
Abstract
Twenty-four oxidovanadium(IV,V) complexes with tridentate Schiff base ligands based on 5-nitrosalicylaldehyde, 5-methoxysalicylaldehyde, or 5-sulfosalicylaldehyde and respective hydrazide were isolated, and characterized using physicochemical and spectroscopic methods. Three of them were structurally characterized by single-crystal X-ray structure determination. The biological activity studies included inhibition of human tyrosine phosphatases, studies on myocyte C2C12, adipocyte 3T3-L1, and human hepatocyte HepG2 cell lines, glucose uptake in myocytes and adipocytes, and cytotoxicity tests. The complexes that were unstable in solutions showed biological activity typical of other V(IV) complexes, while the stable one showed much higher, ligand-dependent, activity.
Similar content being viewed by others


Introduction
Vanadium compounds are of great importance not only in science but also in industry, including the very popular V2O5 used as a catalyst [1,2,3]. Its variety of oxidation states (typically ranging from II to V), coordination numbers, and biological activity result in frequent use of this metal in coordination chemistry, especially because vanadium in different oxidation states forms a range of stable compounds, making synthesis relatively simple. In the course of our investigation connected with the insulin-mimetic properties of vanadium compounds, it is worth noting that both inorganic complexes (including V2O5 and VOSO4) and those with organic ligands are found to be active. Inorganic compounds were studied first, but it was soon shown (by McNeill) that organic compounds of vanadium work much better, and today bis(maltolato)oxidovanadium(IV) (BMOV) is used as a standard for investigation of insulin-mimetic activity [4,5,6,7,8]. Soon, other biological activities were recognized—such as anticancer, antifungal or antibacterial, including activity toward protection from anticancer chemotherapy resistance [9,10,11,12,13,14]. Depending on the type of vanadium complex and ligands used, vanadium compounds exhibit large differences in terms of their structure and properties, and thus show different permeability through the cell membrane.
The best known and most widely described mechanism of the insulin-mimetic action of vanadium and its complexes in activation of cellular glucose uptake is inhibition of protein tyrosine phosphatases, especially PTP1B (but also TCPTP, PTP-MEG2, SHP-1, SHP-2, and LAR) [15,16,17]. Phosphatases are a group of enzymes that hydrolyze monoesters of phosphoric acid to a phosphate group and molecules with a free hydroxyl group. By removing a phosphate group, protein phosphatases can modulate the activity of cellular proteins and regulate cell metabolism by affecting many signal transduction pathways. Protein tyrosine phosphatases (PTPs) play an important role in the pathomechanisms of numerous diseases, thus many of them are potential pharmacological targets. In the glucose uptake process, this leads to maintenance of the phosphorylated state of the insulin receptor and thus to upstreaming signaling, which is similar to the action of the insulin hormone [18]. However, the list of molecular targets identified for vanadium action is much wider. Other enzymes (for example, kinases) in the glucose uptake pathway are also important, but the insulin-like effects of vanadium have not yet been fully clarified [19]. All this leads to modulation of various cellular functions and may be a basis for understanding the therapeutic role of vanadium compounds in metabolic, neoplastic, inflammatory, neurodegenerative, and other diseases.
We present herein the synthesis and physicochemical characterization of vanadium(IV,V) complex with tridentate ONO hydrazone Schiff base ligands, now extended to those based on 5-nitrosalicylaldehyde, 5-methoxysalicylaldehyde, and 5-sulfosalicylaldehyde with a set of hydrazides used by us previously [20,21,22,23,24,25,26,27,28]. As shown earlier, for tridentate contrary to bidentate ligands, a huge change of biological activity is observed, indicating that complexes in unchanged form (or at least with ONO ligand still coordinated to vanadium) pass into cells. Single-crystal X-ray structure analysis of three complexes as well as elemental analysis, magnetic susceptibility measurements, infrared (IR) and ultraviolet–visible (UV–Vis) spectroscopies, thermogravimetric measurements, and cyclic voltammetry are presented and discussed. To increase the stability of the complexes and fill the coordination sphere of V(IV/V), 2,2′-bipyridine (bpy) or 1,10-phenanthroline (phen) was used as co-ligand.
The importance of the PTPs tested in this study (PTP1B, SHP1, SHP2, and LAR) in metabolic disorders and diabetes has been discussed in literature [29, 30]. Due to the low specificity of vanadium compounds in inhibiting the activity of individual PTPs and the potential adverse effects associated with this, the ability to inhibit CD45 phosphatase was examined. This phosphatase is associated with a possible impairment of hematopoietic cells and immune system disturbances [31].
Experimental
Materials and methods
[VO(acac)2], VOSO4aq, 1,10-phenanthroline (phen), 2,2′-bipyridine (bpy), 5-nitrosalicylaldehyde, 5-methoxysalicylaldehyde, 5-sulfosalicylaldehyde, benzhydrazide, nicotinic hydrazide, phenylacetic acid hydrazide, 4-chlorobenzhydrazide, 2-hydroxybenzhydrazide, 4-hydroxybenzhydrazide, 4-methoxybenzhydrazide, and 3-hydroxy-2-naphthoic acid hydrazide were of analytical grade (Aldrich or Alfa Aesar) and used as supplied. bis(maltolato)oxovanadium(IV) (BMOV) was synthesized according to literature method [8]. Ethanol (98%) of pharmaceutical grade was from Polmos and used as supplied. All other solvents were of analytical grade (Aldrich) and used as supplied. BaSO4 was of spectroscopic grade (Japan). Bu4NPF6 was synthesized from Bu4NBr and KPF6 by a standard method and recrystallized from acetone [32]. Microanalysis of carbon, hydrogen, nitrogen, and sulfur was performed using an Elementar Vario MICRO Cube elemental analyzer. IR spectra were recorded on a Bruker EQUINOX 55 FT-IR spectrophotometer in KBr pellets. Electronic absorption spectra were recorded with a Shimadzu UV-3600 UV–Vis–NIR spectrophotometer equipped with a CPS-240 temperature controller. Diffuse reflectance spectra were measured in BaSO4 pellets with BaSO4 as reference on a Shimadzu 2101PC equipped with an ISR-260 integrating-sphere attachment. Magnetic susceptibility measurements were performed on a Sherwood Scientific magnetic susceptibility balance. Cyclic voltammetry measurements were carried out in dimethyl sulfoxide (DMSO) with [Bu4N]PF6 (0.1 M) as supporting electrolyte, using Pt working and counter and Ag/AgCl reference electrodes on an Autolab PGSTAT128N potentiostat/galvanostat. E1/2 values were calculated from the average anodic and cathodic peak potentials as E1/2 = 0.5(Ea + Ec). Redox potentials were calibrated versus ferrocene (0.440 V versus standard hydrogen electrode (SHE)), which was used as an internal potential standard for measurements in organic solvents to avoid the influence of a liquid junction potential; the final values are reported versus SHE. Thermogravimetric measurements were performed on a Mettler Toledo TGA/SDTA 851e microthermogavimeter at scan speed of 10°/min in the 25–755 °C range in air.
Inhibition of human tyrosine phosphatases
For this determination, the ability of the tested compounds to inhibit tyrosine phosphatases human recombinant protein was used. To solution of the tested complexes in black 384-well microplate wells (PerkinElmer), an equal volume of test solution of phosphatase was added in reaction buffer [25 mM 3-(N-morpholino) propanesulfonic acid (MOPS), 50 mM NaCl, 1 mM dithiothreitol (DTT), and 0.05% Tween-20, pH 7.0]. The final concentration of each of phosphatase was as follows: 50 ng/ml, SHP1 400 ng/ml, SHP2 50 ng/ml, LAR 5 ng/ml, and CD45 100 ng/ml. After 10 min, solution of phosphate 6,8-difluoro-4-methyl (DiFMUP) was added to final concentration of 0.1 mM. After 20 min of incubation at room temperature, the fluorescence intensity (excitation at 355 and emission at 560 nm) was measured on a multifunctional PTP1B POLARstar Omega plate reader (BMG Labtech, Germany). Assays were performed in triplicate. Results are expressed as the percent of inhibition of untreated control (enzyme with solvent only) [33].
Cell models
Myocyte C2C12 cell line (ATCC CRL-1772), a subclone of myoblasts from mouse muscles, was cultured according to standard protocol in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum, 100 IU/ml penicillin, and 100 µg/ml streptomycin at 37 °C in 5% CO2. Cells were plated on 96-well microplate and after reaching confluence differentiated in medium with 2% horse serum. Differentiation medium was changed every 72 h.
Adipocyte 3T3-L1 cell line (ATCC CRL-11605), derived from fibroblasts from mouse embryo tissue, was cultured according to standard protocol in DMEM medium supplemented with 10% bovine calf serum, 100 IU/ml penicillin, and 100 µg/ml streptomycin at 37 °C in 5% CO2. Cells were seeded in 96-well poly-d-lysine-coated plates and cultured to reach confluence. The medium was then switched to differentiation medium and changed every 48 h.
Human hepatocyte HepG2 cell line (ATCC HB-8065) was cultured according to standard protocol in DMEM supplemented with 10% fetal bovine serum, 100 IU/ml penicillin, and 100 µg/ml streptomycin at 37 °C in 5% CO2. Cells were plated on 96-well microplate.
Glucose uptake in myocytes and adipocytes
Myocytes C2C12 after 8-day differentiation and adipocytes 3T3-L after 11 days of differentiation were cultured and maintained as described above but plated on 96-well microplate coated with solid-phase scintillator (ScintiPlate, PerkinElmer, USA).
Myocytes were washed, and medium was changed for medium with 0.5% bovine serum albumin (BSA) instead of serum. After 24 h of incubation, tested complexes were added at final concentration of 50 µM. After 2 h of incubation at 37 °C in 5% CO2, medium was changed for low-glucose (1000 mg/l) medium, tested complexes were added again, and further incubation for 4 h was conducted. Next, cells were washed three times with Krebs–Ringer buffer (KRB) without glucose (1 mM MgSO4, 1 mM CaCl2, 136 mM NaCl, 4.7 mM KCl, and 10 mM HEPES; pH 7.4) and KRB with human recombinant insulin at final concentration 100 nM (Sigma-Aldrich) was added for 15 min. Wortmannin at final concentration of 200 nM (Sigma-Aldrich) was used as negative control. After that, cells were washed three times with cold KRB and cytochalasin B (10 µM final) was added to several wells as “no uptake” control, proceeding as described below.
Adipocytes were washed, and medium was changed for basal medium. After 24 h of incubation, tested complexes were added at final concentration of 50 µM. After 2 h of incubation at 37 °C in 5% CO2, medium was changed for DMEM medium without glucose, with 0.1% BSA free from fatty acid, 200 mM l-glutamine, and 100 mM pyruvate. Tested complexes were added again, and further incubation for 4 h was conducted. Next, cells were washed three times with KRB without glucose, and KRB with human recombinant insulin at final concentration of 100 nM (Sigma-Aldrich) was added for 15 min. Wortmannin at final concentration of 200 nM (Sigma-Aldrich) was used as negative control. After that, cells were washed three times with cold KRB, and cytochalasin B (10 µM final) was added for several wells as “no uptake.”
For myocytes and adipocytes processed as described above, 2-deoxy-d-[U-14C]-glucose solution in KRB with total activity of 0.03 µCi was added to each well. After 1 h of incubation at 37 °C and 5% CO2, uptake was blocked by adding cytochalasin B (10 µM final). Radioactivity of samples was measured using a MicroBeta Trilux 1450 scintillation counter (PerkinElmer, USA). Nonspecific radioactivity was subtracted from each result (cpm). Two independent experiments in triplicate were conducted. Final results are expressed as percent of control containing solvent only instead of tested compound.
Viability and cytotoxicity tests
After differentiation of C2C12 myocytes, medium was changed for medium with 0.2% bovine serum albumin, 100 IU/ml penicillin, and 100 µg/ml streptomycin, and after 2 h of incubation medium was changed for fresh medium containing vanadium complexes. After 48 h of incubation of 3T3-L1 adipocytes, medium was changed for standard medium with human recombinant insulin and tested vanadium complexes. Hepatocytes HepG2 were used for experiments after seeding for 18–24 h and after medium exchange. Vanadium complexes were tested at final concentration of 50 µM. After 24 h of incubation with tested compounds at 37 °C in 5% CO2, 10 μl PrestoBlue cell viability reagent (Thermo Fisher Scientific) was added to each microplate well, and the microplate was placed on an orbital shaker for content mixing. Plates were incubated for 20 min, and the fluorescence intensity at 560 nm excitation and 590 nm emission was determined using a POLARstar Omega multimode microplate reader (BMG Labtech, Germany). The results were normalized to untreated control (cells with solvent only), whose fluorescence intensity was taken as 100%. Incubations of vanadium complexes with cells were performed in triplicate, and each compound was tested in three to four independent experiments. Similarly, cytotoxicity tests based on assessment of integrity of HepG2 hepatocyte membrane were performed using ToxiLight cytotoxicity kit (Lonza) according to manufacturer protocol.
Synthesis
Table 1 presents the Schiff base ligand notation and ligand components.
Preparation of [{VO(HL1)(H2O)2}2(SO4)]·1.5H2O (1)
5-Nitrosalicylaldehyde (500 mg, 3 mmol) and benzhydrazide (410 mg, 3 mmol) in 25 ml EtOH were refluxed for 10 min under Ar. Then, VOSO4 (657 mg, 3 mmol) was added, and the mixture was refluxed for 40 min. At this stage, mixture was filtered off and the residue removed. The filtrate was concentrated to 50% of initial volume and cooled, and formed crystals were filtered off. Complex was washed three times with ethanol and dried in air. Yield 0.554 g, 42% MW = 897.50. Anal. Calc. for C28H31N6O19.5SV2: C, 37.47; H, 3.48; N, 9.36; S, 3.61%. Found: C, 37.83; H, 3.55; N, 8.94; S ~ 4%. The complex is paramagnetic µeff = 1.80µB. FT-IR (KBr, cm−1): 3405 (w), 3299 (w), 3193 (w), 3075 (w), 1606 (vs), 1558 (s), 1489 (w), 1388 (m), 1337 (vs), 1317 (vs), 1248 (w), 1192 (w), 1131 (w), 1095 (m), 1041 (w), 959 (m), 904 (w), 837 (w), 808 (w), 751 (w), 705 (m), 659 (w), 569 (w), 514 (w), 475 (w), 451 (w). UV–Vis (DMSO): 324, 390 nm, (MeCN): 227, 290 nm, (EtOH): 317, 371 nm.
Preparation of [VO(L1)(bpy)]·0.5EtOH·0.5H2O (2)
5-Nitrosalicylaldehyde (249 mg, 1.5 mmol) and benzhydrazide (204 mg, 1.5 mmol) in 25 ml EtOH were refluxed for 5 min under Ar. Then, VOSO4 (325 mg, 1.5 mmol) was added and the mixture was refluxed for 50 min. Next, the mixture was filtered, and bpy (236 mg, 1.5 mmol) was added to the filtrate. The complex was filtered off, washed three times with EtOH, and dried in air. Yield 0.417 g, 51% MW = 538.43. Anal. Calc. for C25H21N5O6V: C, 55.77; H, 3.93; N, 13.01%. Found: C, 55.92; H, 3.62; N, 13.28%. The complex is paramagnetic µeff = 1.51µB. FT-IR (KBr, cm−1): 3429 (w, br), 3051 (w), 1600 (s), 1552 (w), 1511 (m), 1491 (m), 1466 (m), 1438 (m), 1357 (w), 1316 (vs), 1245 (w), 1201 (w), 1175 (w), 1101 (m), 1037 (w), 1023 (w), 957 (s), 906 (w), 828 (w), 801 (w), 767 (m), 734 (w), 717 (w), 701 (m), 653 (w), 563 (w), 524 (w), 454 (w). UV–Vis (DMSO): 284, 327, 393 nm, (MeCN): 240, 304, 324, 389 nm, (EtOH): 286, 318, 383 nm.
Preparation of [VO(L1)(phen)]·1.5H2O (3)
5-Nitrosalicylaldehyde (500 mg, 3 mmol) and benzhydrazide (408 mg, 3 mmol) in 50 ml EtOH were refluxed for several minutes under Ar. Then, VOSO4 (666 mg, 3 mmol) was added, and the mixture was refluxed for 40 min. Next, the mixture was filtered and, to the 25 ml of filtrate, phen (272.3 mg, 1.5 mmol) was added. The complex was filtered off, washed two times with EtOH, and dried in air. Yield 0.651 g, 40% MW = 557.40. Anal. Calc. for C26H21N5O6.5 V: C, 56.02; H, 3.62; N, 12.56%. Found: C, 56.32; H, 3.30; N, 12.30%. The complex is paramagnetic µeff = 1.61µB. FT-IR (KBr, cm−1): 3452 (w, br), 3051 (w), 1614 (s), 1598 (s), 1549 (w), 1510 (m), 1491 (w), 1465 (w), 1425 (w), 1360 (w), 1336 (w), 1315 (vs), 1243 (w), 1198 (w), 1141 (w), 1102 (m), 1039 (w), 960 (m), 909 (w), 848 (w), 795 (w), 754 (w), 725 (m), 703 (m), 658 (w), 569 (w), 454 (w), 429 (w). UV–Vis (DMSO): 326, 392 nm, (MeCN): 225, 271, 327, 386 nm, (EtOH): 225, 269, 322, 394 nm.
Preparation of [VO(L2)(EtO)] (4)
5-Nitrosalicylaldehyde (250.7 mg, 1.5 mmol) and 2-hydroxybenzhydrazide (228.2 mg, 1.5 mmol) in 40 ml EtOH were refluxed 15 min under Ar. Then, VOSO4 (333 mg, 1.5 mmol) was added with additional 10 ml EtOH, and mixture was refluxed for 105 min. Next, the mixture was filtered, washed with a small amount of EtOH, and dried in air. Yield 0.356 g, 58% MW = 411.24. Anal. Calc. for C16H14N3O7V: C, 46.73; H, 3.43; N, 10.22%. Found: C, 46.58; H, 3.66; N, 10.08%. The complex is diamagnetic. FT-IR (KBr, cm−1): 3482 (w, br), 2981 (w), 1626 (vs), 1609 (vs), 1580 (w), 1560 (m), 1520 (s), 1484 (m), 1468 (m), 1381 (m), 1341 (vs), 1308 (w), 1284 (m), 1248 (m), 1218 (m), 1160 (m), 1128 (w), 1089 (m), 1065 (w), 1035 (m), 997 (s), 966 (w), 908 (m), 873 (w), 843 (m), 817 (m), 755 (s), 720 (m), 703 (m), 674 (w), 656 (w), 614 (m), 578 (w), 519 (m), 481 (w), 468 (w), 447 (w). UV–Vis (DMSO): 326, 387 nm, (MeCN): 267, 310, 324, 389 nm, (EtOH): 321 nm.
Preparation of [VO(L2)(bpy)]·H2O (5)
5-Nitrosalicylaldehyde (250.7 mg, 1.5 mmol) and 2-hydroxybenzhydrazide (228.2 mg, 1.5 mmol) in 33 ml EtOH were refluxed for 6 min under Ar. Then, VOSO4 (333 mg, 1.5 mmol) was added, and the mixture was refluxed for 10 min. Then, bpy (234 mg, 1.5 mmol) in 2 ml EtOH was added. The mixture was refluxed for an additional 5 min. The mixture was cooled, and the product was filtered off, washed with EtOH, and dried in air. Yield 0.656 g, 81% MW = 540.38. Anal. Calc. for C24H19N5O7V: C, 53.34; H, 3.54; N, 12.96%. Found: C, 53.57; H, 3.34; N, 13.00%. The complex is paramagnetic µeff = 1.46µB. FT-IR (KBr, cm−1): 3476 (w, br), 3057 (w), 1604 (vs), 1551 (w), 1514 (m), 1491 (s), 1469 (m), 1443 (m), 1366 (m), 1315 (vs), 1248 (w), 1160 (w), 1130 (w), 1101 (m), 1060 (w), 1022 (w), 955 (s), 914 (w), 830 (w), 802 (w), 753 (s), 731 (m), 720 (m), 694 (w), 653 (m), 631 (w), 582 (w), 519 (w), 472 (m), 446 (w). UV–Vis (DMSO): 286, 329, 390, 465 nm, (MeCN): 214, 306, 387 nm, (EtOH): 284, 375 nm.
Preparation of [VO(L3)(phen)]·2.5H2O (6)
5-Nitrosalicylaldehyde (250.7 mg, 1.5 mmol) and 4-hydroxybenzhydrazide (225.5 mg, 1.5 mmol) in 40 ml EtOH were refluxed 20 min under Ar. Then, [VO(acac)2] (395 mg, 1.5 mmol) in 10 ml EtOH was added, and the mixture was refluxed for 30 min. The mixture was filtered off, and to the filtrate, phen (273.5 mg, 1.5 mmol) in 10 ml EtOH was added. The mixture was heated for an additional 8 min, and formation of orange precipitate was observed. This was filtered off, washed with a small amount of cold EtOH, and dried in air. Yield 0.476 g, 54% MW = 591.42. Anal. Calc. for C26H22N5O8.5 V: C, 52.80; H, 3.75; N, 11.84%. Found: C, 52.80; H, 3.34; N, 11.67%. The complex is paramagnetic µeff = 1.28µB. FT-IR (KBr, cm−1): 3423 (w, br), 1606 (vs), 1554 (w), 1496 (vs), 1467 (w), 1425 (m), 1379 (m), 1361 (m), 1334 (s), 1313 (vs), 1244 (w), 1197 (w), 1166 (m), 1143 (w), 1103 (m), 1033 (w), 956 (s), 913 (w), 846 (s), 757 (m), 724 (s), 645 (m), 597 (w), 566 (w), 521 (w), 475 (w), 445 (w). UV–Vis (DMSO): 326, 395 nm, (MeCN): 223, 270, 328, 392 nm, (EtOH): 226, 266, 326, 390 nm.
Preparation of [VO(L4)(EtO)]·H2O (7)
5-Nitrosalicylaldehyde (251.0 mg, 1.5 mmol) and phenylacetic acid hydrazide (226.1 mg, 1.5 mmol) in 67 ml EtOH were refluxed 20 min under Ar. Then, [VO(acac)2] (399.0 mg, 1.5 mmol) was added, and the mixture was refluxed for 42 min. This was then evaporated to ca. 20 ml and left aside for crystallization. The next day, brown crystalline product was filtered off, washed with EtOH, and dried in air. Yield 0.340 g, 53% MW = 427.28. Anal. Calc. for C17H18N3O5V: C, 47.79; H, 4.25; N, 9.83%. Found: C, 47.46; H, 4.14; N, 9.69%. The complex is diamagnetic. FT-IR (KBr, cm−1): 3547 (m), 2974 (w), 1612 (s), 1598 (s), 1557 (w), 1526 (s), 1497 (s), 1461 (w), 1334 (vs), 1303 (s), 1201 (w), 1155 (w), 1128 (w), 1088 (m), 1031 (m), 983 (s), 941 (w), 909 (m), 841 (m), 808 (w), 781 (w), 759 (w), 723 (m), 692 (w), 660 (w), 635 (m), 582 (m), 549 (w), 513 (w), 460 (m), 429 (w). UV–Vis (DMSO): 319 nm, (MeCN): 289, 334 nm, (EtOH): 308, 364 nm.
Preparation of [VO(L4)(phen)] (8)
The synthesis was performed for 6. The hydrazide used was phenylacetic acid hydrazide (227.1 mg, 1.5 mmol). Yield 0.649 g, 79% MW = 544.41. Anal. Calc. for C27H19N5O5V: C, 59.57; H, 3.52; N, 12.86%. Found: C, 59.20; H, 3.51; N, 12.62%. The complex is paramagnetic µeff = 1.47µB. FT-IR (KBr, cm−1): 3446 (w, br), 3057 (w), 1616 (s), 1601 (s), 1551 (m), 1520 (vs), 1489 (s), 1467 (w), 1425 (s), 1365 (m), 1313 (vs), 1250 (w), 1197 (w), 1143 (w), 1102 (m), 1028 (w), 957 (s), 941 (m), 908 (w), 840 (s), 800 (w), 759 (w), 722 (s), 689 (w), 661 (w), 647 (w), 587 (w), 558 (w), 539 (w), 517 (w), 473 (m), 428 (w). UV–Vis (DMSO): 313, 380 nm, (MeCN): 270, 316, 375 nm, (EtOH): 362 nm.
Preparation of [VO(L5)(H2O)] (9)
5-Nitrosalicylaldehyde (251 mg, 1.5 mmol) and nicotinic acid hydrazide (206 mg, 1.5 mmol) in 70 ml EtOH were refluxed for 10 min under Ar. Then, [VO(acac)2] (398 mg, 1.5 mmol) was added, and the mixture was refluxed for 20 min. The obtaining product was filtered off, washed with small amount of EtOH, and dried in air. Yield 0.479 g, 75% MW = 369.18. Anal. Calc. for C13H10N4O6V: C, 42.29; H, 2.73; N, 15.18%. Found: C, 42.31; H, 2.72; N, 14.85%. The complex is paramagnetic µeff = 1.39µB. FT-IR (KBr, cm−1): 3417 (w, br), 3070 (w), 1605 (s), 1551 (m), 1520 (m), 1500 (w), 1465 (m), 1365 (m), 1337 (vs), 1312 (m), 1242 (w), 1197 (m), 1154 (w), 1127 (w), 1099 (m), 1056 (w), 1027 (w), 957 (w), 919 (w), 880 (vs), 834 (w), 800 (w), 755 (w), 721 (m), 694 (m), 656 (m), 579 (m), 517 (w), 454 (m). UV–Vis (DMSO): 324, 391 nm, (MeCN): 378, 317 nm, (EtOH): 319 nm.
Preparation of [VO(L6)(EtO)(EtOH)] (10)
5-Nitrosalicylaldehyde (334 mg, 2 mmol) and 3-hydroxy-2-naphthoic acid hydrazide (404 mg, 2 mmol) in 110 ml EtOH were heated on a magnetic stirrer for 15 min under Ar. The temperature was raised to 100 °C. Then, VOSO4 (444 mg, 2 mmol) and 20 ml ethanol was added, and heterogenic mixture was refluxed for ca. 40 min. After that, the volume of the reaction mixture was concentrated to final volume of ca. 40 ml and cooled to room temperature. The precipitate was filtered off, washed with ethanol, and dried in air. Yield 0.652 g, 65% MW = 507.37. Anal. Calc. for C22H22N3O8V: C, 52.08; H, 4.37; N, 8.28%. Found: C, 51.58; H, 4.21; N, 8.17%. The complex is diamagnetic. FT-IR (KBr, cm−1): 3435 (w, br), 3075 (w), 1633 (s), 1612 (s), 1555 (s), 1524 (vs), 1512 (s), 1458 (m), 1378 (m), 1341 (vs), 1302 (w), 1263 (w), 1239 (w), 1198 (w), 1175 (w), 1149 (m), 1128 (w), 1089 (m), 956 (w), 905 (s), 884 (w), 845 (w), 804 (w), 757 (m), 710 (w), 644 (m), 591 (w), 570 (w), 519 (w), 481 (m), 423 (w). UV–Vis (DMSO): 326, 391 nm, (MeCN): 358, 415 nm, (EtOH): 324, 454 nm.
Preparation of [VO(HL7)2]·EtOH (11)
5-Methoxysalicylaldehyde (187 µl, 1.5 mmol) and benzhydrazide (207.3 mg, 1.5 mmol) in 25 ml EtOH were refluxed for 12 min under Ar. Then, [VO(acac)2] (398.6 mg, 1.5 mmol) was added, and the mixture was refluxed for the next 45 min. After that, the solution was evaporated to ca. 10 ml and left cooling in the refrigerator. The product was filtered off, washed with ethanol, and dried in air. Yield 0.170 g, 35% MW = 650.55. Anal. Calc. for C32H31N4O8V: C, 59.17; H, 4.80; N, 8.63%. Found: C, 59.73; H, 4.62; N, 8.50%. The complex is paramagnetic µeff = 1.27µB. FT-IR (KBr, cm−1): 3464 (w, br), 3053 (w), 2915 (w), 2833 (w), 1603 (w), 1575 (m), 1539 (vs), 1503 (m), 1454 (m), 1440 (m), 1410 (w), 1356 (s), 1327 (m), 1275 (s), 1225 (vs), 1196 (w), 1169 (m), 1137 (w), 1119 (w), 1031 (s), 945 (w), 830 (m), 810 (m), 785 (w), 738 (w), 697 (s), 613 (w), 586 (w), 553 (w), 516 (w), 484 (w). UV–Vis (DMSO): 300, 396 nm, (MeCN): 297, 386, 519 nm, (EtOH): 296, 399, 521 nm.
Preparation of [{VO(HL7)}2(SO4)]·H2O (12)
5-Methoxysalicylaldehyde (187 µl, 1.5 mmol) and benzhydrazide (207 mg, 1.5 mmol) in 25 ml EtOH were refluxed for 10 min under Ar. Then, VOSO4 (331.4 mg, 1.5 mmol) was added, and the mixture was refluxed for the next 40 min. After that, the solution was evaporated to ca. 10 ml and left cooling in the refrigerator. The product was filtered off, washed with ethanol, and dried in air. Yield 0.402 g, 68% MW = 788.51. Anal. Calcd. for C30H30N4O13SV2: C, 45.81; H, 3.80; N, 7.12; S, 4.08%. Found: C, 45.77; H, 4.65; N, 7.10; S ~ 4%. The complex is paramagnetic, µ = 1.27µB. FT-IR (KBr, cm−1): 3423 (w, br), 2945 (w), 2845 (w), 1617 (w), 1605 (w), 1581 (w), 1537 (s), 1490 (s), 1448 (w), 1417 (w), 1388 (s), 1346 (s), 1284 (m), 1268 (w), 1243 (s), 1171 (s), 1121 (vs), 1091 (s), 1030 (s), 1002 (m), 968 (s), 897 (m), 828 (m), 793 (w), 700 (s), 671 (w), 629 (w), 609 (w), 584 (w), 560 (w), 507 (w), 471 (w). UV–Vis (DMSO): 331, 424 nm, (MeCN): 326, 419 nm, (EtOH): 327, 416 nm.
Preparation of [VO(L8)(phen)]·4H2O (13)
5-Methoxysalicylaldehyde (187 µl, 1.5 mmol) and 2-hydroxybenzhydrazide (227.4 mg, 1.5 mmol) in 25 ml EtOH were refluxed for 10 min under Ar. Then, VOSO4 (334 mg, 1.5 mmol) was added, and the mixture was refluxed for the next 47 min. Phen (272.9 mg, 1.5 mmol) was added to the transparent solution, and heating was continued for 10 min. This slowly started to crystallize, especially on cooling. The brown crystalline product was filtered off, washed with EtOH, and dried in air. Yield 0.369 g, 38% MW = 703.48. Anal. Calc. for C27H28N4O9V: C, 53.74; H, 4.68; N, 9.28%. Found: C, 53.56; H, 3.86; N, 9.31%. The complex is paramagnetic µeff = 1.41µB. FT-IR (KBr, cm−1): 3429 (m, br), 3064 (w), 1623 (m), 1596 (s), 1537 (s), 1518 (s), 1491 (s), 1466 (w), 1424 (s), 1374 (m), 1340 (w), 1286 (w), 1257 (m), 1223 (m), 1166 (m), 1145 (w), 1107 (w), 1032 (s), 957 (s), 867 (w), 846 (m), 816 (w), 759 (m), 722 (s), 699 (w), 657 (w), 643 (w), 582 (w), 473 (w), 449 (w), 427 (w). UV–Vis (DMSO): 331, 429 nm, (MeCN): 297, 386, 519 nm, (EtOH): 267, 339, 439 nm.
Preparation of [VO(L9)(phen)]·1.5H2O (14)
The synthesis was performed as for 6. The aldehyde used was 5-methoxysalicylaldehyde (187 μl, 1.5 mmol), and the hydrazide used was 4-hydroxybenzhydrazide (226.7 mg, 1.5 mmol). Yield 0.359 g, 43% MW = 558.44. Anal. Calc. for C27H23N4O6.5V: C, 58.07; H, 4.15; N, 10.03%. Found: C, 58.25; H, 4.06; N, 9.77%. The complex is paramagnetic µeff = 1.60µB. FT-IR (KBr, cm−1): 3418 (w, br), 3163 (w, br), 1598 (vs), 1545 (s), 1501 (vs), 1477 (m), 1423 (m), 1374 (w), 1360 (m), 1339 (w), 1279 (s), 1226 (s), 1169 (s), 1104 (w), 1031 (m), 954 (s), 906 (w), 845 (s), 810 (m), 787 (w), 757 (w), 724 (m), 697 (w), 642 (w), 597 (w), 562 (w), 474 (w), 441 (w). UV–Vis (DMSO): 334, 426 nm, (MeCN): 269, 334, 428 nm, (EtOH): 228, 264, 330, 421 nm.
Preparation of [VO(L10)(phen)] (15)
The synthesis was performed as for 14. The hydrazide used was 4-chlorobenzhydrazide (252.1 mg, 1.5 mmol). Yield 0.565 g, 69% MW = 549.86. Anal. Calc. for C27H19ClN4O4V: C, 58.98; H, 3.48; N, 10.19%. Found: C, 58.91; H, 3.47; N, 10.15%. The complex is paramagnetic µeff = 1.53µB. FT-IR (KBr, cm−1): 3452 (w, br), 3051 (w), 2821 (w), 1623 (w), 1602 (s), 1543 (s), 1506 (vs), 1486 (s), 1466 (m), 1423 (s), 1375 (m), 1348 (m), 1293 (m), 1260 (s), 1222 (w), 1179 (w), 1159 (m), 1089 (w), 1053 (w), 1030 (m), 1012 (w), 955 (vs), 906 (w), 867 (w), 851 (m), 811 (m), 774 (w), 741 (w), 729 (m), 680 (w), 644 (w), 583 (w), 467 (w), 436 (w). UV–Vis (DMSO): 331, 428 nm, (MeCN): 267, 337, 447 nm, (EtOH): 228, 264, 326, 430 nm.
Preparation of [VO(L11)(EtO)(EtOH)] (16)
5-Methoxysalicylaldehyde (187 µl, 1.5 mmol) and phenylacetic acid hydrazide (225.2 mg, 1.5 mmol) in 50 ml EtOH were refluxed for 11 min under Ar. Then, [VO(acac)2] (399 mg, 1.5 mmol) was added, and the mixture was refluxed for the next 58 min. This was evaporated in part, but precipitation was observed the next day in refrigerator. This was filtered off, washed with EtOH, and dried in air. Yield 0.410 g, 62% MW = 440.36. Anal. Calc. for C20H25N2O6V: C, 54.67; H, 5.68; N, 6.38%. Found: C, 54.55; H, 5.72; N, 6.36%. The complex is diamagnetic. FT-IR (KBr, cm−1): 3452 (w, br), 2990 (w), 1625 (w) 1600 (s), 1546 (vs), 1514 (vs), 1468 (m), 1421 (m), 1377 (w), 1358 (m), 1326 (w), 1288 (m), 1265 (vs), 1238 (w), 1181 (w), 1160 (m), 1107 (w), 1048 (w), 1026 (m), 991 (vs), 901 (w), 844 (m), 817 (m), 766(w), 726 (vs), 690 (w), 637 (w), 587 (w), 553 (w), 524 (w), 467 (w), 425 (w). UV–Vis (DMSO): 323, 413 nm, (MeCN): 408 nm, (EtOH): 315, 412 nm.
Preparation of [VO(L11)(phen)] (17)
The synthesis was performed as for 14. The hydrazide used was phenylacetic acid hydrazide (225.8 mg, 1.5 mmol). Yield 0.619 g, 78% MW = 529.44. Anal. Calc. for C28H22N4O4V: C, 63.52; H, 4.19; N, 10.58%. Found: C, 63.26; H, 4.11; N, 10.43%. The complex is paramagnetic µeff = 1.59µB. FT-IR (KBr, cm−1): 3135 (w, br), 2966 (w), 1602 (s), 1550 (vs), 1531 (vs), 1476 (s), 1455 (m), 1419 (w), 1383 (w), 1330 (w), 1270 (vs), 1230 (s), 1194 (w), 1164 (m), 1088 (w), 1048 (vs), 1025 (m), 974 (vs), 920 (m), 878 (w), 836 (s), 774 (w), 736 (s), 686 (w), 663 (m), 580 (w), 564 (w), 524 (w), 473 (w), 444 (w), 425 (w). UV–Vis (DMSO): 412, 396 nm, (EtOH): 262 nm.
Preparation of [VO(L12)(H2O)]·0.5H2O (18)
5-Methoxysalicylaldehyde (374 µl, 3 mmol) and nicotinic acid hydrazide (411 mg, 3 mmol) in 45 ml EtOH were refluxed for 15 min under Ar. Then, VOSO4 (666 mg, 3 mmol) was added, and the mixture was refluxed for the next 40 min. The compound was filtered off, washed with EtOH, and dried in air. Yield 0.800 g, 73% MW = 363.20. Anal. Calc. for C14H14N3O5.5 V: C, 46.26; H, 3.85; N, 11.56%. Found: C, 46.06; H, 3.22; N, 11.29%. The complex is paramagnetic µeff = 1.42µB. FT-IR (KBr, cm−1): 3490 (w, br), 3066 (w), 1625 (m), 1597 (s), 1551 (vs), 1481 (s), 1461 (m), 1420 (w), 1340 (s), 1287 (s), 1260 (w), 1219 (s), 1195 (m), 1171 (s), 1031 (s), 949 (s), 932 (m), 872 (s), 827 (s), 786 (m), 707 (s), 673 (m), 614 (s), 574 (s), 477 (m). UV–Vis (DMSO): 333, 429 nm, (MeCN): 254 nm, (EtOH): 249 nm.
Preparation of [VO(L13)(EtO)]·0.5H2O (19)
5-Methoxysalicylaldehyde (187 µl, 1.5 mmol) and 3-hydroxy-2-naphthoic acid hydrazide (303 mg, 1.5 mmol) in 90 ml EtOH were heated on a magnetic stirrer for 10 min under Ar. The temperature was raised to 100 °C. Then, VOSO4 (333 mg, 1.5 mmol) and 10 ml of EtOH was added, and the heterogenic mixture was refluxed for ca. 180 min. After that, the volume of the reaction mixture was concentrated to a final volume of ca. 40 ml and cooled to room temperature. The precipitate was filtered off, washed with ethanol, and dried in air. Yield 0.698 g, 78% MW = 446.33. Anal. Calc. for C21H19N2O6V: C, 56.51; H, 4.29; N, 6.28%. Found: C, 56.08; H, 4.25; N, 6.15%. The complex is diamagnetic. FT-IR (KBr, cm−1): 3453 (w, br), 2932 (w), 1639 (m), 1596 (m), 1572 (w), 1554 (s), 1524 (m), 1473 (m), 1386 (w), 1371 (w), 1341 (w), 1309 (m), 1274 (s), 1239 (m), 1212 (w), 1168 (w), 1145 (w), 1088 (m), 1031 (s), 991 (s), 954 (w), 904 (w), 822 (m), 765 (w), 719 (w), 683 (w), 622 (m), 597 (m), 577 (m), 527 (w), 478 (w). UV–Vis (DMSO): 333, 438 nm, (MeCN): 325, 427 nm, (EtOH): 321, 428 nm.
Preparation of [VO(L14)(phen)]·4.5H2O (20)
Potassium salt of 5-sulfosalicylaldehyde (366 mg, 1.5 mmol) and 4-hydroxybenzhydrazide (228 mg, 1.5 mmol) in 50 ml ethanol were refluxed for 10 min under Ar. Then, [VO(acac)2] (397 mg, 1.5 mmol) was added. The reflux was continued for 15 min under Ar. Next, phen (273 mg, 1.5 mmol) in 10 ml of EtOH was added, and the mixture was heated for an additional 8 min. The next day, formed crystals were filtered off, washed with cold ethanol, and dried in air. Yield: 0.312 g, 30%. MW = 700.60. Anal. Calcd. for C26H27N4O11.5KSV: C, 44.57; H, 3.74; N, 5.58; S, 4.64%. Found: C, 44.70; H, 3.54; N, 7.85; S, 4.31%. The complex is paramagnetic, µeff = 1.26µB. FT-IR (KBr, cm−1): 3436 (vs), 1606 (vs), 1501 (s), 1463 (w), 1423 (m), 1375 (m), 1356 (m), 1280 (w), 1194 (w), 1166 (s), 1112 (m), 1033 (vs), 958 (m), 903 (w), 844 (m), 723 (m), 673 (w), 648 (w), 617 (m), 565 (w), 450 (w). UV–Vis (DMSO): 327, 407 nm, (MeCN): 268, 330, 414 nm, (EtOH): 226, 264, 325, 406 nm.
Preparation of [VO(L15)(phen)]·4H2O (21)
Potassium salt of 5-sulfosalicylaldehyde (367 mg, 1.5 mmol) and 4-methoxybenzhydrazide (250 mg, 1.5 mmol) in 45 ml ethanol were refluxed for 20 min under Ar. Then, [VO(acac)2] (378 mg, 1.5 mmol) was added. The reflux was continued for 20 min under Ar. Next, phen (271 mg, 1.5 mmol) was added. The mixture with precipitate was evaporated to 50% and left for cooling. The mixture was filtered off, washed with ethanol, and dried in air. Yield: 0.599 g, 55%. MW = 705.63. Anal. Calcd. for C27H27N4O11KSV: C, 45.96; H, 3.86; N, 7.94; S, 4.54%. Found: C, 45.65; H, 3.70; N, 7.83; S, 4.40%. The complex is paramagnetic, µeff = 1.34µB. FT-IR (KBr, cm−1): 3436 (vs), 1609 (vs), 1535 (w), 1499 (s), 1461 (m), 1421 (m), 1371 (m), 1352 (s), 1310 (w), 1252 (s), 1175 (vs), 1116 (s), 1040 (vs), 963 (s), 905 (w), 846 (m), 751 (w), 728 (m), 675 (m), 646 (w), 617 (m), 587 (w), 469 (w), 437 (w). UV–Vis (DMSO): 325, 406 nm, (MeCN): 224, 268, 327, 420 nm, (EtOH): 226, 263, 325, 406 nm.
Preparation of [VO(L16)(phen)]·phen·2H2O (22)
Potassium salt of 5-sulfosalicylaldehyde (361 mg, 1.5 mmol) and phenylacetic acid hydrazide (225 mg, 1.5 mmol) in 30 ml ethanol were refluxed for 10 min under Ar. Then, [VO(acac)2] (398 mg, 1.5 mmol) was added. The reflux was continued for 45 min under Ar. Then, phen (270 mg, 1.5 mmol) was added, and reflux was continued for 5 min. The formed precipitate was filtered off, washed with ethanol, and dried in air. Yield: 0.493 g, 42%. MW = 795.71. Anal. Calcd. for C39H32N6O8SV: C, 58.87; H, 4.05; N, 10.56. Found: C, 59.35; H, 4.55; N, 10.75%. The complex is paramagnetic, µeff = 0.93µB. FT-IR (KBr, cm−1): 3050 (w), 1586 (s), 1554 (s), 1501 (vs), 1423 (s), 1400 (s), 1369 (w), 1345 (w), 1318 (w), 1288 (w), 1217 (w), 1173 (w), 1143 (w), 1105 (w), 1073 (w), 1025 (m), 956 (vs), 846 (s), 770 (w), 722 (vs), 703 (w), 654 (w), 608 (w), 522 (w), 469 (w), 423 (w). UV–Vis (DMSO): 367 nm (MeCN): 271, 387 nm, (EtOH): 226, 266, 377 nm.
Preparation of [VO(HL17)(H2O)]∙2H2O (23)
Potassium salt of 5-sulfosalicylaldehyde (480 mg, 2.0 mmol) and nicotinic acid hydrazide (274 mg, 2.0 mmol) in 100 ml ethanol were refluxed for 20 min under Ar. Then, [VO(acac)2] (530 mg, 2.0 mmol) was added. The reflux was continued for 45 min under Ar. The obtained compound was filtered off, washed several times with ethanol, and dried at room temperature. Yield: 0.637 g, 67%. MW = 479.37. Anal. Calcd. for C13H15N3O9KSV: C, 32.54; H, 3.13; N, 8.76; S, 6.70%. Found: C, 33.04; H, 2.77; N, 8.44; S, 6.66%. The complex is paramagnetic, µeff = 1.19µB. FT-IR (KBr, cm−1): 3421 (vs), 1613 (vs), 1534 (m), 1461 (m), 1414 (w), 1384 (m), 1363 (m), 1298 (w), 1193 (s), 1113 (s), 1035 (vs), 912 (m), 883 (m), 832 (w), 720 (w), 671 (w), 619 (s), 535 (w), 458 (w), 415 (w). UV–Vis (DMSO): 326, 409 nm, (MeCN): 242, 325, 405 nm, (EtOH): 320, 399 nm.
Preparation of K4[{VO(L18)}2(SO4)]·2H2O·EtOH (24)
Potassium salt of 5-sulfosalicylaldehyde (361 mg, 1.5 mmol) and 3-hydroxy-2-naphtoic acid hydrazide (303 mg, 1.5 mmol) were dissolved in 60 ml ethanol under Ar flow on a magnetic stirrer. The mixture was refluxed for 20 min with constant stirring, and the temperature was raised to 100 °C. Then, VOSO4 (333 mg, 1.5 mmol) in 15 ml ethanol was added. The reflux was continued for 180 min under Ar. After that, the volume of the reaction mixture was concentrated to final volume of ca. 20 ml and cooled to room temperature. The obtained compound was filtered off, washed several times with ethanol, and dried in air. Yield: 0.766 g, 85%. MW = 1197.06. Anal. Calcd. for C38H32K4N4O21S3V2: C, 37.01; H, 2.61; N, 4.54; S, 7.79%. Found: C, 37.35; H, 2.67; N, 4.66; S, 8.54%. The complex is diamagnetic. FT-IR (KBr, cm−1): 3471 (w, br), 3290 (w), 3081 (w), 1629 (m), 1600 (s), 1578 (w), 1540 (vs), 1462 (w), 1375 (s), 1229 (s), 1138 (vs), 1106 (vs), 1039 (s), 977 (s), 918 (w), 886 (vs), 835 (w), 761 (w), 727 (w), 617 (s), 583 (w), 505 (w), 454 (w), 429 (w). UV–Vis (DMSO): 324, 414 nm, (EtOH): 225, 274, 326 nm.
Crystallographic data collection and structure refinement
Diffraction intensity data for a single crystal of three new compounds were collected at 293 K on an Oxford Diffraction Super Nova diffractometer using monochromatic Mo Kα (10, 16) or Cu Kα (4) radiation at λ = 0.71073 Å and λ = 1.5418 Å, respectively. Cell refinement and data reduction were performed using firmware [34]. Positions of all nonhydrogen atoms were determined by direct methods using SIR software (4, 10: SIR92 [35], 16: SIR97 [36]. All nonhydrogen atoms were refined anisotropically using weighted full-matrix least-squares on F2. Refinement and further calculations were carried out using SHELXL software [37] (4, 10: ver 2017/1, 16: ver 2016/1]. All hydrogen atoms joined to carbon atoms were positioned with idealized geometries and refined using a riding model with Uiso(H) fixed at 1.2 Ueq(Carom). In structure 10, two carbon atoms of the coordinating ethanol molecule, C31 (methylene) and C32 (methyl), show disorder and were modeled (split) into two sets of positions A and B with their geometries restrained, with the refined occupancies being 0.54(5) and 0.46(5), respectively. The positions of all hydrogen atoms were constrained for all compounds using AFIX (SHELXL) commands. CCDC 1871461, 1871462, and 1871642 contain the supplementary crystallographic data for 4, 10, and 16. These data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif. The crystallographic data and detailed information on the structure solution and refinement are presented in Table 2. The bond distances and angles are listed in Supplementary Tables S1–S3.
Results and discussion
General remarks on the synthesis
As a result of the syntheses, 24 oxidovanadium(IV,V) complexes were obtained by reaction of VOSO4aq or [VO(acac)2] with in situ formed tridentate Schiff base ligands. Six of these (4, 7, 10, 16, 19, and 24) are V(V) complexes, while the others are V(IV) complexes. In some syntheses, co-ligand in the form of 2,2′-bipyridine or 1,10′-phenanthroline was used to precipitate the compounds. As vanadium source, VOSO4aq or [VO(acac)2] was used. In general, [VO(acac)2] was found to be more suitable due to solubility; however, the product composition in most cases was similar, independently of the complex used. Only in three cases (1, 12, and 24) was SO 2−4 group noticed to be present in the complex and could not be removed by simple washing. The analysis indicates that one ion per two vanadium centers is present, in all the performed syntheses. Thus, the dimeric form of the complex is suggested.
Usually, in our previous investigations with vanadium complexes based on Schiff base ligands, oxidation of V(IV) to V(V) when co-ligand (for example, phen) was not used [22, 23, 25, 27, 28] was observed. In this part of the investigation, the oxidovanadium(IV) complexes (1, 9, 11, 12, 18, 23) were obtained, among others, for which there was no change in the oxidation state and for which a stabilizing ligand for the V(IV) oxidation state was not used in the synthesis. It must be underlined, however, that those complexes are not as stable in time as those with co-ligands, especially with phen.
As the syntheses were performed under Ar atmosphere, it was difficult to dry the samples to constant mass due to their air sensitivity. Moreover, the samples were weighed in air for elemental analysis. This caused the analyses not to fit perfectly (within 0.4%) in several cases to a proposed formula (in two cases the largest mistake was on hydrogen, but still less than 0.9%).
Description of the structures
Compound 4 crystallizes in the triclinic space group P-1 with an asymmetric cell unit (Fig. 1) containing the monomeric complex of [VO(L2)(EtO)]. The central vanadium atom has a five-coordinate environment with VO4N composition, taking the shape of an almost perfect square pyramid. The square base of the polyhedron is made of the atoms of a tridentate L2 ligand and consists of phenolic oxygen O8, imine nitrogen N4, enol oxygen O1, and an oxygen atom O24 from deprotonated ethoxide oxygen. The apical position of the square pyramid is occupied by the terminal oxido (O2-) group O23. The short V1–O23 distance of 1.566(5) Å indicates the presence of a double vanadium–oxygen bond (V=O). The central vanadium V1 atom is located above the basal plane of O24, O1, N4, and O8 by 0.450 Å and is directed toward the apical oxido ligand. Both L2 aromatic rings are slightly bent downwards in relation to the plane they create, and the highest deviation relates to the C17 and C11 atoms and amounts to 0.341 Å and 0.233 Å, respectively (Fig. 1). The square pyramid is slightly distorted, in which the individual angles N4–O1–O24, O1–O24–O8, O24–O8–N4, and O8–N4–O1 are 97.16°, 85.10°, 87.69°, and 89.96°, respectively, where the highest deviation from the ideal 90° is 7.16°. The observed distortion is due to the intramolecular structural tension caused by the presence of both five- and six-membered rings in the square plane of the pyramid. Packing analysis of the crystal structure reveals that the complex molecules are held together by two π…π stacking and four weak intermolecular interactions of C–H…O type, as summarized in Supplementary Tables S4 and S5.

Structure of 4 with marked coordination polyhedron and atom labeling scheme (left), showing the planarity of the ligand and donor atoms with plane constructed on N4–O1–O24 atoms with 30% probability ellipsoids (right). Hydrogen atoms omitted for clarity
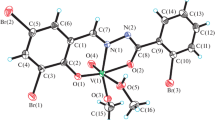
The structure of 10 with atom numbering scheme and coordination polyhedron of vanadium is presented in Fig. 2. The vanadium atom is surrounded by ONO donor atoms of ligand L, two oxygens from EtOH and EtO− molecules, and O2− anion. The central vanadium atom has six-coordinated surrounding with ethoxy and L ligand in plane and EtOH and O2− as apexes. The vanadium atom is shifted out of plane (constructed on N15–O13–O27 atoms) toward the oxygen atom by 0.286 Å. Contrary to 4, in 10 aromatic rings are shifted below this plane (hydrazide ones) and above the plane (from aldehyde moieties) with the longest distances to plane for C7 (0.676 Å) and C21 (0.387 Å). The octahedron is slightly distorted, with O33–V1–O30 angle of 174.28°. Again, as in 4, distortion of the square-pyramidal plane N15–O13–O27–O23, caused by the presence of five- and six-membered rings, is observed.

Structure of 10 with marked coordination polyhedron and atom labeling scheme. The packing in 10 with 30% probability ellipsoids (right). Hydrogen atoms omitted for clarity
Similarly to 4, in the cell unit there is an equimolar mixture of two isomers, but in the case of 10, four molecules are present in the unit cell. The packing is presented in Fig. 2 (right).
In 10, both intra- and intermolecular hydrogen bonds are present. The intramolecular one is between N14 and O11, similarly to 4, with a distance of 2.577(2) Å, slightly shorter than in 4. The intermolecular hydrogen bond is formed between O11 and O30, with a distance equal to 2.784(2) Å (Supplementary Table S6).
The structure of 10 consists of separated layers, as presented in Fig. 3. While the layers are supported by a net of hydrogen bonds, only very weak intramolecular interactions are present between layers.

Packing scheme in 10; the net of hydrogen bonds and short contacts are visualized. Hydrogen atoms omitted for clarity, with 30% probability ellipsoids shown
The structure of the asymmetric parts of the unit cell of complex 16 and the adopted atomic numbering scheme are shown in Fig. 4. This complex crystallizes in the triclinic space group P-1 with the asymmetric unit cell containing one molecule of [VO(L11)(EtO)(EtOH)] complex. Oxidovanadium cation (VO2+) is coordinated with the fully deprotonated tridentate hydrazone ligand (L11) through phenolate oxygen (O7), azomethine nitrogen (N11), and enolate oxygen (O21) and additionally with two oxygen atoms, one of which is deprotonated ethoxide oxygen (O22), while the second oxygen (O25) comes from the ethanol molecule. Thus, the coordination environment of the central atom V adopts the geometry of an elongated octahedron with the scheme O5N. The basic plane is defined by the atoms O7, O21, O22, and N11, while the central vanadium atom is above this plane by 0.3037(8) Å and is shifted toward the O28 atom (the V–O distance equals 1.583(2) Å). In the discussed complex (16), the lengths of V–O bonds have different values, the V=Ooxido bond being the shortest and the V–Oenolate = 2.383(2) Å (O25 of ethanol) bond being the longest. The oxygen (O28) atom and atom O25 of the ethanol molecule occupy the apical sites of the octahedron with a trans angle of 174.7(6)°. The phenyl ring (C15–C20) of the ligand forms a dihedral angle of 112.6(3)° with the 2-hydroxy-5-methoxyphenyl ring. This causes intracomplex tension and, consequently, is an additional cause of the octahedral deformation of the geometry of the coordination environment of the central vanadium atom. Packing analysis of the crystal structure of 16 reveals that the complex molecules are stabilized by one typical hydrogen interaction of O–H…N type [O(25)–H(25)…N(12) [−x + 1, −y + 1, −z + 1], d(D–H) = 0.76(2) Å, d(H…A) = 2.07(2) Å, d(D…A) = 2.823(2) Å, and ∠(DHA) = 172(2)°] (Supplementary Table S7). The long-distance packing scheme is presented in Fig. 4 (right).

Structure of 16 with marked coordination polyhedron and atom labeling scheme (left). Packing scheme in 16, with 30% probability ellipsoids (right). Hydrogen atoms omitted for clarity
A comparison of the V=O distances in 4, 10, and 16, of 1.568, 1.582, and 1.583 Å, respectively, reflects the presence of EtOH ligand in 10 and 16, resulting in an increase in the bond distance by ca. 0.014 Å compared with 4. The V=O bond distance in 4, 10, and 16 is shorter than that observed in V(IV) analogs (1.604, 1.606, 1.593, 1.598, and 1.594 Å), and similar shortening is observed also for the V–OL (L = Schiff base ligand) bond [21, 22]. In contrast, the V–NL distance is longer in V(V) compared with V(IV) complexes (bond distances 2.046, 2.058, 2.039, 2.038, and 2.031 Å) [21, 22].
IR spectra
IR spectral measurements confirmed the presence of characteristic groups in the synthesized oxidovanadium complexes for both V(IV) and V(V). All the vibrational bands are listed in the “Experimental” section. The most characteristic strong band located in the range of 955–965 cm−1 is associated with ν(V=O) vibration in V(IV) complexes. This is a direct difference from the V(V) complexes, where weak bands in this range or intense bands at 983 cm−1 are observed. The investigated complexes also show a band at ca. 1600 cm−1 that can be assigned to ν(C=N) vibration in a Schiff base ligand. For complexes with phen or bpy as ligand, the characteristic bands at ca. 1490, 1425, and 725 cm−1 are related to the vibration in these ligands and were also observed in other d-electron metal complexes with phen or bpy [38].
UV–Vis spectra
The positions of the bands for the compounds in three selected solvents (DMSO, MeCN, and EtOH) were presented above. The UV–Vis spectra for all the complexes in organic solvents [DMF, DMSO, EtOH, MeOH, MeCN, dichloroethane (EtCl2), and n-octanol] and in solid state as well as their stability at neutral pH and at pH 2.00 are presented in the Supplementary Electronic Material. In the UV part of the electronic spectra, the bands attributed to the coordinated Schiff base ligand and co-ligand (phen or bpy) where present are observed as separate peaks. In the visible part of the spectra, the band located at ca. 400 nm can be assigned to a ligand-to-metal charge-transfer (LMCT) transition between the metal center and Schiff base ligand. This band is relatively intense with molar extinction coefficient (mol−1 cm−1 dm3) in DMSO of 2.56 × 104 (1, 390 nm), 2.89 × 104 (2, 393 nm), 2,61 × 104 (3, 399 nm), 4.97 × 104 (4, 387 nm), and 2.55 × 104 (5, 390 nm).
The reflectance spectra of representative complexes [oxidovanadium(IV) complexes with bpy and phen as co-ligand and oxidovanadium(V) complex] are presented in Fig. 5. Complexes 2 and 3 show absorption above 700 nm, which is typical for d1 configuration of V(IV) and is in accordance with the magnetic measurements presented above. Due to low solubility of all complexes, the d–d transition can be observed only in reflectance spectra, while in solution it is obscured by low intensity. For 10, no band appears in the range above 500 nm, confirming the V(V) oxidation state and d0 configuration. This situation was also observed for the other V(V) complexes (4, 7, 10, 16, 19, and 24).

Reflectance spectra of complexes 2 (black, solid line), 3 (red, dashed line), and 10 (blue, dotted line) in the range 200–900 nm after Kubelka–Munk transformation. BaSO4 as internal (white) reference
Due to problems with the insolubility of the obtained compounds in water, the complexes were studied in DMSO. This solvent was selected because of its usefulness in, e.g., medicine, as it supports transfer of substances through biological membranes. The stability of the selected complexes was measured by UV–Vis spectra versus time. The compounds showed stability in the solvent tested and were dissolved in DMSO-H2O mixture (20 µl + 3 ml, respectively) at neutral pH and at pH 2.00. The UV–Vis spectra were measured over time for each mixture. Most of the complexes tested were stable in natural pH but gradually decomposed in pH 2.00 (1, 3, 6, 8–10, 12–15, 18–21, 23, 24). Complexes 2, 4, 17, and 22 were unstable at natural pH and pH 2.00. Only complexes 7 and 16 were stable in natural pH as well as pH 2.00. Exemplary spectral changes for complexes 2 and 3 are presented in Fig. 6.

UV–Vis spectra of complex 2 (left side) and 3 (right side) in DMSO-H2O mixture (20 µl + 3 ml, respectively) at natural pH (above) and pH 2.00 (below). T = 37 °C, d = 1 cm, 15 spectra measured at 340-s intervals. The arrow shows the direction of changes in spectra
Cyclic voltammetry
Cyclic voltammetry measurements were performed in DMSO using Pt working and counter and Ag/AgCl reference electrodes in 0.1 M Bu4NPF6 as supporting electrolyte. Exemplary voltammograms were selected for oxidovanadium(IV) complexes with phen (Fig. 7) and bpy (Fig. 8) ligands. In general, for oxido complexes of vanadium, irreversible redox processes are observed. Only for complexes with phen or bpy as co-ligand were reversible peaks, attributed to reduction of these ligands, observed in the negative part of the voltammograms. The presence of two reversible processes for 3 (Fig. 7) in this part is a result of the observed instability of 3 in DMSO. Thus, we observe “free” and coordinated phen reversible redox processes. As a result of complex stability, Fig. 8 presents the voltammogram of 5 which, contrary to 3, is stable in DMSO. The presented voltammograms are very similar to those described by us earlier [20,21,22,23,24,25,26].

Cyclic voltammogram of 3 in DMSO. 0.1 M Bu4NPF6 as electrolyte, Pt working and counter and Ag/AgCl reference electrodes, potentials versus NHE. Scan speed 100 mV/s (for main curve in full range); insets: scans in selected voltage range with scan speeds in the range of 20–1000 mV/s

Cyclic voltammogram of 5 in DMSO. 0.1 M Bu4NPF6 as electrolyte, Pt working and counter and Ag/AgCl reference electrodes, potentials versus NHE. Scan speed 100 mV/s (for main curve in full range); insets: scans in selected voltage range with scan speeds in the range of 20–1000 mV/s
Thermogravimetry
Thermal decomposition studies were performed in air atmosphere for complexes 2, 3, and 5. The TG curve together with SDTA results are presented in Figs. 910, and 11. The numerical data are presented in Table 3. In general, all the presented complexes are of V(IV) type with co-ligand (bpy or phen). Complicated decomposition processes overlapping with each other were observed. Similarly, as in our previous study, we observed that Schiff base ligand decomposed partly, with hydrazide component released first. A high temperature of solvent molecules release indicates their coordination as ligands. The ligand release in 3 is the most typical example. The most volatile hydrazide is released first, while the high-molecular-mass phen ligand is released second. The thermograms of 2 and 5 with relatively similar composition (the only difference in the benzhydrazide or hydroxybenzhydrazide component of ONO ligand) are very interesting, as they show very different thermal behavior, as presented in Figs. 9 and 11.

TG (left axis) and SDTA (right axis) curves for 2. Scan speed 10°/min, air

TG (left axis) and SDTA (right axis) curves for 3. Scan speed 10°/min, air

TG (left axis) and SDTA (right axis) curves for 5. Scan speed 10°/min, air
As the thermograms were measured in air, the final product is a V2O5 molecule. The formation of vanadium(V) oxide increases the mass of the final product by 4.46% for 2 and by 4.31% for 3, and this should be corrected from the final results presented in Table 3, for example from the calculated 88.35% to 84.05%, which is very close to the experimental value of 83.50%.
Pharmacological activity
Most of the tested complexes at concentration of 1 μM showed similar (2, 4, 9) or weaker (12, 13, 16) activity than BMOV used as a comparator of activity. These results may be explained by structural differences between these complexes. Complex 3 (Table 4) showed significantly lower ability to inhibit all the tested PTPs. This complex is characterized by the greatest differences between the inhibition potency of PTPs associated with antidiabetic activity (PTP1B, SHP1, and SHP2) and the inhibition of CD45 phosphatase. To assess the antidiabetic activity of 3, pharmacological activity studies were performed in cell models (Table 5). The antidiabetic activity of this complex has the same cellular profile and activity as BMOV. Radiolabeled glucose uptake by C2C12 myocytes is 126% for 3 and 125% for BMOV. The effect on glucose uptake in 3T3-L1 adipocytes is negligible for both complexes. In addition, both complexes do not show significant changes in myocyte and adipocyte viability and are not cytotoxic to HepG2 hepatocytes.
The studies of the pharmacological activity of the vanadium complexes showed that they are characterized by diverse activity in inhibiting tyrosine phosphatases. The degree of inhibition of tyrosine phosphatases described as the primary mechanism of action of vanadium compounds need not be crucial for antidiabetic activity such as PTP1B, SHP1, or SHP2. Due to the above, vanadium compound activity studies should include tests in cell models, not just using isolated enzymes.
Conclusions
The results presented herein indicate that the type of tridentate Schiff base ligand has a huge influence on the biological activity of the studied complexes. In previous publications, we presented a series of Schiff base ONO ligands with the same set of hydrazide component of ligands, with one or two selected aldehydes. Very different biological activity was observed with various hydrazides, but compared with data from other publications, it is obvious that the type of aldehyde used in the synthesis is of similar importance. Here, we present 24 new complexes synthesized with three different aldehydes. In most cases, we used phen as co-ligand to stabilize the oxidation state of V(IV), and here we use also the more flexible bpy co-ligand, with good stabilization effect. In general, however, more flexible ligand complexes were found to be less stable in solutions. Comparing the biological activity of complexes 2 and 3, the only difference is the co-ligand used (bpy or phen). It was found that complex 3 with the more rigid phen ligand shows similar activity to BMOV, while complex 2 with flexible bpy shows greater activity. In contrast, ca. 32% of complex 2 remains in DMSO-H2O mixture at 60 min after solution preparation, while complex 3 decomposes almost completely after this time. Similarly, at pH 2, complex 2 is slightly more stable than 3. All this may indicate that the biological activity of 3 is similar to that of BMOV, as both types of complex decompose after injection into simple VO2+ ions. Complex 2, being more stable, is more effective due to its only partial decomposition, supporting the observed fact that the type of L ligand influences the biological activity and in some cases V(IV) complexes with ONO ligands are threefold more effective than BMOV and human insulin [23].
References
Tuenter G, Van Leeuwen WF, Snepvangers L (1986) Ind Eng Chem Prod Res Dev 25:633–636
Bröer S, Hammer T (2000) Appl Catal B 28:101–111
Kamata H, Ohara H, Takahashi K, Yukimura A, Seo Y (2001) Catal Lett 73:79–83
McNeill JH, Yuen VG, Dai S, Orvig C (1995) Increased potency of vanadium using organic ligands. In: Srivastava AK, Chiasson J-L (eds) Vanadium compounds: biochemical and therapeutic applications. Springer, Boston, pp 175–180
Yuen VG, McNeill JH, Orvig C (1995) Can J Physiol Pharmacol 73:55–64
Yuen VG, Orvig C, McNeill JH (1993) Can J Physiol Pharmacol 71:263–269
Yuen VG, Vera E, Battell ML, Li WM, McNeill JH (1999) Diabetes Res Clin Pract 43:9–19
Orvig C, Caravan P, Gelmini L, Glover N, Herring FG, Li H, McNeill JH, Rettig SJ, Setyawati IA, Shuter E, Sun Y, Tracey AS, Yuen VG (1995) J Am Chem Soc 117:12759–12770
Ebrahimipour SY, Sheikhshoaie I, Kautz AC, Ameri M, Pasban-Aliabadi H, Rudbari HA, Bruno G, Janiak C (2015) Polyhedron 93:99–105
D’Cruz OJ, Uckun FM (2002) Expert Opin Investig Drugs 11:1829–1836
Crans DC, Henry L, Cardiff G, Posner BI (2019) Met Ions Life Sci 19:203–230
Torabi S, Mohammadi M, Shirvani M (2018) Trends Pharm Sci 4:87–94
Liu EC, Li W, Cheng XS (2019) Acta Chim Slov 66:971–977
Qian HY (2017) Russ J Coord Chem 43:780–786
Krishnan N, Konidaris KF, Gasser G, Tonks NK (2018) J Biol Chem 293:1517–1525
Koren S, Fantus IG (2007) Best Pract Res Clin Endocrinol Metab 21:621–640
Niu X, Xiao R, Wang N, Wang Z, Zhang Y, Xia Q, Yang X (2016) Curr Top Med Chem 16:811–822
Rehder D (2013) Dalton Trans 42:11749–11761
Scior T, Guevara-Garcia JA, Do QT, Bernard P, Laufer S (2016) Curr Med Chem 23:2874–2891
Gryboś R, Szklarzewicz J, Jurowska A, Hodorowicz M (2018) J Mol Struct 1171:880–887
Szklarzewicz J, Jurowska A, Hodorowicz M, Gryboś R, Matoga D (2019) J Mol Struct 1180:839–848
Szklarzewicz J, Jurowska A, Hodorowicz M, Gryboś R, Kruczała K, Głuch-Lutwin M, Kazek G (2020) J Coord Chem 73:986–1008
Szklarzewicz J, Jurowska A, Matoga D, Kruczała K, Kazek G, Mordyl B, Sapa J, Papież M (2020) Polyhedron 185:114589
Szklarzewicz J, Jurowska A, Hodorowicz M, Gryboś R (2019a) Sci Tech Innov 4:1–8
Szklarzewicz J, Jurowska A, Hodorowicz M, Matoga D, Gryboś R, Filipek B, Sapa J, Głuch-Lutwin M, Mordyl B, Kazek G (2019) Sci Tech Innov 4:9–20
Jurowska A, Szklarzewicz J, Hodorowicz M, Gryboś R (2019) Sci Tech Innov 4:21–29
Szklarzewicz J, Jurowska A, Hodorowicz M, Gryboś R (2019b) Sci Tech Innov 4:30–36
Szklarzewicz J, Jurowska A, Olszewska A, Hodorowicz M, Gryboś R, Kruczała K (2019) Sci Tech Innov 4:37–46
Hale AJ, Ter Steege E, den Hertog J (2017) Dev Biol 428:283–292
Knobler H, Elson A (2014) J Biomed Res 28:157–168
Courtney AH, Shvets AA, Lu W, Griffante G, Mollenauer M, Horkova V, Lo WL, Yu S, Stepanek O, Chakraborty AK, Weiss A (2019) Sci Signal 12:8151–8165
Dümmling S, Eichhorn E, Schneider S, Speiser B, Würde M (1996) Curr Sep 5:53–56
Pastula C, Johnson I, Beechem JM, Patton WF (2003) Comb Chem High Throughput Screen 6:341–346
Rigaku Oxford Diffraction (2015) CrysAlis PRO. Rigaku Oxford Diffraction, Yarnton, England
Altomare A, Cascarano G, Giacovazzo C, Guagliardi A, Burla MC, Polidori G, Camalli M (1994) J Appl Cryst 27:435
Altomare A, Burla MC, Camalli M, Cascarano G, Giacovazzo C, Guagliardi A, Moliterni AGG, Polidori G, Spagna R (1999) J Appl Cryst 32:115–119
Sheldrick GM (2015) Acta Cryst C 71:3
Schilt A, Taylor RC (1959) J Inorg Nucl Chem 9:211–221
Acknowledgments
This work was partly financed by the European Regional Development Fund under the Innovative Economy Programme 2007–2013 (WND POIG.01.03.01-174/09).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third-party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Szklarzewicz, J., Jurowska, A., Hodorowicz, M. et al. Characterization and antidiabetic activity of salicylhydrazone Schiff base vanadium(IV) and (V) complexes. Transit Met Chem 46, 201–217 (2021). https://doi.org/10.1007/s11243-020-00437-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-020-00437-1