
Abstract
Comprehending the corrosion mechanism of magnesium is of major interest in diverse fields. Typically, hydrogen evolution reaction is considered as the only cathodic reaction during Mg corrosion. However, recent works demonstrate importance of considering oxygen reduction reaction (ORR) as a second cathodic process at specific conditions. With oxygen micro-optode, we show that ORR rate was higher on slower corroding ultra-high-purity Mg (UHP-Mg), while lower on faster corroding commercially pure Mg (CP-Mg), where massive hydroxide layer impeded oxygen permeation. These findings shed light on yet another facet of complex mechanism of Mg corrosion.
Similar content being viewed by others


Magnesium, owing to its low density and high strength-to-weight ratio, has become a fascinating technological material attracting enormous academic and industrial interest. However, limited corrosion resistance of Mg confines its structural application including aerospace, automotive, and biomedical fields. Hence, promoting the understanding of the fundamental principles of corrosion behavior paves the path to further enhancement of corrosion protection for Mg and extends its industrial application.1,2,3
It has been extensively acknowledged that hydrogen evolution reaction (HER) is the main cathodic reaction during Mg corrosion at neutral or even alkaline conditions unlike other metals (zinc, iron etc.), where oxygen reduction reaction (ORR) is the main cathodic process.1,4,5,6,7,8 A convincing explanation for the predominance of HER is the highly negative corrosion potential exhibited by Mg.9
Even though compelling evidence supports HER as the primary cathodic reaction, ORR, as a secondary cathodic reaction, has progressively earned its value in the corrosion behavior of Mg and its alloys. It has been reported that ORR might participate in the formation of corrosion products of AZ91D during NaCl induced atmospheric corrosion.10 Wang et al.11 found that AZ31 exhibited different potentiodynamic behaviors in NaCl under deaerated and aerated conditions, where ORR caused higher current density. Recently, dissolved oxygen (DO) was found to be consumed on the surface of Mg coupled to Al and Cu, which certainly was not expected.12 Afterwards, direct DO consumption was measured by Silva et al. in active, and even relatively passive, corrosion regions of CP-Mg in NaCl solution. Simultaneous measurements of local DO concentration and current density (by SVET) was performed.13 Moreover, Strebl et al.14,15 provided the evidence for the contribution of ORR to cathodic processes on Mg alloy during atmospheric corrosion. In spite of these compelling evidences collected by different scientific groups, varying conditions and methods, the essential role of ORR during corrosion of Mg remains largely unexplored and poses a number of questions.
In Mg corrosion research, impurities, especially Fe particles embedded in Mg matrix have been considered as suitable cathodically active sites.16,17,18 Thus, cathodic reaction and growth kinetic of corrosion products have been significantly influenced by size, content, and distribution of impurities, which probably have potential relevance to ORR. The relation between impurities and oxide growth kinetic on Mg surface under ambient exposure condition was reported by McIntyre and Chen19 two decades ago. They found that Mg containing impurities of iron or manganese oxidized more rapidly than pure Mg and intensifying oxidation was found around impurities. DO consumption was detected by Xavier et al.20 during corrosion process occurring on Fe/Mg couple immersed in 0.1 M NaCl aqueous solution by setting different tip potential of scanning electrochemical microscopy. Höche et al. discovered that Fe and other noble metal impurities could re-deposit on Mg surface and enlarge the cathodic area.18 Further experimental evidence for Fe re-deposition was supplied by Mercier et al.21 A similar mechanism for Cu re-deposition during AZ91 corrosion was proposed and conclude that Cu could not be protected by Mg and tended to be oxidized due to the involvement of ORR.22 However, it has not been clarified until now whether the contribution of ORR to cathodic process is constant or it differs for Mg of varying purity grades and degradation rates.
In this work, oxygen consumption on Mg of two different purity grades was systematically investigated by in situ monitoring DO concentration in a mildly aggressive medium with localized techniques. This comparative study aimed at understanding the correlation between the intensity of ORR and impurity level. The evolution of Mg corrosion was elaborated regarding the variation of oxygen consumption particularly at the initial stage prior to visible filiform corrosion.
Counterintuitively high rate ORR on slowly corroding UHP-Mg
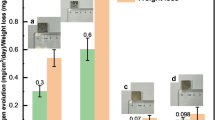
Figure 1a shows the distribution of local concentration of DO over the surfaces of CP-Mg and UHP-Mg immersed in 0.05 M NaCl electrolyte recorded by a micro-optode DO sensor. Clear consumption of DO was observed on both specimens, and DO consumption was higher in the middle of the sample typical for a diffusion-controlled ORR. This primarily indicates that ORR occurred all over Mg surface rather than exclusively on cathodic impurities. The lowest detected DO concentration was 2.5 ppm on UHP-Mg while 5.1 ppm on CP-Mg. This was unexpected because the corrosion rate of UHP-Mg was much lower than that of CP-Mg as also shown by the results of hydrogen evolution and accompanying weight loss measurements (Table 1). At first sight, one could expect higher rate of oxygen reduction during cathodic process on CP-Mg.

a the visual appearance and distribution of oxygen concentration above the surface. b the evolution of local DO concentration in the midpoint of each sample.
Considering the variation of DO concentration is also time-dependent, DO concentration was continuously measured in the midpoint of the sample in order to capture the time-varying process and avoid the edge effect (Fig. 1b). Once the specimens encountered the electrolyte, DO concentrations close to the magnesium/electrolyte interface sharply decreased due to the suddenly fierce ORR. The minimum DO concentration on UHP-Mg remained longer than that on CP-Mg, indicating that ORR rate maintained persistently higher on UHP-Mg. Subsequently, DO consumption was impeded, and DO concentration above CP-Mg started to increase and reached ~6.8 ppm in 1 h. By contrast, a moderate increase of DO concentration was displayed on UHP-Mg resulting in 4.6 ppm after 1 h, manifesting incessant DO consumption on the surface of UHP-Mg. The linear increment of DO concentration demonstrated that ORR rate decreased constantly and reach the plateau in 1 h due to the oxygen diffusion control. The difference of ORR rate observed for two grades of Mg was explained by different permeability of formed Mg(OH)2 to O2 dissolved in NaCl electrolyte.
O2 permeation limited by Mg(OH)2 and H2 evolved at Mg interface
Figure 2 presents the cross-section STEM micrographs of CP-Mg and UHP-Mg after 30 min immersion in air saturated 0.05 M NaCl electrolyte. High content of impurities embedded in CP-Mg accelerated the dissolution of Mg substrate, quickly generating a large amount of Mg2+ and OH− to intersect the Mg2+/Mg(OH)2 equilibrium line,23 causing a fast growth of Mg(OH)2 with a thick corrosion products layer (thickness ≈280 nm) and obviously larger hydroxide flakes.24 On the contrary, a low impurity level in UHP-Mg significantly decreased the corrosion rate accompanied by slower formation of Mg(OH)2. The thickness of corrosion products layer on UHP-Mg was near 70 nm, which was four times thinner than that on CP-Mg. Therefore, the formation of Mg(OH)2 on the surface of CP-Mg could certainly limit the transport of oxygen molecules, but water molecules continued infiltrating though hydroxide layer and combining the electrons left on the surface thus generating hydrogen gas. Furthermore, a considerable fraction of CP-Mg surface could be covered by generated H2 bubbles that blocked the cathodically active sites for ORR (as indicated by the visual appearance of CP-Mg shown in Fig. 1a). Meanwhile, numerous sites on CP-Mg were capable of supporting HER, promoting H2 evolution (as seen in Table 1 where the HER rate was ca. 53 times faster on CP-Mg than on UHP-Mg). Consequently, it is also reasonable that convection associated with H2 evolution helped replenishing dissolved O2 above the surface of CP-Mg.

a CP-Mg, b UHP-Mg. The cross-section STEM bright-field/dark-field images and EDX elemental distribution of each sample after 30 min of immersion in air saturated 0.05 M NaCl electrolyte.
Considerable cathodic ORR contribution on slowly corroding Mg
The results of this work promote better understanding of underlying mechanism of magnesium corrosion. Commonly, hydrogen evolution test is a typical measurement to quantify the corrosion rate of Mg. However, for magnesium possessing low degradation rate, ORR plays a significant role in total cathodic process. Strebl et al.15 estimated that during atmospheric corrosion of AZ91 alloy, ORR contribution to total cathodic reaction may reach 60% at certain relative humidity. Concomitantly, benefiting from the sealed construction of the eudiometer, ORR contribution to the total cathodic process was calculated based on the evolved hydrogen and weight loss after 24 h (Table 1). For slowly corroding UHP-Mg, the contribution of oxygen reduction reaction to the total cathodic process was reaching up to ~16.5%, while for faster corroding CP-Mg this contribution was at least ten times smaller, accounting only to 1.3%. Even though these values were calculated using H2 collection and weigh loss measurements which are certainly subjected to experimental errors probably of the order of those attributed to the ORR, in particular for the systems with low corrosion rates, measuring only HER (by H2 evolution test) leads to underestimated values of corrosion rates for slowly corroding Mg and its alloys. The most reliable test method in this case remains the weight loss measurement. The difference between the corrosion rate derived from hydrogen evolution and weight loss measurements has been reported before.25,26 Typically, this difference was attributed to rather high solubility of hydrogen gas in test electrolyte.27,28 This, obviously, remains a valid argument, but significant contribution of ORR should be also taken into account. Understanding the function of dissolved oxygen would not only provide insights into the fundamental research but also enlighten innovations in corrosion control strategies.
In summary, this work demonstrated that a significant amount of oxygen was consumed for ORR during corrosion of Mg in 0.05 M NaCl electrolyte. Most interesting, the higher level of oxygen consumption was revealed for slower corroding ultra-high-purity Mg, compared to lower rate of oxygen consumption by commercially pure Mg. Although the corrosion rate of two materials correlated with the level of impurities, ORR rate was two times higher for UHP-Mg at the initial corrosion stage (remaining DO concentration 2.5 ppm for UHP-Mg vs 5.1 ppm for CP-Mg in Fig. 1a). This counterintuitive behavior was explained by different morphology of main corrosion product, Mg(OH)2 formed on Mg surface immediately upon immersion. Thinner Mg(OH)2 generally formed on slowly dissolving UHP-Mg (ca. 70 nm) allowing diffusion of dissolved oxygen to the Mg/MgO interface. Stacked Mg(OH)2 formed on rapidly corroding CP-Mg (ca. 280 nm thick) restricted diffusion of oxygen molecules to CP-Mg interface. Moreover, CP-Mg interface could also be covered by H2 bubbles, hampering surface interaction with O2 molecules. These results provided additional experimental evidence for the strong role of ORR in corrosion of magnesium. The corrosion rates estimated based on H2 evolution measurements underestimate the actual corrosion damage, more precisely shown by weight loss analysis, and the difference is especially high for slowly corroding magnesium.
Methods
Samples
Commercially pure Mg (CP-Mg) and ultra-high-purity Mg (UHP-Mg; the elemental composition shown in Table 2) were machined in the rod shape (Φ = 2 mm) and embedded in epoxy resin with a round face exposed as a working surface (Fig. 1). The specimens were abraded successively with SiC papers from 1000 grit to 4000 grit and then cleaned in absolute ethanol. The test electrolyte was 0.05 M NaCl aqueous solution (pH = 5.8) prepared using deionized water and analytical grade reagent.
Instrumentation
A FireStingO2 fiber-optic oxygen micro-sensor coupled with an oxygen concentration meter, both from PyroscienceTM were employed to monitor the DO concentration. A needle-type retractable micro-optode with the diameter of 50 μm detected the DO concentration locally, 50 μm above the specimen surface. The detection limit is 0.01 ppm. For the distribution of DO concentration, a sample-centered area (3000 μm × 3000 μm) was scanned with the rate of 75 μm per step and the sampling interval 2 s. The total time of one map (40 × 40 grid) was ~1 h including the time needed to move the DO micro-optode.
DO micro-optode was integrated in a commercial SVET-SIET system from Applicable ElectronicsTM for movement and data acquisition by LV4 software from SciencewaresTM in parallel with PyroOxygenLogger from PyroscienceTM. The volume of 0.05 M NaCl electrolyte was 5 mL with the height of 5 mm in a custom made cell designed for local measurement.
Hydrogen evolution tests were carried out in a hermetically sealed eudiometers coupled with an electronic balance constantly recording the weight of the displaced water (described in Mei et al.29). The plates of ground CP and UHP-Mg with the surface area of 3.84 cm2 (1.2 cm × 1.2 cm × 0.2 cm) were placed in a container filled with 500 mL 0.05 M H2-presaturated NaCl electrolyte27,28 constantly agitated by a magnetic stirrer. The weight loss measurements were performed after removing corrosion products by chromic acid (aqueous solution of CrO3, 200 g L−1). All the experiments were repeated for at least three times. The average and standard deviation of the parallel tests are shown in the figures.
The cross-section morphology of corrosive specimen was characterized under a dual beam SEM/FIB platform (LYRA3 TESCAN) equipped with an EDX system (Oxford Inca with a silicon drift detector). The specimens were pre-corroded by immersing CP-Mg and UHP-Mg in air saturated 0.05 M NaCl electrolyte for 30 min, then cleaned in absolute ethanol and sparked with gold. For optimal resolution, the interest area was sputtered with a protective layer of platinum. FIB milling was subsequently adapted to produce a thin foil (~100 nm thick) via the standard in situ lift-out sample preparation routine in order to produce electron transparent lamellae.30 The cross-section micrographs were taken in STEM mode with the accelerating voltage of 30 kV.
Data availability
The authors declare that all data supporting the findings of this study are available within the paper.
References
Esmaily, M. et al. Fundamentals and advances in magnesium alloy corrosion. Prog. Mater. Sci. 89, 92–193 (2017).
Liu, M., Guo, Y., WangJ. & Yergin, M. Corrosion avoidance in lightweight materials for automotive applications. NPJ Mater. Degrad 2, 24 (2018).
Mei, D., Lamaka, S. V., Lu, X. & Zheludkevich, M. L. Selecting medium for corrosion testing of bioabsorbable magnesium and other metals – a critical review. Corros. Sci. 171, 108722 (2020).
Światowska, J., Volovitch, P. & Ogle, K. The anodic dissolution of Mg in NaCl and Na2SO4 electrolytes by atomic emission spectroelectrochemistry. Corros. Sci. 52, 2372–2378 (2010).
Chu, P.-W., Le Mire, E. & Marquis, E. A. Microstructure of localized corrosion front on Mg alloys and the relationship with hydrogen evolution. Corros. Sci. 128, 253–264 (2017).
Laurent, C., Scenini, F., MonettaT., Bellucci, F. & Curioni, M. The contribution of hydrogen evolution processes during corrosion of aluminium and aluminium alloys investigated by potentiodynamic polarisation coupled with real-time hydrogen measurement. NPJ Mater. Degrad 1, 6 (2017).
Fajardo, S., Bosch, J. & Frankel, G. S. Anomalous hydrogen evolution on AZ31, AZ61 and AZ91 magnesium alloys in unbuffered sodium chloride solution. Corros. Sci. 146, 163–171 (2019).
Williams, G., Kousis, C., McMurray, N. & Keil, P. A mechanistic investigation of corrosion-driven organic coating failure on magnesium and its alloys. NPJ Mater. Degrad 3, 41 (2019).
Cain, T., Bland, L. G., Birbilis, N. & Scully, J. R. A compilation of corrosion potentials for magnesium alloys. Corrosion 70, 1043–1051 (2014).
Jönsson, M., Persson, D. & Thierry, D. Corrosion product formation during NaCl induced atmospheric corrosion of magnesium alloy AZ91D. Corros. Sci. 49, 1540–1558 (2007).
Wang, L., Shinohara, T. & Zhang, B. P. Influence of deaerated condition on the corrosion behavior of AZ31 magnesium alloy in dilute NaCl solutions. Mater. Trans. 50, 2563–2569 (2009).
Snihirova, D., Taryba, M., Lamaka, S. V. & Montemor, M. F. Corrosion inhibition synergies on a model Al-Cu-Mg sample studied by localized scanning electrochemical techniques. Corros. Sci. 112, 408–417 (2016).
Silva, E. L., Lamaka, S. V., Mei, D. & Zheludkevich, M. L. The reduction of dissolved oxygen during magnesium corrosion. ChemistryOpen 7, 664–668 (2018).
Strebl, M. & Virtanen, S. Real-time monitoring of atmospheric magnesium alloy corrosion. J. Electrochem. Soc. 166, C3001–C3009 (2019).
Strebl, M., Bruns, M. & Virtanen, S. Editors’ choice—respirometric in situ methods for real-time monitoring of corrosion rates: part I. Atmospheric corrosion. J. Electrochem. Soc. 167, 021510 (2020).
Fajardo, S. & Frankel, G. S. Effect of impurities on the enhanced catalytic activity for hydrogen evolution in high purity magnesium. Electrochim. Acta 165, 255–267 (2015).
Yang, L. et al. Effect of iron content on the corrosion of pure magnesium: Critical factor for iron tolerance limit. Corros. Sci. 139, 421–429 (2018).
Höche, D. et al. The effect of iron re-deposition on the corrosion of impurity-containing magnesium. Phys. Chem. Chem. Phys. 18, 1279–1291 (2016).
Mcintype, N. S. & Chen, C. Role of impurities on Mg surfaces under ambient exposure conditions. Corros. Sci. 40, 1697–1709 (1998).
Joseph Raj, X. & Nishimura, T. Studies on galvanic corrosion of iron–magnesium couple by scanning electrochemical microscopy in 0.1 M NaCl solution. J. Ind. Eng. Chem. 41, 141–150 (2016).
Mercier, D., Światowska, J., Zanna, S., Seyeux, A. & Marcus, P. Role of segregated iron at grain boundaries on Mg corrosion. J. Electrochem. Soc. 165, C42–C49 (2018).
Li, J., Sun, W., Hurley, B., Luo, A. A. & Buchheit, R. G. Cu redistribution study during the corrosion of AZ91 using a rotating ring-disk collection experiment. Corros. Sci. 112, 760–764 (2016).
Santucci, R. J., McMahon, M. E. & Scully, J. R. Utilization of chemical stability diagrams for improved understanding of electrochemical systems: evolution of solution chemistry towards equilibrium. NPJ Mater. Degrad 2, 1 (2018).
Maltseva, A., Shkirskiy, V., Lefèvre, G. & Volovitch, P. Effect of pH on Mg(OH)2 film evolution on corroding Mg by in situ kinetic Raman mapping (KRM). Corros. Sci. 153, 272–282 (2019).
Zainal Abidin, N. I. et al. The in vivo and in vitro corrosion of high-purity magnesium and magnesium alloys WZ21 and AZ91. Corros. Sci. 75, 354–366 (2013).
Hofstetter, J. et al. Assessing the degradation performance of ultrahigh-purity magnesium in vitro and in vivo. Corros. Sci. 91, 29–36 (2015).
Bland, L. G., King, A. D., Birbilis, N. & Scully, J. R. Assessing the corrosion of commercially pure magnesium and commercial AZ31B by electrochemical impedance, mass-loss, hydrogen collection, and inductively coupled plasma optical emission spectrometry solution analysis. Corrosion 71, 128–145 (2015).
King, A. D., Birbilis, N. & Scully, J. R. Accurate electrochemical measurement of magnesium corrosion rates; a combined impedance, mass-loss and hydrogen collection study. Electrochim. Acta 121, 394–406 (2014).
Mei, D. et al. The role of individual components of simulated body fluid on the corrosion behavior of commercially pure Mg. Corros. Sci. 147, 81–93 (2019).
Taheri, M., Danaie, M. & Kish, J. R. TEM examination of the film formed on corroding Mg prior to breakdown. J. Electrochem. Soc. 161, C89–C94 (2014).
Acknowledgements
Mr. Cheng Wang, Mr. Di Mei, Ms. Linqian Wang, and Mr. Min Deng thank China Scholarship Council for the award of fellowship and funding (No. 201806310128, 201607040051, 201706370183, and 201606370031). The technical support of Mr. Daniel Streath, Mr. Volker Heitmann, and Mr. Ulrich Burmester during this work is gratefully acknowledged.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
C.W. conducted the experiments, data analysis, plotted figures, and wrote the first draft of the manuscript. D.M. and G.W. conducted experiments. D.M., L.W., and M.D. contributed to data analysis and plotting figures. S.V.L. contributed to establishing experimental approaches, plotting figures and writing text. S.V.L. and M.L.Z. contributed to developing the overall research strategy, data analysis, manuscript review, and supervised the work. All coauthor read and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, C., Mei, D., Wiese, G. et al. High rate oxygen reduction reaction during corrosion of ultra-high-purity magnesium. npj Mater Degrad 4, 42 (2020). https://doi.org/10.1038/s41529-020-00146-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41529-020-00146-1
This article is cited by
-
Effect of surface modification on interfacial behavior in bioabsorbable magnesium wire reinforced poly-lactic acid polymer composites
npj Materials Degradation (2023)
-
Oxygen-sensitive nanoparticles reveal the spatiotemporal dynamics of oxygen reduction during magnesium implant biodegradation
npj Materials Degradation (2022)
