
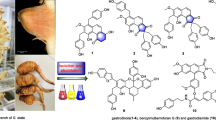
Abstract
Myrcauones A–D (1–4), four new phloroglucinol–terpene adducts were isolated from the leaves of Myrciaria cauliflora. Their structures with absolute configurations were elucidated by combination of spectroscopic analysis, single crystal X-ray diffraction, and electronic circular dichroism (ECD) calculations. Compound 1 was a rearranged isobutylphloroglucinol–pinene adduct featuring an unusual 2,3,4,4a,10,11-hexahydro-1H-3,11a-methanodibenzo[b,f]oxepin backbone. Compound 4 showed moderate antibacterial activity against Gram-positive bacteria including multiresistant strains.
Graphic Abstract

Similar content being viewed by others


1 Introduction
The plant Myrciaria cauliflora is an evergreen shrub and widely distributed in southern and central Brazil [1]. This plant has been traditionally used as a folk medicine to treat asthma, diarrhea, and gastrointestinal diseases [2, 3]. Previous phytochemical investigations on this plant only reported essential oils and flavonoids [4,5,6]. As a part of our efforts to search for structural unique and bioactive constituents from Myrtaceae plants [7,8,9,10], four new phloroglucinol–terpene adducts, myrcauones A–D (1–4), were isolated from the leaves of M. cauliflora. Their structures and absolute configurations were determined by means of 1D and 2D NMR spectroscopy, X-ray diffraction analysis, and electronic circular dichroism (ECD) calculations. Compound 1 is a rearranged isobutylphloroglucinol–pinene adduct featuring an unusual 2,3,4,4a,10,11-hexahydro-1H-3,11a-methanodibenzo[b,f]oxepin backbone. All isolates were evaluated for their antibacterial activities. Herein, we describe the isolation, structural elucidation, and antibacterial activities of these myrcauones A–D (1–4).
2 Results and Discussion
2.1 Structural Elucidation
Compound 1 was obtained as yellow gum. The molecular formula of 1 was established as C22H32O3 by its HRESIMS data (m/z 345.2423 [M+H]+, calcd for C22H33O3: 345.2424). The UV spectrum displayed absorption maximum at 206 nm. The IR spectrum showed characteristic absorptions for hydroxyl group (3475 cm−1) and aromatic ring (1611 and 1488 cm−1). The 1H NMR spectrum of 1 suggested the presence of an olefinic proton [δH 6.23 (1H, s, H-5′)], a hydroxyl group [δH 4.75 (1H, s, 2′-OH)], a methoxy group [δH 3.76 (3H, s, H3-11′)], an isopropyl moiety [δH 1.97 (1H, m, H-8′), 0.52 (3H, d, J = 6.8 Hz, H3-9′), and 1.03 (3H, d, J = 6.8 Hz, H3-10′)], and three tertiary methyls [δH 2.06 (3H, s, H3-12′), 0.90 (3H, s, H3-9), and 0.79 (3H, s, H3-8)]. The 13C NMR and DEPT spectra of 1 exhibited 22 carbon signals including 7 quaternary carbons (5 olefinic ones), 5 methines (an oxygenated and an olefinic ones), 4 methylenes, and 6 methyls (an oxygenated one). The aforementioned data implied that 1 could be an isobutylphloroglucinol–monoterpene adduct (Fig. 1) [11].

Chemical structures of 1–4
A comparison of the NMR data of 1 with those of melaleucadine A [11] indicated the presence of an uncommon rearranged β-pinene unit (part 1a), which was further confirmed by the two spin systems (H-2 to H-6 and H-10 to H-9′/H-10′) in its 1H–1H COSY spectrum (Fig. 2) and the HMBC correlations between H3-8/H3-9 and C-1/C-4/C-7 and between H2-10 and C-1/C-2/C-6/C-7. In addition, the HMBC correlations between H-5′ and C-1′/C-3′/C-4′/C-6′, between H3-12′ and C-2′/C-3′/C-4′, between H3-11′ and C-4′, and between H-7′ and C-1′/C-2′/C-6′, allowed the establishment of an isobutylphloroglucinol moiety (part 1b). Furthermore, the HMBC correlations between H-7′ and C-1, and between H2-10 and C-1′ defined the connection of 1a and 1b via C-7′–C-10 bond. Finally, the leftover oxygen atom was anticipated to connect C-2 (δC 88.6) with C-6′ (δC 159.4) to form an uncommon 2,3,4,5-tetrahydrooxepine ring on the basis of the HMBC correlation between H-2 and C-6′ as well as the molecular formula information.

Key 1H–1H COSY and HMBC correlations of 1, 2, 4
The relative configuration of 1 was established by a NOESY experiment (Fig. 3). The NOE correlations between H-10β and H-2/Me-8/H-8′ indicated that H-2, Me-8, and the isopropyl group (C-9′/8′/10′) were β-oriented. Meanwhile, the correlations between H-10α and H-6a/H-7′ as well as between H-6a and Me-9 suggested that Me-9 and H-7′ were α-oriented. To determine the absolute configurations of 1, a comparison of its experimental and calculated ECD data was performed. The experimental ECD spectrum of 1 exhibited negative Cotton effects at 211 (Δε + 6.2) and 277 (Δε + 0.7) nm, and a negative one at 238 (Δε − 0.5) nm, which were similar with those in the calculated CD spectrum for 1R,2R,4S,7′S-isomer (Fig. 4). Thus, the absolute configuration of 1 was determined as 1R, 2R, 4S, and 7′S.

Key NOESY correlations of 1–4

Calculated and experimental ECD spectra of 1–3
The molecular formula of compound 2 was determined as C22H32O4 by its HRESIMS data (m/z 361.2386 [M+H]+, calcd for C22H33O4: 361.2373). The IR spectrum showed absorptions of hydroxyl (3357 cm−1), carbonyl group (1656 cm−1), and double bonds (1605 and 1462 cm−1). The 1H NMR data (Table 1) for an olefinic proton [δH 5.33 (1H, s, H-3′)], a methoxy group [δH 3.75 (3H, s, H3-11′)], an isopropyl moiety [δH 2.86 (1H, m, H-8′), 0.61 (3H, d, J = 6.8 Hz, H3-9′), and 0.93 (3H, d, J = 6.8 Hz, H3-10′)], and three tertiary methyls [δH 1.51 (3H, s, H3-12′), δH 1.23 (3H, s, H3-9), and 0.96 (3H, s, H3-10)] indicated that 2 could be an isobutylphloroglucinol–monoterpene adduct [12].
The 1H–1H COSY spectrum of 2 revealed the presence of two spin systems (H-2 to H-6 and H-7 to H-9′/H-10′) (Fig. 2). Accordingly, a β-pinene unit (part 2a) could be established by the HMBC correlations between H-2 and C-4/C-6/C-7, between H2-3/H2-5 and C-1/C-8, and between H3-9/H3-10 and C-2/C-4/C-8. Furthermore, comparison of its NMR data with those of the known compound baeckfrutone H indicated the existence of an isobutyrylphloroglucinol moiety (part 2b), which was further confirmed by the HMBC correlations between H-3′ and C-1′/C-2′/C-4′/C-5′, between H3-12′ and C-4′/C-5′/C-6′, between H3-11′ and C-4′, and between H-7′ and C-1′/C-2′/C-6′ [12]. The closure mode of dihydropyran ring which connected the two fragments (2a and 2b) could be deduced on the basis of the molecular formula information and the downfield chemical shift at C-1 (δC 85.4).
In the NOESY spectrum, the correlations between H-3b and H-2/H-4/Me-9, between H-2 and H-7′, between H-7β and Me-10/H-7′, as well as between H-3a and Me-12′ indicated that these protons were all β-oriented (Fig. 3). The absolute configuration of 2 was determined by ECD calculation. The experimental ECD spectrum of 2 displayed positive cotton effects at 244 (+ 11.7) and 296 (+ 8.1) nm, and negative ones at 203 (− 19.8) and 337 (− 1.8) nm, which were similar to those in the calculated spectrum for 1R,2R,4S,5′S,7′R-2 (Fig. 4). Thus, the absolute configuration of 2 was identified as 1R, 2R, 4S, 5′S, 7′R.
Compound 3 possessed the same molecular formula as 2 on the basis of its HRESIMS data (m/z 361.2391 [M+H]+, calcd for C22H33O4, 361.2373). Analyses of the NMR data of 3 and comparison with those of 2 indicated that these two compounds had the same planar structure but differed in their relative configurations. The downfield chemical shifts of C-2 (from δC 45.2 to 53.0) and C-7 (from δC 32.7 to 33.7), as well as the upfield chemical shifts of C-6 (from δC 30.8 to 27.4), C-7′ (from δC 33.6 to 32.2) revealed that 3 was a C-7′ epimer of 2. This deduction was confirmed by the NOE correlations between H-3b and H-2/H-4/Me-9, between H-2 and H-7β, between H-7β and H-8′/Me-9′/Me-10, between H-3a and Me-12′, and between H-7α and H-7′ (Fig. 3). Finally, the agreement of the ECD curve of 3 with those of the calculated 1R,2R,4S,5′S,7′S-3 (Fig. 4) allowed the assignment its absolute configuration.
Compound 4 was obtained as colorless blocks. Its molecular formula was determined to be C27H40O4 by its HRESIMS data at m/z 429.2996 [M+H]+ (calcd for C27H41O4: 429.2999). Comparison of the NMR data of 4 with those of 3 suggested that they had the same isobutyrylphloroglucinol moiety (part 4b) (Fig. 2). The remaining NMR signals for 15 carbons implied the presence of a sesquiterpene moiety. The spin systems (from H-3 to H-10 and from H-7 to H-9′/H-10′) established by the 1H–1H COSY spectrum as well as the HMBC correlations between H3-14/ H3-15 and C-1/C-10, between H2-13 and C-7/C-9, and between H3-12 and C-3/C-5 indicated the presence of a caryophyllene unit (part 4a) (Fig. 2), which was further confirmed by comparison of the NMR data of 4 with those of myrtucommulone K [10]. Furthermore, the HMBC correlations between H-7′ and C-4/C-5/C-1′ indicated the connection of parts 4a and 4b via a C-5 and C-7′ bond. Finally, the closure mode of dihydropyran ring which connected the two fragments (4a and 4b) could be deduced on the basis of the molecular formula information and the downfield chemical shift at C-4 (δC 85.8).
In the NOESY spectrum, the correlations between H-5 and H-1/H-7′, between H-1 and Me-15, between H-9 and Me-14, as well as between Me-12 and H-8′/Me-12′ suggested that the relative configurations of C-1, C-4, C-5, C-9, and C-7′ were identical to those of myrtucommulone K (Fig. 3). Additionally, the structure and absolute configuration of 4 was unambiguously determined by X-ray crystallographic analysis using Cu Kα radiation with the Flack parameter [0.08 (13)] (Fig. 5). Hence, the absolute configuration of 4 was defined as 1R, 4R, 5S, 9S, 5′S and 7′R.

X-ray ORTEP drawing of 4
2.2 Bioactivity Evaluation
The antibacterial activities of compounds 1–4 against Gram-positive strains Staphylococcus aureus ATCC 43300 (MRSA), S. aureus ATCC 700699 (VISA), S. aureus ATCC 25923 and Enterococcus faecalis ATCC 29212 and Gram-negative strains Pseudomonas aeruginosa ATCC 27853, Escherichia coli ATCC 25922 and Klebsiella pneumoniae ATCC 700603 were measured by broth microdilution method. As a result, compound 4 exhibited moderate antibacterial activity against all Gram-positive strains with MIC value of 32 μg/mL (Table 2).
3 Experimental
3.1 General Methods
Melting points were obtained on a Buchi melting point B-545 apparatus (Buchi Instrument, Switzerland) and are uncorrected. Optical rotations were measured on a JASCO P-2000 digital polarimeter (Jasco Co., Ltd., Tokyo, Japan) at room temperature. IR spectra were determined on a JASCO FT/IR-4600 plus Fourier transform infrared spectrometer (Jasco Co., Ltd., Tokyo, Japan) using KBr pellets. UV spectra were recorded on a JASCO V-550 UV/Vis spectrophotometer (Jasco Co., Ltd., Tokyo, Japan). CD spectra were obtained on a ChirascanqCD (Applied Photophysics Ltd., Surrey, UK). HRESIMS spectra were acquired on an Agilent 6210 LC/MSD TOF mass spectrometer (Agilent Technologies, CA, USA). NMR spectra were measured on Bruker AV-500 or AV-400 spectrometers (Bruker, Switzerland) with TMS as internal standard, and chemical shifts were denoted in δ values (ppm). X-ray crystallographic analyses were carried out on an Agilent Gemini S Ultra CCD diffractometer with Cu Kα radiation (λ = 1.54178 Å). Silica gel (200–300 mesh; Qingdao Marine Chemical, Inc., Qingdao, People’s Republic of China), Sephadex LH-20 (Pharmacia Biotech AB, Uppsala, Sweden), and reversed-phase C18 silica gel (YMC, Kyoto, Japan) were used for column chromatography (CC). Preparative HPLC was carried out on an Agilent 1260 Chromatograph equipped with a G1311C pump and a G1315D photodiode array detector (Agilent Technologies, CA, USA) with a semi-preparative C18 reversed-phase column (Cosmosil, 10 mm × 250 mm, 5 μm). All solvents used in CC and HPLC were of analytical grade (Shanghai Chemical Plant, Shanghai, People’s Republic of China) and chromatographic grade (Fisher Scientific, New Jersey, USA), respectively.
3.2 Plant Material
The leaves of M. cauliflora were collected from Nanning city, Guangxi Province of People’s Republic of China, in July of 2018. A voucher specimen (No. 2018070607) identified by Professor Guang-Xiong Zhou (Jinan University) was deposited in the Institute of Traditional Chinese Medicine and Natural Products, Jinan University, Guangzhou, People’s Republic of China.
3.3 Extraction and isolation
The air-dried leaves of M. cauliflora (15 kg) were powdered and extracted with 95% EtOH (v/v, 50 L) at room temperature. The extract (2.2 kg) was suspended in H2O and extracted with petroleum ether (PE, b.p. 60–90 °C). The PE extract (673.2 g) was subjected to a silica gel column chromatography using cyclohexane–EtOAc (100:0 → 0:100, v/v) as eluent to afford 10 fractions (Frs. A–J). Fr. G (48.3 g) was further separated by silica gel column using a gradient cyclohexane–EtOAc (100:0 → 0:100, v/v) to give 8 subfractions (Frs. G1–G8). Subfraction G5 (10.7 g) was chromatographed on Sephadex LH-20 (CH2Cl2/MeOH, 1:1, v/v) to obtain three subfractions (Frs. G5A–G5C). Subfraction G5B (7.3 g) was subjected to ODS column using MeOH/H2O (50:50 → 100:0, v/v) and further purified by semi-preparative reversed-phase HPLC (MeOH/H2O, 70:30, v/v, 3 mL/min) to afford 1 (12.5 mg, tR 41.8 min), 3 (11.3 mg, tR 33.7 min) and 4 (15.7 mg, tR 49.3 min). Subfraction G6 (5.3 g) was separated on Sephadex LH-20 (CH2Cl2/MeOH, 1:1, v/v) to obtain 2 (7.3 mg).
Compound 1 yellow gum (CH3OH); [α] 25D = + 119 (c = 0.50, MeOH); UV (MeOH) λmax (log ε) 206 (3.73) nm; IR (KBr) νmax 3475, 2977, 2954, 2876, 1611, 1584, 1488, 1445, 1415, 1385, 1307, 1236, 1199, 1132, 1084, 1035, 1014, 982, 904, 831 cm−1; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz), see Table 1; HRESIMS m/z 345.2423 [M+H]+ (calcd for C22H33O3: 345.2424); ECD (MeCN, Δε) 211 (+ 6.2), 238 (− 0.5), 277 (+ 0.7) nm.
Compound 2 yellow oil (CH3OH); [α] 25D = + 59 (c = 0.50, MeOH); UV (MeOH) λmax (log ε) 202 (3.85), 245 (3.92), 296 (3.50) nm; IR (KBr) νmax 3357, 2957, 2933, 2871, 1656, 1605, 1462, 1385, 1353, 1236, 1137, 1092, 998, 983, 893, 843 cm−1; 1H NMR (CDCl3, 400 MHz) and 13C NMR (CDCl3, 100 MHz), see Table 1; HRESIMS m/z 361.2386 [M+H]+ (calcd for C22H33O4: 361.2373); ECD (MeCN, Δε) 203 (− 19.8), 244 (+ 11.7), 296 (+ 8.1), 337 (− 1.8) nm.
Compound 3 yellow oil (CH3OH); [α] 25D = − 63 (c = 0.50, MeOH); UV (MeOH) λmax (log ε) 202 (3.71), 245 (3.77), 297 (3.32) nm; IR (KBr) νmax 3359, 2958, 2924, 2871, 1656, 1603, 1463, 1388, 1372, 1259, 1232, 1136, 1089, 989, 899, 842 cm−1; 1H NMR (CDCl3, 400 MHz) and 13C NMR (CDCl3, 100 MHz), see Table 1; HRESIMS m/z 361.2391 [M+H]+ (calcd for C22H33O4: 361.2373); ECD (MeCN, Δε) 203 (+ 18.0), 245 (− 20.5), 293 (− 9.6), 333 (+ 2.5) nm.
Compound 4 colorless blocks (CH3OH); m.p. 162–163 ℃; [α] 25D = − 42 (c = 0.50, MeOH); UV (MeOH) λmax (log ε) 205 (3.86), 247 (3.87), 310 (3.44) nm; IR (KBr) νmax 3336, 2957, 2870, 1663, 1615, 1462, 1386, 1364, 1284, 1259, 1233, 1176, 1140, 999, 941, 886, 844 cm−1; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz), see Table 1; HRESIMS m/z 429.2996 [M + H]+ (calcd for C27H41O4: 429.2999).
3.4 X-Ray Analysis
Crystal data for 4 C27H40O4, monoclinic, space group P21, a = 10.2148 (3) Å, b = 24.2552 (4) Å, c = 20.4139 (5) Å, α = 90°, β = 97.254 (2)°, γ = 90°, V = 5017.3 (2) Å3, T = 293 (2) K, Z = 2, Dcalcd = 1.135 g cm−3, F(000) = 1872, 20761 reflections measured (2.18° ≤ θ ≤ 73.86°), 13408 unique (Rint = 0.0389, Rsigma = 0.0574) which were used in all calculations. The final R1 was 0.0916 [I > 2σ(I)] and wR2 was 0.2884 (all data). CCDC-1998470 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
3.5 Antibacterial Activity Assay
Staphylococcus aureus ATCC 43300 (methicillin-resistant S. aureus, MRSA), S. aureus ATCC 700699 (vancomycin-intermediate S. aureus, VISA), S. aureus ATCC 25923, E. faecalis ATCC 29212, P. aeruginosa ATCC 27853, E. coli ATCC 25922 and K. pneumoniae ATCC 700603 were standard isolates from ATCC (Manassas, VA, USA). The MIC values were measured using a previously reported method [7]. Ciprofloxacin and vancomycin were used as positive controls.
References
S.B. Wu, C. Long, E.J. Kennelly, Food Res. Int. 54, 148–159 (2013)
K.A. Reynertson, A.M. Wallace, S. Adachi, R.R. Gil, H. Yang, M.J. Basile, J. D’Armiento, I.B. Weinstein, E.J. Kennelly, J. Nat. Prod. 69, 1228–1230 (2006)
M. Giraldi, N. Hanazaki, Acta Bot. Bras. 24, 395–406 (2010)
I. Plagemann, U. Krings, R.G. Berger, M.R. Marostica Jr., J. Essent. Oil Res. 24, 45–51 (2012)
C.A. Moura, D. Luckmann, J.C.U. Laureth, D. Christ, G.C. Braga, D. Paulus, S.R.M. Coelho, L.F. Francisco, Afr. J. Agric. Res. 11, 3154–3161 (2016)
N.C.T. Mariani, G.H.A. Teixeira, K.M.G. de Lima, T.B. Morgenstern, V. Nardini, L.C.C. Júnior, Food Chem. 174, 643–648 (2015)
J.Q. Cao, X.J. Huang, Y.T. Li, Y. Wang, L. Wang, R.W. Jiang, W.C. Ye, Org. Lett. 18, 120–123 (2016)
F. Liu, W.J. Lu, N.P. Li, J.W. Liu, J. He, W.C. Ye, L. Wang, Fitoterapia 128, 93–96 (2018)
M. Chen, L.F. Chen, M.M. Li, N.P. Li, J.Q. Cao, Y. Wang, Y.L. Li, L. Wang, W.C. Ye, RSC Adv. 7, 22735–22740 (2017)
C. Liu, S. Ang, X.J. Huang, H.Y. Tian, Y.Y. Deng, D.M. Zhang, Y. Wang, W.C. Ye, L. Wang, Org. Lett. 18, 4004–4007 (2016)
X. Xie, L. Wu, Z.R. Cui, M.H. Yang, Y. Yin, J. Luo, L.Y. Kong, Tetrahedron Lett. 60, 1011–1013 (2019)
X.J. Qin, Y.E. Zhi, H. Yan, Y. Zhang, H. Liu, Q. Yu, S. Wang, Q. Zhao, L. He, X. Ma, L.K. An, H.Y. Liu, Tetrahedron 74, 6658–6666 (2018)
Acknowledgements
This work was supported by the National Key R&D Program of China (No. 2017YFC1703800), the Program for the National Natural Science Foundation of China (Nos. 81822042, 81630095), the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (2017BT01Y036), Key-Area Research and Development Program of Guangdong Province (2020B1111110004), and the High-performance Computing Platform of Jinan University.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, M., Cao, JQ., Wang, WJ. et al. Four New Phloroglucinol-Terpene Adducts from the Leaves of Myrciaria cauliflora. Nat. Prod. Bioprospect. 11, 111–118 (2021). https://doi.org/10.1007/s13659-020-00288-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-020-00288-4