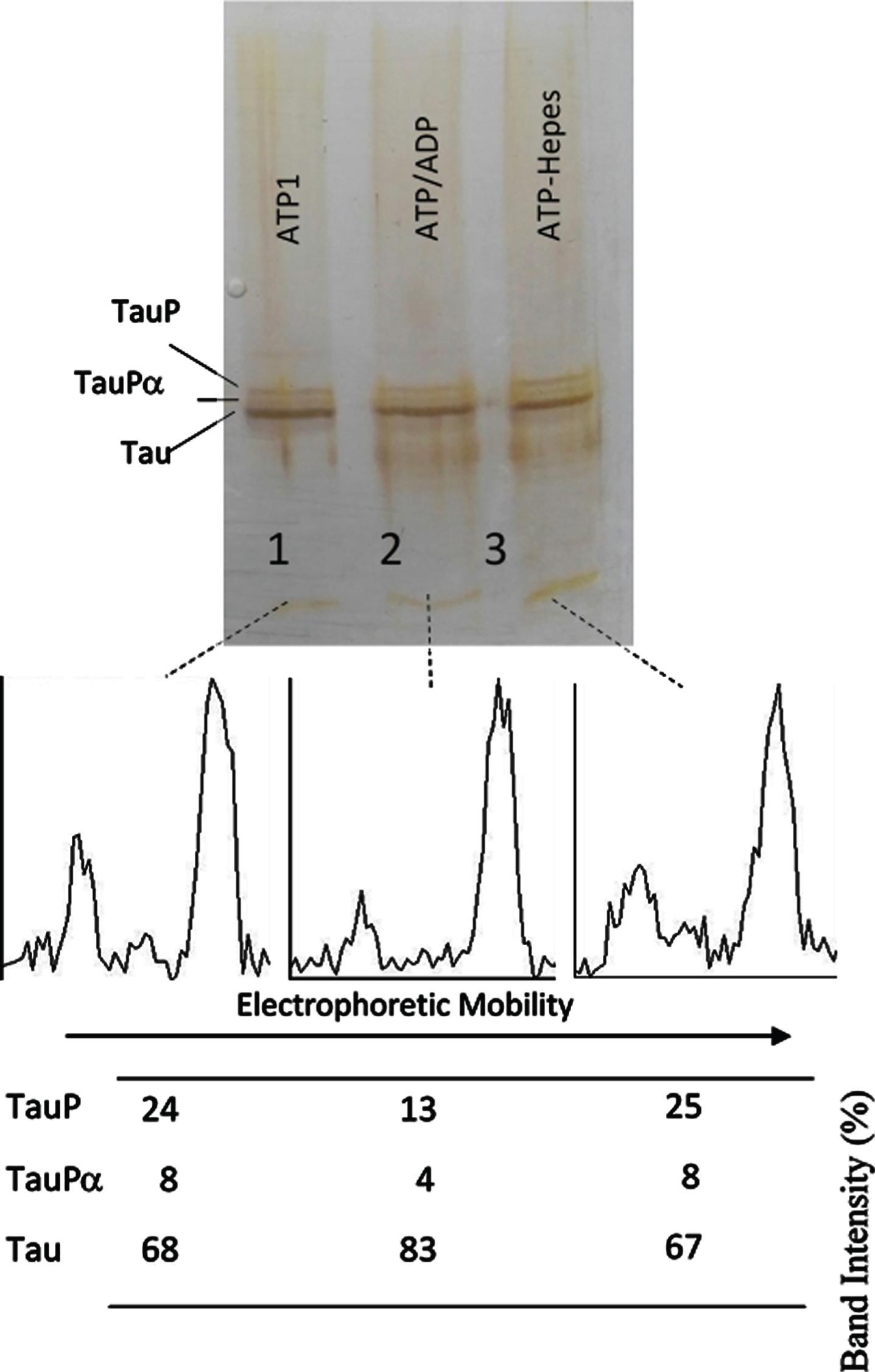
The results shown in Figs. 3 and 4, on the one hand, and Figs. 5 and 6 on the other hand, indicate that the phosphorylated forms of tau showing the maximum EM retard (TauP) are rather poor substrates for C-PKA. Several days of TauP incubation with ADPMg2+ were needed so that the reversion of the EM retard of TauP could be observed, whereas the phosphorylation of tau seems to be complete in hours. However, the reversion of the EM retard of fresh samples having a low percentage of phosphorylation occurred at about the same rate as the EM retard induced by phosphorylation (Figs. 5 and 6). These results suggest the possibility that tau protein may suffer, upon phosphorylation, some conformational change that makes it a worse substrate for the protein kinase.
Fig. 7
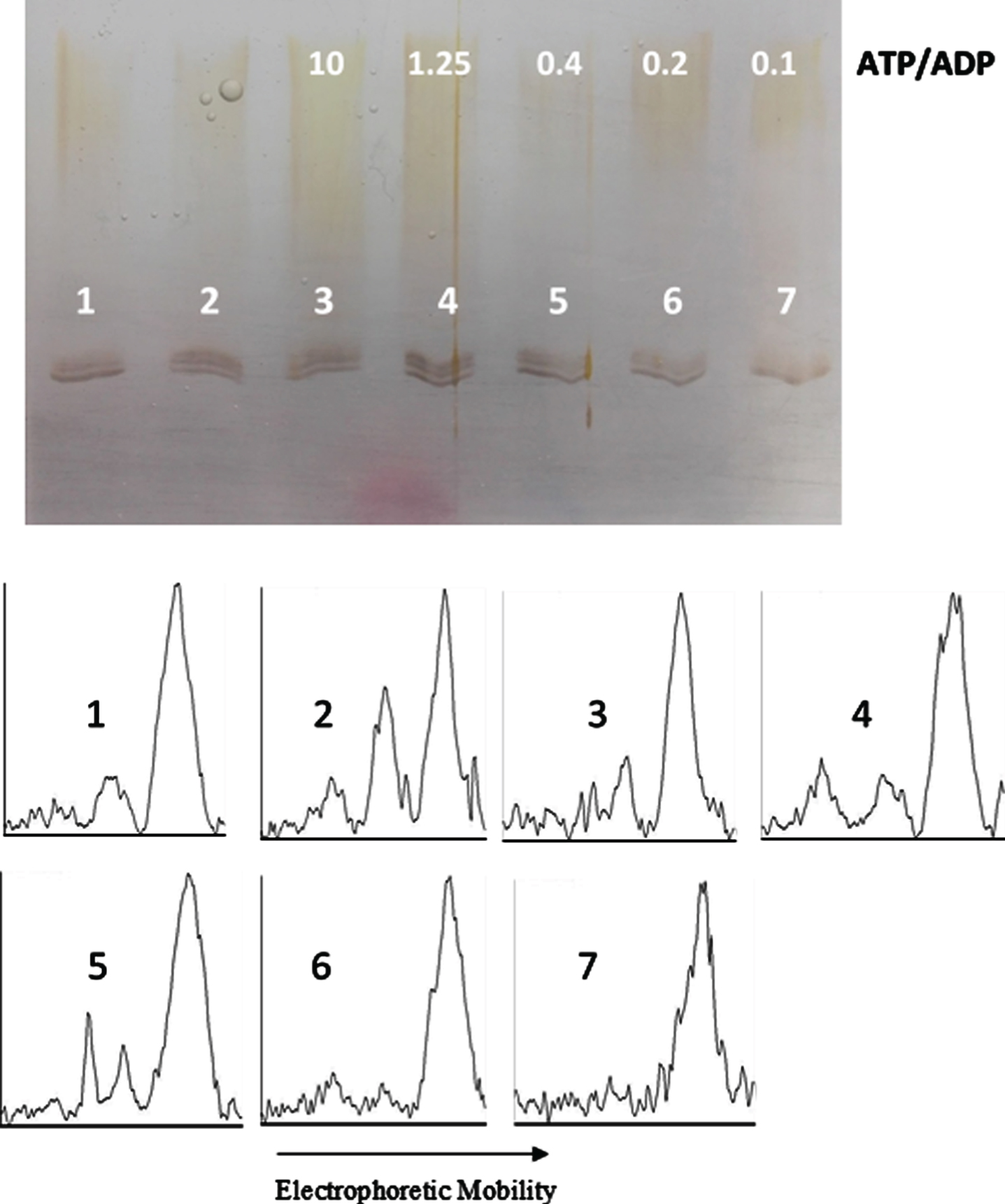
ATP/ADP ratio-dependency of tau protein phosphorylation as probed by electrophoretic mobility. The mixture of tau protein and C-PKA, prepared as described in Materials and Method (Tau-C-PKA) was diluted 1/11.5 in a buffer composed of 15 μl of 10 mM Hepes and 0.1 M NaCl at pH 7, and 10 μl of phosphate buffer containing 250 mM phosphate and 0.1 mg/ml Ampicillin at pH 7. This reaction mixture contained 15 mM MgCl2, 0.25 mM EGTA, 0.25 mM 2-mercaptoethanol, ATP 0.1 mM and the following ADP concentrations (mM): 0 (lane 2), 0.01 (lane 3), 0.08 (lane 4), 0.25 (lane 5), 0.5 (lane 6), and 1 (lane 7). Lane 1 is a control in the absence of nucleotides. All samples were incubated at 20°C for 22 h. Scanning results are included in Table 1. The rolling ball radius used for subtraction of background was of 11 pixels.
Equilibrium constant of tau phosphorylation
In an attempt to find the equilibrium constant of phosphorylation, tau protein was incubated at 20°C with C-PKA and a mixture of nucleotides in an ATP/ADP-ratio dependent manner. In a first experiment, the ADP concentration ranged from 0.01 mM to 1 mM, while the ATP concentration was kept constant at 0.1 mM (Fig. 7). The results of scanning the bands are included in Fig. 9. In a second experiment, the ADP concentration ranged from 0.087 mM to 8.7 mM, while the ATP concentration was kept constant at 0.87 mM (Fig. 8). The incubation time was 22.5 h in the experiment at low ATP concentration and 22 h in the experiment at high ATP concentration. In both experiments, at low and high ATP concentration, the extent of phosphorylation, as proved by scanning the bands with ImageJ, was essentially the same for all the ATP/ADP ratio (Table 1). In a separate experiment, it was found that after 21.5 h of incubation at 20°C in the presence of 0.1 mM ATP and 0.5 mM ADP, the extent of tau phosphorylation did not change for an additional period of 5 h.
Fig. 8
ATP/ADP ratio-dependency of tau protein phosphorylation as probed by electrophoretic mobility. The mixture of tau protein and C-PKA prepared as described in Materials and Method (Tau-C-PKA) was diluted 1/10 in a buffer composed of 35 μl of 10 mM Hepes and 0.1 M NaCl at pH 7, and 23 μl of phosphate buffer containing 250 mM phosphate and 0.1 mg/ml Ampicillin at pH 7. This reaction mixture contained 13 mM MgCl2, 0.22 mM EGTA, 0.25 mM 2-mercaptoethanol, 0.87 mM ATP and the following ADP concentrations (mM): 0 (lane 2), 0.087 (lane 3), 0.7 (lane 4), 2.2 (lane 5), 4.4 (lane 6), and 8.7 (lane 7). Lane 1 is a control in the absence of nucleotides. All samples were incubated at 20°C for 22 h. The rolling ball radius used for subtraction of background was of 30 pixels.
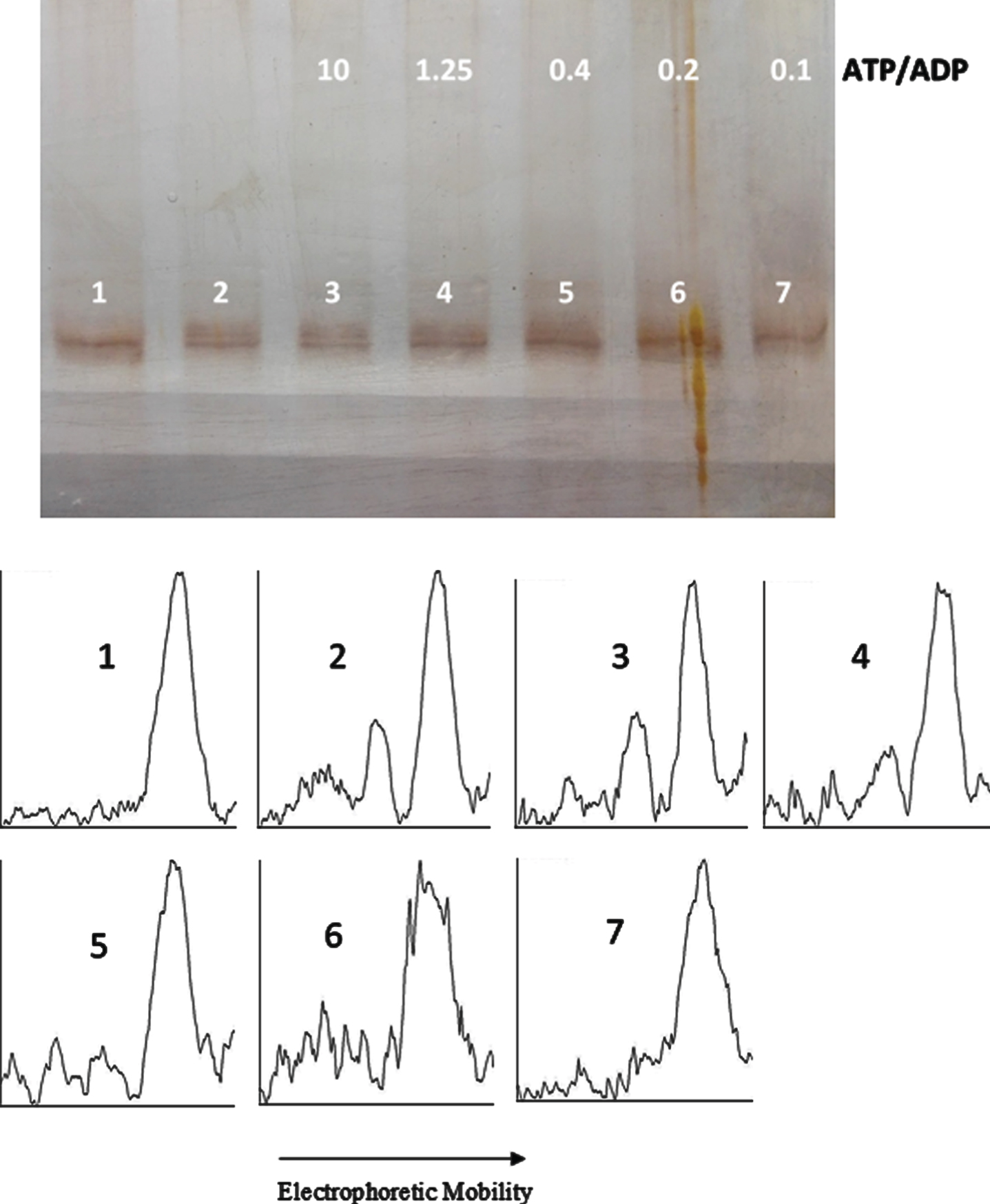
Table 1
Fractional saturation of phosphorylation sites as a function of the [ATP]/[ADP] ratio
| [ATP] = 0.1 mM | [ATP] = 0.87 mM | ||
| [ATP]/[ADP] | Ya | [ATP]/[ADP] | Yb |
| 0.1 | 0.1 | 0.1 | 0.11 |
| 0.2 | 0.17 | 0.2 | 0.22 |
| 0.4 | 0.22 | 0.4 | 0.25 |
| 1.25 | 0.31 | 1.25 | 0.29 |
| 10 | 0.32 | 10 | 0.42 |
a,b Y values obtained as 1 –(IN/(IR+IN)), according to equation (4), from results shown in Figs. 7 (a) and 8 (b).
Phosphorylation data in Figs. 7 and 8 were processed under the following assumptions: a) tau protein becomes phosphorylated according to the following set of successive equilibria:
We can define the fractional saturation of phosphorylation sites on tau, Y, as:
(2)
Following assumption b), fractional saturation, Y, is given by the next rectangular hyperbola:
(3)
IR
(4)
where IR and IN stand for the intensity of the retarded and non-retarded electrophoresis bands, respectively.
Figure 9 shows the data of fractional saturation (obtained from equation (4) and the scanning of bands in Figs. 7 and 8, to obtain IR and IN) represented as a function of the [ATP]/[ADP] ratio. As can be observed, the data do not follow the rectangular hyperbola corresponding to equation (3); rather they fit what would be a negative cooperativity of phosphorylation. It means that phosphorylation of the initial sites may induce a diminution in the equilibrium constant, K, for the next sites.
Fig. 9
Fractional saturation of phosphorylation sites on tau, following the data of Table 1. The fractional saturation as a function of the [ATP]/[ADP] ratio, keeping [ATP] constant at 0.1 mM (part A) or 0.87 mM (part B). The insets show the inverse plots corresponding to equation (5).
![Fractional saturation of phosphorylation sites on tau, following the data of Table 1. The fractional saturation as a function of the [ATP]/[ADP] ratio, keeping [ATP] constant at 0.1 mM (part A) or 0.87 mM (part B). The insets show the inverse plots corresponding to equation (5).](https://content.iospress.com:443/media/jad/2021/79-3/jad-79-3-jad201077/jad-79-jad201077-g009.jpg)
The insets in Fig. 9 show the inverse plots of equation (3):
(5)
The inverse plots at both high and low ATP concentration yields a curve which extrapolates to a straight line at low phosphorylation levels. The microscopic equilibrium constant in both cases is close to 1 (1.1 and 1.2). The phosphorylation reaction seems to be reversible with a microscopic equilibrium constant close to 1 at low level of phosphorylation. However, at high ATP concentration, when the extent of phosphorylation becomes higher, the equilibrium constant in the forward direction of phosphorylation becomes apparently much lower. This result would mean that, once tau protein is phosphorylated, the equilibrium constant for the transfer of phosphate from the protein to ADP to form ATP must be very large. However, the results shown in Figs. 3 and 4 indicate that the action of C-PKA on phosphorylated tau protein is very slow. These results, together with those shown in Fig. 9, strongly suggest that, after reaching some phosphorylated state, tau protein becomes a very poor substrate for C-PKA, obviously for both catalytic processes of phospho- and dephosphorylation, giving place to the apparent negative cooperativity shown in Fig. 9 and to the slow dephosphorylation of phosphorylated tau shown in Figs. 3 and 4.
DISCUSSION
About 25 sites on tau protein are susceptible of phosphorylation by PKA, many of them found in brains of AD (reviewed by Martin et al. [19]). However, just a maximum of 3±1 moles of phosphate per mol of tau protein are reported by Raghunandan and Ingram [33] to be found after incubation of tau with PKA for 4 h. This number is reduced to 1.3 moles per mol of tau when the protein substrate is previously incubated with the proline-directed kinase Erk2. The reverse effect is also reported [33]; prior phosphorylation of tau protein with PKA reduces the number of moles of phosphate per mol of tau observed after incubation with Erk2 from 14 to 5. We are aware of the lack of experimental proofs of an exact correlation between the extent of phosphorylation and the extent of mobility retard. Nevertheless, the results of Raghunandan and Ingram concerning the number of residues the phosphorylation of which is catalyzed by C-PKA and the observation that pre-phosphorylation of tau usually induces some inhibition of a subsequent phosphorylation suggest that the results shown in Figs. 7–9 are, at least qualitatively, correct and that, according to the phosphorylated forms of tau recognized by AT8 and 12E8, we are probably following the phosphorylation of a reduced number of phosphorylation sites, from about two (Ser 202 or Thr 205 and Ser 262 or Ser 356) to four.
Two different groups have reported NMR spectroscopy results showing conformational changes of tau after phosphorylation of some of the residues the phosphorylation of which induces mobility retard. Antibodies AT8 and 12E8, which were used to confirm that the electrophoretic retard was a consequence of tau phosphorylation, were very appropriate to detect phosphorylatable residues involved in conformational changes in tau protein. Tau is an intrinsically disordered protein that lacks a well-defined secondary (and tertiary) structure. It has been described as conformed by a collective of long-range interactions [47]. It has been reported by Bibow et al. [48] that conversion of some serine and threonine (between them, Ser202 and Thr205) into glutamic acid to mimic phosphorylation, produces a weakening of those long-range interactions which contribute to the tau conformation. Similarly, a turn conformation has been reported to be stabilized of the AT8 epitope by phosphorylation of Thr205 and the subsequent hydrogen bond formation with Gly207 [49]. The results shown in Figs. 7–9 suggest that phosphorylation of tau by C-PKA can be reverted by the same kinase, displaying an intrinsic equilibrium constant close to 1. This result agrees with those describing the reversibility of the phosphorylation reaction, catalyzed by the same protein kinase which catalyzes the phosphorylation [20–27]. However, at larger extent of phosphorylation, the phosphorylation reaction slows down, probably as a consequence of conformational changes induced by the phosphorylation itself. That this is a kinetic problem is suggested by the observation that the dephosphorylation of phosphorylated tau is catalyzed by C-PKA only very slowly, leading therefore to the conclusion that phosphorylated forms of tau are very poor substrates for PKA, for both the phosphorylation and the dephosphorylation reactions. This agrees with the low stoichiometry of phosphorylation found in AD tau. In the tangles of PHFs associated to AD, tau is found phosphorylated with 8 to 10 moles of phosphate per mol of protein. Indeed, this is hyperphosphorylation as compared with normal tau [2, 8–12]. However, if we consider that this protein contains 85 putative sites of phosphorylation, and that more than 8 different kinases may catalyze the transfer of phosphate from ATP, we have to conclude that, as an average, initial phosphorylation sites must induce conformational changes in the protein, most of the times decreasing the capacity of being phosphorylated (and of course of being dephosphorylated). It is worthy of note, however, that in some cases phosphorylation by one kinase can produce a better substrate for a different kinase, as for example when phosphorylation by PKA facilitates the subsequent phosphorylation by GSK3 [50].
The reversibility of the phosphorylation reaction catalyzed by PKA emphasizes the plausible role that the ATP/ADP ratio may have in regulating protein phosphorylation, in addition to the role of phosphatases, even more if we consider that, according to the existing data [20–27], this reversibility might be extensible to the rest of kinases catalyzing tau phosphorylation. From a thermodynamic point of view, the reaction of transfer of a phosphate group from ATP to an - OH residue (from serine, threonine, or tyrosine) catalyzed by a protein kinase can be considered in general, as the sum of the next two half-reactions:
(6)
(7)
———————————————–
(8)
The first one (reaction (6)) corresponds to the hydrolysis of ATP; the second one (reaction (7)) corresponds to the incorporation of phosphate into a protein. The free energy for the reaction of protein phosphorylation (reaction (8)), ΔG°P, will be
ΔG°P =ΔG°ATP +ΔG°Pr
ΔG°P will be close to zero whenever the equilibrium constant of phosphorylation is close to 1. Therefore ΔG°Pr must be about the same order of magnitude and of opposite sign to the value of hydrolysis of ATP, ΔG°ATP, and, obviously, the free energy of hydrolysis of the phosphoprotein (–ΔG°Pr) will be approximately equal to the free energy of hydrolysis of ATP, ΔG°ATP. It is worth noting that the chemical reaction catalyzed by phosphatases is not exactly the reverse of reaction (8), but the hydrolysis of the phosphoprotein (the inverse of reaction (7)). According to the phosphoryl potentials of phosphoproteins described above, reaction (7) takes place in the presence of phosphatase with an equilibrium constant so large as to evade the control of the ATP/ADP ratio. For example, using the free energy of hydrolysis of ATP [45], the equilibrium constant for the hydrolysis of a phosphoserine catalyzed by phosphatase, at 25°C is: K = exp(- (–32490)/RT) = 5×105
This value means that in the presence of an active phosphatase more than 99.999% of the protein would dephosphorylate independently of the ATP or ADP concentrations, therefore eluding the energetic state of the system. The possibility of reversion by the kinase itself, however, would allow a better regulation of the phosphorylation extent, controlled by the ATP/ADP ratio, and therefore, of the homeostasis mechanisms dependent of the energy situation of the neuron, avoiding the unregulated shift of the equilibria toward the total dephosphorylation, as it would occur under the action of a phosphatase catalysis.
Oxidative stress and impairment of mitochondria are associated to aging and the initial events in neurodegeneration, with the corresponding deprivation of the energy supplied by oxidative phosphorylation, in the form of ATP [51, 52]. Human brain requires about 20% of the body’s energy income and most of this energy is consumed in the synaptic activity, where mitochondria supplies the needed ATP [51]. Beside stabilizing microtubules, tau is involved in the axonal transport of mitochondria in a phosphorylation-dependent manner. In those areas containing dysfunctional mitochondria, the ATP/ADP ratio may be moved to deprivation of ATP; the phosphorylation state of tau may be then altered, due to the reversible action of the different protein kinases, therefore perturbing the axonal transport of mitochondria to the synapse and eventually causing synapse and neuronal loss [53, 54].
Hyperphosphorylation of tau is considered as one of the main hallmarks of tauopathies and in particular of Alzheimer’s disease. Nevertheless, the detriment of ATP associated to the impairment of mitochondria may cause a gain in unphosphorylated forms of tau, the toxic properties of which are also found in different contexts of the tau related diseases: A clear phosphatase-independent hypophosphorylation of tau expressed in mouse P301L tau knockin-in neurons has been reported to be associated to alterations in axonal mitochondrial transport [55]; dephosphorylated forms of tau has been found involved in the propagation of toxicity caused by the spreading of the protein [56–58]; oligomeric forms of tau, both phosphorylated and dephosphorylated have been suggested as pre-filament toxic species [59]. The well-established association of GSK3 phosphorylation at defined sites on tau to detaching of the protein from the microtubules, together with the hyperphosphorylation found on tau associated to PHFs [60, 61] has led to assume phosphorylation as a negative functional regulator of the tau activity. The same GSK3 catalyzes a tau phosphorylation—different of that involved in the interaction with microtubules—that is associated to the anterograde transport of mitochondria. In this case, however, the dephosphorylated tau is the negative regulator, while the phosphorylated form is the positive one [62], emphasizing the idea that it is the phosphorylation state of tau, in different sites and different neuronal functions, the functional regulator. The reversible character of the phosphorylation catalyzed by protein kinases would place the compartmentalized ATP/ADP ratio and the mitochondrial-functions disruption upstream of the tau-related pathologies. The average concentration of ATP in the cell cytosol is within the mM range [63], and the ATP/ADP ratio in brain tissue has been reported to be about 10; however, that ratio can fall to values close to 1 under conditions of ATP depletion as hypoglycemia [64] or ischemia and hypoxia [65] in brain tissue or mouse neuroblastoma cells. Similar results are reported when the adenylate energy charge (AEC), as defined by Atkinson [66] as ([ATP] + ½[ADP])/([ATP] + [ADP] + [AMP]), is used to express the energy status in the cell [65, 67]. As far as we know, there are no data of ATP concentration in the close proximity to synapses. However, the dynamics of mitochondria determines decreasing gradients of ATP in the vicinity of synapses, together with the corresponding increasing gradient of ADP [68]. Therefore, it seems reasonable the assumption that the ATP/ADP ratio may play, especially in those cases of mitochondria disruption, a role in determining the spatial variation of tau phosphorylation.
ACKNOWLEDGMENTS
Financial support has been provided by a grant from the Spanish Government PGC2018_096177_B_100.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/20-1077r1).
REFERENCES
[1] | Weingarten MD , Lockwood AH , Hwo SY , Kirschner MW ((1975) ) A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A 72: , 1858–1862. |
[2] | Grundke-Iqbal I , Iqbal K , Tung YC , Quinlan M , Wisniewski HM , Binder LI ((1986) ) Abnormal phosphorylation of the microtubule-associated protein (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A 83: , 4913–4917. |
[3] | Drubin DG , Kirschner MW ((1986) ) Tau protein function in living cells. J Cell Biol 103: , 2739–2746. |
[4] | Ebneth A , Godemann R , Stamer K , Illenberger S , Trinczek B , Mandelkow E ((1998) ) Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: Implications for Alzheimer’s disease. J Cell Biol 143: , 777–794. |
[5] | Ávila J , Jiménez JS , Sayas CL , Bolós M , Zabala JC , Rivas G , Hernández F ((2016) ) Tau structures. Front Aging Neurosci 8: , 262. |
[6] | Feinstein SC , Wilson L ((2005) ) Inability of tau to properly regulate neuronal microtubule dynamics: A loss-of-function mechanism by which tau might mediate neuronal cell death. Biochim Biophys Acta 1739: , 268–279. |
[7] | Ávila J ((2006) ) Tau phosphorylation and aggregation in Alzheimer’s disease pathology. FEBS Lett 580: , 2922–2927. |
[8] | Takashima A ((2012) ) GSK-3β and memory formation. Front Mol Neurosci 5: , 47. |
[9] | Lee VM , Balin BJ , Otvos L Jr , Trojanowski JQ ((1991) ) A68: A major subunit of paired helical filaments and derivatized forms of normal tau. Science 251: , 675–678. |
[10] | Tepper K , Biernat J , Kumar S , Wegmann S , Timm T , Hübschmann S , Redecke L , Mandelkow EM , Müller DJ , Mandelkow E ((2014) ) Oligomer formation of tau protein hyperphosphorylated in cells. J Biol Chem 289: , 34389–34407. |
[11] | Köpke E. , Tung YC , Shaikh S , Alonso AC , Iqbal K , Grundke-Iqbal I ((1993) ) Microtubule-associated protein tau. Abnormal phosphorylation of a nonpaired helical filament pool in Alzheimer disease. J Biol Chem 268: , 24374–24384. |
[12] | Ksiezak - Reding H , Liu WK , Yen SH ((1992) ) Phosphate analysis and dephosphorylation of modified tau associated with paired helical filaments. Brain Res 597: , 209–219. |
[13] | Hernández F , Ávila J ((2007) ) Tauopathies. Cell Mol Life Sci 64: , 2219–2233. |
[14] | von Bergen M , Barghorn S , Biernat J , Mandelkow EM , Mandelkow E ((2005) ) Tau aggregation is driven by a transition from random coil to beta sheet structure. Biochim Biophys Acta 1739: , 158–166. |
[15] | Kuret J , Congdon EE , Li G , Yin H , Yu X , Zhong Q ((2005) ) Evaluating triggers and enhancers of tau fibrillization. Microsc Res Tech 67: , 141–155. |
[16] | Wang JZ , Gong CX , Zaidi T , Grundke-Iqbal I , Iqbal K ((1995) ) Dephosphorylation of Alzheimer paired helical filaments by protein phosphatase-2A and -2B. J Biol Chem 270: , 4854–4860. |
[17] | Wang JZ , Grundke-Iqbal I , Iqbal K ((1996) ) Restoration of biological activity of Alzheimer abnormally phosphorylated τ by dephosphorylation with protein phosphatase -2A -2B and -1. Mol Brain Res 38: , 200–208. |
[18] | Wang JZ , Grundke-Iqbal I , Iqbal K ((2007) ) Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur J Neurosci 25: , 59–68. |
[19] | Martin L , Latypova X , Wilson CM , Magnaudeix A , Perrin M-L , Yardin C , Terro F ((2013) ) Tau protein kinases: Involvement in Alzheimer’s disease. Ageing Res Rev 12: , 289–309. |
[20] | Rabinowitz M , Lipmann F ((1960) ) Reversible phosphate transfer between yolk phosphoprotein and adenosine triphosphate. J Biol Chem 235: , 143–1050. |
[21] | Shizuta Y , Beavo JA , Bechtel PJ , Hofmann F , Krebs EG ((1975) ) Reversibility of adenosine 3’:5’-monophosphate-dependent protein kinase reactions. J Mol Biol 250: , 6891–6896. |
[22] | El Maghrabi MR , Haston WS , Flockhart DA , Claus TH , Pilkis SJ ((1980) ) Studies on the phosphorylation and dephosphorylation of L-type pyruvate kinase by the catalytic subunit of cyclic AMP-dependent protein kinase. J Biol Chem 255: , 668–675. |
[23] | Shizuta Y , Khandelwal RL , Maller JL , Vandenheede JR , Krebs EG ((1977) ) Reversibility of phosphorylase kinase reaction. J Biol Chem 252: , 3408–3413. |
[24] | Fukami Y , Lipmann F ((1983) ) Reversal of Rous sarcoma-specific immunoglobulin phosphorylation on tyrosine (ADP as phosphate acceptor) catalyzed by the src gene kinase. Proc Natl Acad Sci U S A 80: , 1872–1876. |
[25] | Belunis CJ , Bae-Lee M , Kelley MJ , Carman GM ((1988) ) Purification and characterization of phosphatidylinositol kinase from Saccharomyces cerevisiae. J Biol Chem 263: , 18897–18903. |
[26] | Litwin CME , Gendreau M , Wang JH ((1992) ) p56lyn catalyzes a reversible autophosphorylation reaction and a nucleoside diphosphate kinase reaction. FEBS Lett 309: , 275–278. |
[27] | Argetsinger LS , Shafer JA ((1992) ) The reversible and irreversible autophosphorylations of insulin receptor kinase. J Biol Chem 267: , 22095–22101. |
[28] | Lindwall G , Cole RD ((1984) ) Phosphorylation affects the ability of tau protein to promote microtubule assembly. J Biol Chem 259: , 5301–5305. |
[29] | Baudier J , Cole RD ((1987) ) Phosphorylation of tau proteins to a state like that in Alzheimer’s brain is catalyzed by a calcium/calmodulin-dependent kinase and modulated by phospholipids. J Biol Chem 262: , 17577–17583. |
[30] | Ihara Y , Nukina N , Miura R , Ogawara M ((1986) ) Phosphorylated tau protein is integrated into paired helical filaments in Alzheimer’s disease. J Biochem 99: , 1807–1810. |
[31] | Steiner B , Mandelkow EM , Biernat J , Gustke N , Meyer HE , Schmidt B , Mieskes G , Söling HD , Drechsel D , Kirschner MW , Goedert M , Mandelkow E ((1990) ) Phosphorylation of microtubule-associated protein tau: Identification of the site for Ca2+-calmodulin dependent kinase and relationship with tau phosphorylation in Alzheimer tangles. EMBO J 9: , 3539–3544. |
[32] | Scott CW , Spreen RC , Herman JL , Chow FP , Davison MD , Young J , Caputo CB ((1993) ) Phosphorylation of recombinant tau by cAMP-dependent protein-kinase. J Biol Chem 268: , 1166–1173. |
[33] | Raghunandan R , Ingram VM ((1995) ) Hyperphosphorylation of the cytoskeletal protein tau by the MAP-Kinase PK40erk2: Regulation by prior phosphorylation with cAMP-dependent protein kinase A. Biochem Biophys Res Commun 215: , 1056–1066. |
[34] | Wegener AD , Jones LR ((1984) ) Phosphorylation-induced mobility shift in phospholamban in sodium dodecyl sulfate-polyacrylamide gels. J Biol Chem 259: , 1834–1841. |
[35] | Hofmann F , Beavo JA , Bechtel PJ , Krebs EG ((1975) ) Comparison of adenosine 3’:5’- monophosphate-dependent protein kinases from rabbit skeletal and bovine heart muscle. J Biol Chem 250: , 7795–7801. |
[36] | Ahmad Z , DePaoli-Roach AA , Roach PJ ((1982) ) Purification and characterization of a rabbit liver calmodulin-dependent protein kinase able to phosphorylate glycogen synthase. J Biol Chem 257: , 8348–8355. |
[37] | Hemmings BA , Yellowlees D , Kernohan JC , Cohen P ((1981) ) Purification of glycogen synthase kinase 3 from rabbit skeletal muscle. Eur J Biochem 119: , 443–451. |
[38] | Lee CR. Park YH , Kim YR , Peterkofsky A , Seok YJ ((2013) ) Phosphorylation-dependent mobility shift of proteins on SDS-PAGE is due to decreased binding of SDS. Bull Korean Chem Soc 34: , 2063–2066. |
[39] | Novak M , Jakes R , Edwards PC , Milstein C , Wischik CM ((1991) ) Difference between the tau protein of Alzheimer paired filament core and normal tau revealed by epitope analysis of monoclonal antibodies 423 and 7.51. Proc Natl Acad Sci U S A 88: , 5837–5841. |
[40] | Goedert M , Jakes R , Vanmechelen E ((1995) ) Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci Lett 189: , 167–170. |
[41] | Seubert P , Mawal-Dewan M , Barbour R , Jakes R , Goedert M , Johnson GVW , Litersky JM , Schenk D , Lieberburg I , Trojanowski JQ , Lee VMY ((1995) ) Detection of phosphorylated Ser262 in fetal tau, adult tau, and paired helical filament tau. J Biol Chem 270: , 18917–18922. |
[42] | Lindwall G , Cole RD ((1984) ) The purification of tau protein and the occurrence of two phosphorylation states of tau in brain. J Biol Chem 259: , 12241–12245. |
[43] | Schneider CA , Rasband WS , Eliceiri KW ((2012) ) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: , 671–675. |
[44] | Sheng XR , Li X , Pan XM ((1999) ) An iso-random Bi Bi mechanism for adenylate kinase. J Biol Chem 274: , 22238–22242. |
[45] | Alberty RA , Goldberg RN ((1992) ) standard thermodynamic properties for the adenosine 5’-triphosphate series. Biochemistry 31: , 10610–10615. |
[46] | Jiménez JS , Kupfer A , Gani V , Shaltiel S ((1982) ) Salt-induced conformational changes in the catalytic subunit of adenosine cyclic 3’,5’-phosphate dependent protein kinase. Use for establishing a connection between one sulfhydryl group and the γ-P Subsite in the ATP site of this subunit. Biochemistry 21: , 1623–1630. |
[47] | Mukrasch MD , Bibow S , Korukottu J , Jeganathan S , Biernat J , Griesinger C , Mandelkow E , Zweckstetter M ((2009) ) Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol 7: , 0399–0414. |
[48] | Bibow S , Ozenne V , Biernat J , Blackledge M , Mandelkow E , Zweckstetter M ((2011) ) Structural impact of proline-directed pseudophosphorylation at AT8, AT100, and PHF1 epitopes on 441-residue tau. J Am Chem Soc 133: , 15842–15845. |
[49] | Gandhi NS , Landrieu I , Byrne C , Kukic P , Amniai L , Cantrelle FX , Wieruszeski JM , Mancera RL , Jacquot Y , Lippens G ((2015) ) A phosphorylation-induced turn defines the Alzheimer’s disease AT8 antibody epitope on the tau protein. Angew Chem Int Ed 54: , 6919–6823. |
[50] | Liu SJ , Zhang JY , Li HL , Fang ZY , Wang Q , Deng HM , Gong CX , Grundke-Iqbal I , Iqbal K , Wang JZ ((2004) ) Tau becomes a more favorable substrate for GSK-3 when it is phosphorylated by PKA in rat brain. J Biol Chem 279: , 50078–50088. |
[51] | Pickett EK , Rose J , McCrory C , McKenzie CA , King D , Smith C , Gillingwater TH , Henstridge CM , Spires-Jones TL ((2018) ) Region-specific depletion of synaptic mitochondria in the brains of patients with Alzheimer’s disease. Acta Neuropathol 136: , 747–757. |
[52] | Lejri I , Agapouda A , Grimm A , Eckert A ((2019) ) Mitochondria- and oxidative stress. Targeting substances in cognitive decline-related disorders: From molecular mechanisms to clinical evidence. Oxid Med Cell Longev 2019: , 9695412. |
[53] | Spires-Jones TL , Stoothoff WH , de Calignon A , Jones PB , Hyman BT ((2009) ) Tau pathophysiology in neurodegeneration: A tangled issue. Trends Neurosci 32: , 150–159. |
[54] | Konzack S , Thies E , Marx A , Mandelkow EM , Mandelkow E ((2007) ) Swimming against the tide: Mobility of the microtubule-associated protein tau in neurons. J Neurosci 27: , 9916–9927. |
[55] | Rodríguez-Martín T , Pooler AM , Lau DHW , Mórotz GM , De Vos KJ , Gilley J , Coleman MP , Hanger DP ((2016) ) Reduced number of axonal mitochondria and tau hypophosphorylation in mouse P301L tau knockin neurons. Neurobiol Dis 85: , 1–10. |
[56] | Díaz-Hernández M , Gómez-Ramos A , Rubio A , Gómez-Villafuertes R , Naranjo JR , Miras-Portugal MT , Ávila J ((2010) ) Tissue-nonspecific alkaline phosphatase promotes the neurotoxicity effect of extracellular tau. J Biol Chem 285,: , 32539–32548. |
[57] | Pooler AM , Phillips EC , Lau DHW , Noble W , Hanger DP ((2013) ) Physiological release of endogenous tau is stimulated by neuronal activity. EMBP Rep 14: , 389–394. |
[58] | Gerson JE , Sengupta U , Lasagna-Reeves CA , Guerrero-Múñoz MJ , Troncoso J , Kayed R ((2014) ) Characterization of tau oligomeric seeds in progressive supranuclear palsy. Acta Neuropathol Commun 2: , 1–9. |
[59] | Cowan CM , Quraishe S , Mudher A ((2012) ) What is the pathological significance of tau oligomers. Biochem Soc Trans 40: , 693–697. |
[60] | Drewes G , Ebneth A , Preuss U , Mandelkow EM , Mandelkow E ((1997) ) MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell 89: , 297–308. |
[61] | Schneider A , Biernat J , von Bergen E , Mandelkow E , Mandelkow EM ((1999) ) Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry 38: , 3549–3558. |
[62] | Tatebayashi Y , Haque N , Tung YC , Iqbal K , Grundke-Iqbal I ((2004) ) Role of tau phosphorylation by glycogen synthase kinase-3b in the regulation of organelle transport. J Cell Sci 117: , 1653–1663. |
[63] | Mörikofer-Zwez S , Walter P ((1989) ) Binding of ADP to rat liver cytosolic proteins and its influence on the ratio of free ATP/free ADP. Biochem J 259: , 117–124. |
[64] | Ereciñska M , Silver IA ((1994) ) Ions and energy in mammalian brain. Prog Neurobiol 43: , 37–71. |
[65] | Chen Y , Xing D , Wang W , Ding Y , Du L ((2007) ) Development of an ion-pair HPLC method for investigation of energy charge changes in cerebral ischemia of mice and hypoxia of Neuro-2a cell line. Biomed Chromatogr 21: , 628–634. |
[66] | Atkinson DE ((1968) ) The energy charge of the adenylate pool as a regulatory parameter. Interaction with feedback modifiers. Biochemistry 7: , 4030–4034. |
[67] | De la Fuente IM , Cortés JM , Valero E , Desroches M , Rodrigues S , Malaina I , Martínez L ((2014) ) On the dynamics of the adenylate energy system: Homeorhesis vs homeostasis. PLoS One 9: , e108676. |
[68] | Mironov SL ((2009) ) Complexity of mitochondrial dynamics in neurons and its control by ADP produced during synaptic activity. Cell Biol 41: , 2005–2014. |

