
Abstract
Rumen microbiota play a key role in the digestion and utilization of plant materials by the ruminant species, which have important implications for greenhouse gas emission. Yet, little is known about the key taxa and potential gene functions involved in the digestion process. Here, we performed a genome-centric analysis of rumen microbiota attached to six different lignocellulosic biomasses in rumen-fistulated cattle. Our metagenome sequencing provided novel genomic insights into functional potential of 523 uncultured bacteria and 15 mostly uncultured archaea in the rumen. The assembled genomes belonged mainly to Bacteroidota, Firmicutes, Verrucomicrobiota, and Fibrobacterota and were enriched for genes related to the degradation of lignocellulosic polymers and the fermentation of degraded products into short chain volatile fatty acids. We also found a shift from copiotrophic to oligotrophic taxa during the course of rumen fermentation, potentially important for the digestion of recalcitrant lignocellulosic substrates in the physiochemically complex and varying environment of the rumen. Differential colonization of forages (the incubated lignocellulosic materials) by rumen microbiota suggests that taxonomic and metabolic diversification is an evolutionary adaptation to diverse lignocellulosic substrates constituting a major component of the cattle’s diet. Our data also provide novel insights into the key role of unique microbial diversity and associated gene functions in the degradation of recalcitrant lignocellulosic materials in the rumen.
Similar content being viewed by others


Introduction
As the main sources of energy for their growth and lactation, ruminants depend largely on the end products of the forage digestion, i.e., energy accessible short chain fatty acids (SCFAs) [1]. These animals lack enzymes that are required to break down the complex lignocellulosic carbohydrates as the major component of the forage [2, 3]. For this, they rely on microbial communities inhabiting their gastrointestinal tract particularly the rumen. Rumen microbes are thus equipped with diverse carbohydrate active enzymes (CAZymes) for breaking down the complex structure of plant cell-wall materials. They produce a complex cocktail of enzymes for the efficient degradation of plant polysaccharides, including diverse glycoside hydrolases (GHs), carbohydrate esterases (CEs), and polysaccharide lyases (PLs) together with their accessory non-catalytic carbohydrate-binding modules (CBMs), enabling their targeted attachment to lignocellulosic substrates. However, little is known about specific adaptations of microbes to the complex and dynamic rumen environment.
Rumen microbes utilize different mechanisms to invade complex plant polysaccharide substrates and thus there may be some specialization in substrate types utilized and functional capabilities across different members of the rumen microbial community. Some rumen bacteria including species of Ruminococcus have evolved multienzyme assemblies, the cellulosomes, in which multiple CAZymes bind via dockerin domains to cell surface localized scaffolding proteins containing repeated cohesin domains [4]. These cohesin–dockerin interactions will enable a concerted action of multiple different CAZymes with varying substrate specificities on lignocellulosic substrates and thus enhance the degradation of recalcitrant polysaccharides, e.g., cellulose and hemicellulose, by orders of magnitude [5]. However, Bacteroidetes as abundant members of the rumen microbiome, lack the cellulosome. In these bacteria, all genes required for utilizing specific glycan substrate are physically localized on the same portion of their genomes featured by the presence of a susC/D gene pair. These special arrangements of CAZymes are known as polysaccharide utilization loci (PULs) [6, 7]. Since each PUL is dedicated to the break down of specific complex glycan substrate, the combination of enzymes present in PULs determines the carbohydrate degrading capacity of the bacteria [8].
In addition, to degrade plant materials efficiently, rumen microbes must be able to colonize and to attach feed particulates. Particle-associated microbes that are embedded in a biofilm matrix play central role in rumen digestion. The attachment of rumen microbes to feed particles promotes their prolonged residence in the rumen and enables their enzymes to be in contact with the substrates [9,10,11]. Bacterial attachment to feed particles is initiated within 5 min of feed entry and gets established within 1 h of rumen incubation [11, 12]. In this regard, the physicochemical properties of feeds proved to be the major factors determining microbial colonization and digestion in the rumen [12,13,14,15]. Particularly, rumen microbial communities are distinct between forage-fed and concentrate-fed animals [16, 17]. Despite these advances, we still have little information about the diversity of rumen microbiota colonizing forages of different lignocellulosic compositions as well as their dynamic changes along the digestion process in the rumen.
The rumen microbial communities contribute to variations in feed efficiency in the host [18, 19]. Several 16S rRNA gene-based metabarcoding studies report that rumen microbes show significant variations with respect to their ability to attach to feed particles [12, 14, 15, 20]. However, little is known about the genomic constituents of particle-attached microbiota and the variations in their CAZyme repertoires which determine their lignocellulose degrading potential in the rumen. Most particle adhering microbes (>70%) are unable to grow in routine culture conditions [21]. Culture-independent metagenomics has enabled us to gain insight into the functional significance of these microorganisms and their contribution to lignocellulose degradation in the rumen. This, together with recent advances in bioinformatics analyses, has now made it possible to recover near complete and draft genome sequences of uncultured rumen bacteria and archaea from metagenome-assembled contigs [22,23,24,25,26]. A recent study, for example, recovered 4941 metagenome-assembled genomes with >80% completeness from the cattle rumen, many of them (>70%) had <95% nucleotide identity with the existing genomes, suggesting their novel entities [22]. Metagenome analyses have also revealed that rumen microbiota could be a potential source of novel enzymes for biotechnological applications including biofuel, paper, textile, and food industries [23, 27]. Knowledge about the diversity and functional capabilities of rumen microbiota may provide opportunities to improve feed digestion efficiency of domestic ruminants through microbiome engineering for an enhanced lignocellulose degrading capacity.
Here, we analyzed 333 gigabase pairs (Gbp) of metagenome sequencing data from rumen microbes attached to six different lignocellulosic biomasses following their in situ incubation in the cattle rumen. Our aim was to determine the key taxa and associated gene functions involved in the digestion of different lignocellulosic substrates and their potential shifts during the digestion process. In our previous 16S rRNA gene metabarcoding [15], we showed that rumen microbiota had differential preferences for attachment to forages of varying lignocellulose compositions. Here we demonstrate how this differential colonization is reflected in their genomic potential for the degradation of different plant cell-wall polymers. We also hypothesized that changes in community of forage-attached microbiota along the length of rumen incubation reflect their lignocellulose degrading potential. To evaluate this hypothesis, we sequenced and de novo assembled the whole metagenomes of fiber-attached rumen microbiota from three rumen-fistulated cattle and reconstructed the genome sequences of rumen microbiota involved in the break down of different lignocellulosic biomasses with varying physicochemical properties.
Material and methods
Sample collection
The details of three Taleshi bulls, rumen cannulation technique, the physiochemical properties, preparation, and sampling of the six common lignocellulosic forages including camelthorn (AP), common red (CR), date palm (DP), rice straw (RS), Kochia (KS), and Salicornia (SC) have been presented in our previous study [15] and briefly summarized in Supplementary Note and Fig. S1. The experiment was approved by the Ethics Committee for Animal Experiments of the Animal Science Research Institute of Iran.
DNA extraction and metagenome sequencing
Metagenome DNA extraction from microbial cells tightly attached to the feed particulates was performed as described previously [15, 28]. Metagenome library preparation was performed according to TruSeq DNA Library Preparation Kit (Illumina, San Diego, CA, USA). The quantity of each library was evaluated using a Qubit fluorimeter (Invitrogen, Carlsbad, CA, USA). Metagenome sequencing was performed at the Novogene Inc. (Beijing, China) using Illumina HiSeq 2500 system with 150 bp paired-end sequencing of ~340 bp fragments.
De novo metagenome assembly and binning
All metagenome sequences were co-assembled based on all 24 samples across the six forages (co-assembly) using MEGAHIT v1.2.9 [29] with the following options: --k-min 31, --k-max 141, --k-step 10, --min-count 2, and --min-contig-len 200. We also assembled reads generated for each forage separately (forage-specific assemblies) using SPAdes v3.13.1 [30] with k-mers 31, 51, 71, 91, 111, and 127. To obtain the coverage profiles of assembled contigs, reads were mapped back to the filtered contigs (e.g., contigs longer than 2000 bp) using Bowtie2 v2.3.5 [31]. Samtools v1.9 [32] was used to convert and sort BAM files. Genome bins were reconstructed using MetaBat2 v2.14 [33] and MaxBin2 v2.2.6 [34] on the co-assembly and forage-specific assemblies. MetaBat2 was run on the co-assembly and forage-specific assemblies considering contigs with minimum length 2000 (--minContig 2000) and their coverage profile across either all 24 samples (for the co-assembly) or the four sampling points (for each forage-specific assembly). MaxBin2 was also run on the co-assembly and forage-specific assemblies with options ‘--min_contig_length 2000 and --max_iteration 50’. All genome bins obtained from the co-assembly and forage-specific assemblies were aggregated and dereplicated separately using DAS_Tool v1.1.1 [35] in order to ensure that the dereplicated genomes did not have any shared contigs. DAS_Tool was run under default setting using diamond as a search engine. Genome bins remained after the dereplication using DAS_Tool from the co-assembly (646 bins) and forage-specific assemblies (1020 bins) were finally aggregated and dereplicated using dRep v2.3.2 [36]. In this dereplication step, checkm v1.0.18 [37] was used to evaluate for genome completeness and contamination, and only bins with ≥75% completeness and ≤10% contamination were retained. The final cluster representatives n = 538 (hereafter called Rumen-Uncultured Genomes, RUGs) were further analyzed in this study.
Taxonomy and functional analysis of RUGs
Taxonomic classification of RUGs was performed using GTDB-Tk v1.3.0 [38]. PhyloPhlAn v0.97 [39] was used to place RUGs into a phylogenetic tree and to further infer their taxonomic labels using >400 universally conserved proteins encoded by 3171 genomes. The closest cultured relatives from the Hungate1000 collection [3] were also included. The tree was visualized using iTol v5 [40], and phylum-level taxonomies imputed by GTDB-Tk were assigned to leaves or tips. To identify novel and known genomes, the RUGs were compared to the Hungate1000 collection (n = 408) and the rumen metagenome-assembled genomes (n = 4941) from Stewart et al. [22] using fastANI v1.1 with the option ‘–fragLen 2000’. Coding sequences (CDS) and clustered regularly interspaced short palindromic repeats were predicted during Prokka v1.14.0 annotation [41]. tRNAs were predicted using tRNAscan-SE v2.0.6 [42]. Ribosomal RNAs were predicted using barrnap v0.9 (https://github.com/tseemann/barrnap).
Quantification and differential abundance of RUGs
The abundance of RUGs across samples were estimated by mapping reads to binned contigs using quant_bins module from metaWRAP [43]. RUGs were clustered based on their abundance profiles into heatmap using average linkage method and spearman-rank correlation distance matrix in heatmapper [44]. The differentially abundant RUGs between forages and across sampling intervals were identified using DESeq2 [45] with an FDR-corrected p value cutoff < 0.01.
Microbial community diversity based on RUGs
The microbial community structure of the samples across forages and sampling intervals was explored using Weighted Unifrac distance matrix. Permutational multivariate analysis of variance (PERMANOVA) was used to test for significant differences among the forages and across the sampling intervals using the Adonis function of vegan v2.5–5 [46]. Group-wise differences were tested using pairwiseAdonis. PERMDISP was also used to test for the homogeneity of dispersions using the beta-disper function of vegan.
Identification and annotation of CAZymes in RUGs
Prodigal v2.6.3 [47] was used in metagenome mode to predict protein coding genes. The predicted proteins were scanned for candidate CAZymes using hmmscan module from HMMER v3.2.1 and dbCAN database v6 as input [48]. The hmmscan output was parsed by applying a minimum query coverage 35% and an e-value cutoff 1e−5. PULs were identified using PULpy [49]. The predicted CAZymes were BLASTp [50] searched against the NCBI nr database (November 2019) using an e-value cutoff of 1e−30 and a maximum target sequence of 30. Signal peptides were predicted using SignalP v.4.1 [51] based on both gram-positive and gram-negative models.
Metabolic reconstitution of RUGs
To infer the metabolic potential of RUGs, the predicted proteome of each RUGs was scanned for enzymes involved in specific metabolic pathways including glycolysis, pentose phosphate (PPP), and Embden–Meyerhof–Parnas (EMP). The search was performed using the hmmscan module of HMMER software against Pfam domains representing key marker genes of each metabolic pathway. Pfam domains were retrieved from Pfam v32 and TIGRFAMs v15 databases (see Supplementary Data 1 for the details of pfam domains). The ability to metabolize monomeric carbohydrates including mannose, rhamnose, xylose, galactose, arabinose, fucose, and glucose requires the presence of key kinase, isomerase, and epimerase enzymes catalyzing critical steps in their metabolism as carbon source.
The ability of RUGs to ferment carbohydrate monomers into SCFAs was also determined. Butyrate production was approved by the presence of genes for phosphate butyryltransferase (ptb), butyrate kinase (buk), and butyryl-CoA:acetate CoA-transferase (but). Lactate dehydrogenase (ldh) was used as the marker of lactate production. Acetate production was checked by the presence of genes encoding phosphate acetyltransferase (pta) and acetate kinase (ackA). Malic enzyme (men), fumarate hydratase (fum), and fumarate reductase (frd) were used as markers of succinate production. Propionate production was also approved by the presence of three key enzymes methylmalonyl-CoA decarboxylase (mmdA), methylmalonyl-CoA mutase (mutA), and methylmalonyl-CoA epimerase (mce). The potential of degradation of carbohydrate polymers was further evaluated by enumerating key enzymes for the degradation of cellulose, xylan, xyloglucan, chitin, pectin, and starch.
Results and discussion
Metagenome assembly and genome bin selection
In this study, we sequenced the whole metagenome of 24 rumen samples, resulted in a total of 333 Gbp of sequencing data. A combined coverage and tetra nucleotide frequency-based binning resulted in 538 RUGs having >75% completeness and <10% contamination (Supplementary Data 2 and Fig. S2A, B). Among the RUGs, 205 had >90% completeness and <5% contamination and 21 could be considered as high-quality draft MAGs based on the criteria defined by Bowers et al. [52] (completeness > 90%, contamination < 5%, and the presence of the 23S, 16S, and 5S rRNA genes and at least 18 tRNAs). In addition, 468 (87%) of RUGs also had a quality score (completeness − 5 × contamination) > 50%, indicating most of the RUGs to be high quality. Among the 538 RUGs, 351 had at least 18 out of the 20 standard tRNAs (Fig. S2C) and 53 had at least a full-length and 117 a partial 16S rRNA gene. The sizes of RUGs ranged from 606 kilobases (kb) to 5.95 megabases (Mb), with N50 values from 4 to 387 kb. The smallest RUG was a mycoplasma (RUG208), followed by four Saccharimonadaceae bacteria with genome sizes <0.8 Mb and an average 78% genome completeness. The number of contigs in RUGs ranged from 13 to 703 with an average of 189 contigs.
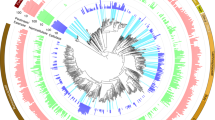
By comparing the RUGs with Hungate1000 collection (n = 408) [3], the set of RUGs (n = 4941) reported by Stewart et al. [22], and the GTDB-Tk representative genomes (n = 31,910) [38] we found that most of our RUGs were novel and lacked any representative in available databases (Fig. 1A). To test whether these low ANI similarities were likely resulted from genome incompleteness, we correlated our RUGs completeness and their ANI with Stewart MAGs and found no clear relationship (Fig. S3). Only 92 RUGs (~17%) were reliably assigned to known species with an ANI >95% over ≥65% of alignment fraction, congruent with their placement in GTDB-Tk reference tree. Seventy-eight RUGs were remained unannotated at the genus level, six at the family level, and one at the order and the class levels (Supplementary Data 2). A total of 243 RUGs (45%) were taxonomically assigned to Firmicutes and 190 (35%) to Bacteroidota (Figs. 1B, S2D). Spirochaetota were the third most abundant group of bacteria in 26 RUGs (4.8%), followed by Proteobacteria (15), Verrucomicrobiota (12), Synergistota (7), Patescibacteria (6), Elusimicrobiota (5), Cyanobacteria (5), Desulfobacterota (4), and Fibrobacterota (4) (Fig. S2D). The high number of Spirochaetota genomes recovered in this study agreed with an early study on moose rumen, in which Spirochaetota were found to play a major role in pectin degradation [25]. All Spirochaetota genomes belonged to the genus Treponema, known as the major hemicellulose degrader in the gut microbiota [53].

A The average nucleotide identity (ANI) and aligned fraction (AF) of the 538 RUGs compared with rumen cultured genomes from the Hungate1000 genome project [3], the cattle rumen metagenome-assembled genomes [22], and the GTDB-Tk representative genomes (release 95) [38]. The horizontal dashed line (purple) shows accepted AF threshold (65%), vertical dashed lines (green and blue) show 95% and 99% ANI thresholds, respectively. B The phylogenetic tree of the 538 RUGs along with 62 closest relatives from the Hungate1000 collection. The tree was constructed based on the concatenated multiple sequence alignments of 400 universally conserved proteins using PhyloPhlAn [39] and visualized using iTOL [40]. Clades are colored based on phylum-level taxonomies inferred by GTDB-Tk [38]. The Hugnate1000 representative genomes are labeled by their taxonomic names.
Overall, 15 RUGs were taxonomically assigned to the archaeal families Methanomethylophilaceae (n = 11), Methanobacteriaceae (3), and Methanomicrobiaceae (1). Among Methanomethylophilaceae, eight RUGs were affiliated to ISO4-G1, a methylotrophic methanogen first isolated from the sheep rumen [54]. Methanobacteriaceae genomes were assigned to the genus Methanosphaera (RUG228) and Methanobrevibacter (RUG389 and RUG508). The single genome recovered from Methanomicrobiaceae was assigned to Methanomicrobium mobile, which is one of the major metanogenic archaeon detected in the rumen of sheep (accounting for 54% of the total methanogens) [55] and cattle (>106 cells/mL in rumen digesta) [56]. The details of taxonomies inferred by GTDB-Tk are presented in Supplementary Data 2.
The differential colonization of forages by RUGs
The six lignocellulosic biomasses (AP, CR, DP, KS, RS, and SC) were incubated in three rumen-fistulated bulls for a period of 96 h with 24 h sampling intervals. The samples taken from the three bulls at each sampling interval (e.g., 24, 48, 72, and 96 h) were pooled and processed for whole metagenome sequencing of fiber-attached microbiota. Clustering samples at the four sampling intervals and across the six forages based on the relative abundances of the recovered RUGs revealed a clear separation of the samples at 24 h of rumen incubation (Fig. 2). The distribution of RUGs across forages varied as the incubation time was extended. At 48 h, only AP, CR, DP, and SC clustered together. There was no apparent clustering of forages at 72 and 96 h, indicating that the digestion during early incubation stages converted feed particulates into unfavored substrates for most of the RUGs, a pattern in agreement with our 16S rRNA metabarcoding data [15].

Heatmap clustering shows a clear separation of samples collected at 24 h from those at later stages of the rumen incubation. The RUGs were hierarchically clustered based on their relative abundances estimated by recruiting reads to contigs in the binned genomes using an average linkage method and spearman-rank correlation distance measurement using Heatmapper [44]. AP camelthorn, CR common reed, DP date palm, KS Kochia, RS rice straw, and SC Salicornia.
Microbial community diversity and composition analyses also showed significant differences between the forages and sampling intervals (PERMANOVA P < 0.001; Fig. 3), which could be mainly ascribed to community changes in AP compared to CR, RS, and SC (P < 0.05). The distribution of RUGs across sampling intervals also showed a clear separation of 24 h samples (pairwise PERMANOVA P < 0.003) from all other stages which exhibited overlapped clustering (Fig. 3). There were higher relative abundances of Firmicutes (26 RUGs), Fibrobacterota (2 RUGs), Bacteroidota (2 RUGs), and Spirochaetota (2 RUGs; FDR-corrected P < 0.01) attached to SC (the forage with the highest hemicellulose and the lowest ADF concentrations) compared with other forages. Ruminococcaceae (3 RUGs) were significantly enriched in the microbial community attached to RS (the forage with the lowest levels of ADF and ADL) compared with other forages. Verrucomicrobiota (2 RUGs) were represented by higher abundance in CR (the forage with the highest cellulose content) compared to AP and SC (two forages with the lowest cellulose concentration). This finding indicated the key role of Verrucomicrobiota in cellulose degradation, and was in agreement with a recent analysis of mice microbiota with Bacteroidetes to be accumulated in the presence of bamboo shoot while Verrucomicrobiota to be significantly increased with crystalline cellulose supplementation [57].

A PCoA plot shows a significant separation of RUGs based on the forages and following rumen incubation lengths. Forages are indicated by colors and sampling intervals with shapes. Statistically significant differences in rumen microbial community attached to the forages and between the incubation lengths were tested by permutation multivariate analysis of variance (PERANOVA). The homogeneity of group dispersions between forages (B) and sampling intervals (C) were tested using the beta-disper function in R package vegan. AP camelthorn, CR common reed, DP date palm, KS Kochia, RS rice straw, and SC Salicornia.
Temporal analysis of microbial communities colonizing the forages also revealed the dominances of Bacteroidetes, Firmicutes, and Proteobacteria during initial stage of rumen incubation when maximal DM degradation took place (e.g., 24 h), while Verrucomicrobia accumulation started later when most DM got disappeared (Supplementary Data 3). Bacteroidetes, Firmicutes, and Proteobacteria are known as copiotrophic taxa with potentially rapid growth rates and their adaptability to the condition of high carbon availability [58, 59]. On the other hand, oligotrophic bacteria such as Verrucomicrobia are able to grow on low quality and recalcitrant substrates with slow growth rates but a higher substrate utilization efficiency [59]. This is functionally relevant as upon feed entry into the rumen surface accessible carbohydrates in feed particulates attract the copiotrophic taxa first, but the digestion during early hours of rumen incubation turns forage residues into favored substrates for oligotrophic taxa. The shift from copiotrophic to oligotrophic taxa is likely critical for the digestion of recalcitrant substrates in the rumen.
Carbohydrate degrading potential of RUGs
In total, 1,185,394 protein coding genes were predicted in the 538 RUGs, of which 5.1% were annotated as CAZymes, including 27,328 (43.8%) identified as GHs, 1444 as PLs (2.3%), 9965 as CEs (16%), 6249 as CBMs (10%), 387 as dockerin (0.6%), 168 as cohesin (0.26%), 1305 as AAs (2.1%), 732 as SLHs (1.17%), and 14,731 as GTs (23.6%) (Supplementary Data 4). A comparison to the number of sequences annotated as CAZyme in the GTDB representative genomes (n = 31,910) revealed a significantly higher enrichment of CAZymes (5.1% vs. 3.7%) particularly those encoding GHs (43.8% vs. 34.4%), CBMs (10% vs. 7.6%), cohesin (0.26% vs. 0.14%), dockerin (0.6% vs. 0.23%), and SLHs (1.17% vs. 0.92%), but a lower enrichment of CEs (16% vs. 18.2%), AAs (2.1% vs. 5.6%), and GTs (23.6% vs. 30.8%) in our RUGs. This finding suggests that rumen microbes have been naturally selected and evolutionary adapted with a high genomic potential for efficient lignocellulose degradation in the rumen.
The top 10 most abundant GH families were GH3, GH31, and GH97 glucosidases, GH2 galactosidases, GH109 α-N-acetylgalactosaminidases, GH28 polygalacturonases, GH23 and GH25 lysozymes, GH51 endoglucanases, and GH127 arabinofuranosidases. Pectin degrading enzymes including PL11 rhamnogalacturonan endolyases, PL1_2 pectate lyases, CE1 acetyl xylan esterases, and CE10 arylesterases were among the most abundant PL and CE families. Only 418 CAZymes showed 100% identity while 3089 (4.96%) shared ≥95% identity with proteins in the NCBI-NR database (Fig. 4A). The AA domain-containing proteins showed the highest (∼74%) but dockerin domain-containing proteins had the lowest (∼45%) average amino acid identity with proteins in the database. The average sequence identities were even lower when compared to CAZyme database (Fig. 4B), suggesting that the majority of the predicted CAZymes are novel, with no well-characterized homolog in the current databases.

The average percentage identity of CAZymes (n = 62,309) predicted in the RUGs with proteins deposited in the NCBI nonredundant protein database (A) and CAZyme database (B). AAs axillary activity enzymes (n = 1305), CBMs carbohydrate-binding modules (n = 6249), CEs carbohydrate esterases (n = 9965), cohesins (n = 168), dockerins (n = 387), GHs glycoside hydrolases (n = 27,328), GTs glycoside transferases (n = 14,731), PLs polysaccharide lyases (n = 1444), and SLHs surface layer homology domains (n = 732). Center lines indicate the median value; red triangles show mean; boxes show the interquartile range; and whiskers extend to the most extreme data point. The distribution (C) and density (D) of the predicted CAZyme families across 528 RUGs taxonomically classified at the class level.
On average, >94% of CAZymes were encoded by seven bacterial classes including Bacteroidia (48.9%), Clostridia (33.6%), Spirochaetia (3.9%), Kiritimatiellae (3.1%), Bacilli (2.4%), Lentisphaeria (1.7%), and Fibrobacteria (1.2%; Fig. 4C). However, normalizing the number of predicted CAZymes to the number of RUGs identified in each taxon (class level) revealed that Kiritimatiellae (phylum Verrucomicrobia), followed by Lentisphaeria, Fibrobacteria, and Bacteroidia, were mainly enriched for GHs, CEs, PLs, and CBMs (Fig. 4D), suggesting Kiritimatiellae, Lentisphaeria, and Fibrobacteria to be the likely key lignocellulose degraders in the rumen regardless of their lower abundance compared with Bacteroidia and Clostridia. The capacity to degrade hemicellulose is widely encoded across the members of Verrucomicrobia [60]. Previous single-cell genome sequencing of certain phylotypes of Verrucomicrobia also revealed the enrichment of genes encoding for CAZymes, sulfatases, and peptidases in their genomes, potentially allowing these bacteria to degrade diverse carbohydrate polymers [61].
The production of SLH domain-containing proteins was mainly restricted to Clostridia, Vampirovibrionia, Negativicutes, and Synergistia. These proteins anchor cellulosome complexes to the cell surface and thus facilitate access to the degraded products from the complexes by the bacterium. Among CAZymes, cohesin and dockerin domain encoding genes were mainly encoded by Bacteroidia and Clostridia. However, genes encoding for these two proteins mainly co-occurred in members of Oscillospirales and certain Christensenellales known as main cellulosome producers in the rumen [62]. The key feature of cellulosome complexes is the multimodular scaffoldin proteins containing multiple tandemly repeated cohesin domains, serving as docking site for the attachment of dockerin-bearing CAZymes [62,63,64]. In addition, they bear cellulose-binding or carbohydrate-binding modules to attach lignocellulosic substrates. Further analysis of proteins with such configuration revealed 36 proteins in 29 RUGs with 2 (n = 20) or more (n = 16) cohesin domains (Supplementary Data 5). Proteins with greater than four cohesin domains were only detected in the families Ruminococcaceae (7 RUGs) and unknown Christensenellales (2 RUGs). In particular, two Ruminococcus genomes of RUG452 and RUG21 each encoded one ortholog protein with 10 and 5 tandemly repeated cohesin domains, respectively. Based on a BLAST search, these two proteins matched to the scaffoldin B from Ruminococcus flavefaciens with 51.4 and 52.3% identity, respectively, suggesting them to be novel candidate scaffoldins (Fig. S4).
In cellulosome complexes, lignocellulose degrading enzymes bind via their dockerin domains to cohesin domains in the scaffoldin proteins, these cohesin–dockerin interactions are fundamental to cellulosome assembly [64, 65]. We identified 126 proteins in 31 RUGs which contained at least one dockerin and one catalytic CAZyme module to be the putative components of cellulosome complexes as they potentially encoded acetyl xylan esterases (families CE1, CE2, CE3, CE4, and CE6), α-amylases (GH13, GH13_15, GH13_28, GH13_6, GH13_7, and GH97), xylanases (GH10, GH11, GH16, GH30_8, GH5_21, GH5_35, and GH98), cellulases (GH124, GH128, GH5_2, GH5_4, GH5, and GH9), β-galactanases (GH2, GH53, GH43_24, and GH30_5), and pectate lyases (PL1, PL1_2, PL1_8, PL11, PL9_4, and PL9). The presence of dockerin-bearing α-amylases implies the existence of cohesin-based amylosomes, which are known to be formed in certain members of Ruminococcus including R. bromii in human gut in response to resistant starches [66, 67]. In addition, arabinanases (GH43_37 and GH43_4), L-arabinofuranosidases (GH43_10, GH43_29, and GH127) and α-L-fucosidases (GH95 and GH141) were also represented among the dockerin-bearing enzymes. Many of these proteins also contained CBMs capable of binding to either cellulose, xylan, starch or galactans, highlighting the key role of cellulosomes in lignocellulosic polymer degradation in the rumen [25].
The process of plant polysaccharide degradation by rumen microbiota requires lignocellulose degrading enzymes being secreted into the external milieu. Inspection of CAZyme sequences for the presence of secreted signal revealed that a significant portion of predicted CAZymes in RUGs affiliated to Fibrobacteria (∼53%), Bacteroidia (∼50%), and Kiritimatiellae (∼49%) were secreted and/or periplasmic proteins. While for non-CAZyme sequences only 9.8%, 11.9%, and 14.4% of sequences in Fibrobacteria, Bacteroidia, and Kiritimatiellae contained signal peptide, respectively, indicating a significant enrichment of signal peptide in CAZymes and thus the critical role of these taxa in lignocellulose degradation in the rumen (see Supplementary Note and Supplementary Data 6 for greater details). Even though that Clostridia were among abundant members of the rumen community, only ∼20% of their CAZymes were predicted to be secreted, suggesting that they have been largely tailored to break down cellobiose products of polysaccharide degradation inside of the cell.
Classifying RUGs according to their polysaccharide degrading capacities showed that Bacteroidota and Firmicutes can potentially degrade a wide range of carbohydrate polymers (Supplementary Data 1), which were also found in different ruminants including camel [26], moose [25, 68] and cattle [22, 24]. Fibrobacterota were particularly adapted to degrade glucan, cellulose, mannan, arabinan, xylan, xyloglucan, chitin, starch, and pectin but lacked genes for laccase and those for fucose and rhamnose cleavage. The ability to deconstruct pectin seemed to be widely present among the members of Ruminococcus, Fibrobacteres, and Bacteroidetes, consistent with the finding in moose [25]. Both Verrucomicrobiota and Spirochaetota also showed high lignocellulose degrading capacity but the former lacked genes for laccase which are required for lignin metabolism while the latter didn’t demonstrate their potential to break down focuse, rhamnose, and mannan. In termite hindgut microbiota, Spirochaetota were found to play a key role in hemicellulose degradation as they carry genes for targeting cellulose-xyloglucan polysaccharide complexes [53, 69]. In the rumen microbiome, however, Spirochaetota were shown to be associated with pectin degradation, particularly under a pectin rich diet [25, 70]. Treponema (Spirochaetota), Fibrobacter, Clostridia, and Bacteroidia widely encoded enzymes involved in chitin metabolism (domain DUF4849 and GH18; Supplementary Data 1). Since chitin is not a component of plant cell wall, the presence of chitin degrading enzymes is likely required for the degradation of host-derived glycoproteins such as mucin [71, 72]. Only single Verrucomicrobium bin (RUG64) in this study had genes for chitin degradation while in human gut, Verrucomicrobia including Akkermansia muciniphila are known as mucin-degraders [73]. The remaining RUGs including those belonging to Synergistota, Proteobacteria, Elusimicrobiota, Desulfobacterota, Chloroflexota, and archaea had limited polysaccharide degrading capacities, yet they may play an accessory or complementary role in carbohydrate degradation and metabolism in the rumen. The variability in lignocellulose degrading capacity across different lineages may enable bacteria to tolerate fluctuations in physicochemical properties of the forages and to ensure nutrient supply from different lignocellulosic substrates in the rumen.
The organization of CAZymes in PULs in Bacteroidota
In Bacteroidota, all genes required for perception, cleavage, and utilization of a specific polysaccharide glycan are localized on the same portion of their genomes characterized by the presence of a susC/D gene pair which encode for sugar transporters [7]. This special organization of CAZymes is known as PULs. The PUL profiles can be used to predict the ecological niche and substrate specificity of Bacteroidota as the most abundant rumen microbes. To identify potential PULs, RUGs were screened for tandem susC-susD gene pair followed by identification of the occurrence of genes encoding CAZymes around the susC/D pair [6]. A total of 1669 potential PULs were predicted in 154 RUGs (out of the 190 Bacteroidota RUGs), of which 708 lacked any known CAZyme gene, consistent with previous findings (Supplementary Data 7) [8, 68]. The lack of CAZymes in a large set of PULs suggests that PULs may also contribute to molecular processes other than polysaccharide degradation [8]. The occurrence of CAZymes in predicted PULs are detailed in Supplementary Note.
Several abundant and functionally important lignocellulose degrading enzymes including families GH11 endo-β-1,4-xylanases, GH43_33 α-L-arabinofuranosidases, GH43_8 β-D-galactofuranosidases, GH30_5 endo-β-1,6-galactanases, GH4 and GH5_26 cellulases, and GH59 β-galactosidases were not found in the PULs. In addition, GH9, GH5_2, GH8 cellulases, GH13_38 α-glucosidases, GH74 endoglucanases, GH30_2 β-xylosidases, GH35 β-galactosidases, GH29 α-L-fucosidases, GH106 α-L-rhamnosidases, GH43_29 α-1,2-L-arabinofuranosidases, GH43_35, and GH43_10 β-xylosidases were also rarely present in the PULs. The phenomenon that not all lignocellulose degrading enzymes are organized into PULs suggests that they may have been developed for the decomposition of complex polysaccharides with heterogeneous structures, which require a combination of enzymes for their degradation. Similar to our finding, no PUL for the digestion of crystalline cellulose, which is largely composed of β-1-4 linked D-glucopyranose units, has been reported [8]. This is also the case for starch and glycogen made of glucose subunits linked by either α-1-4 or α-1-6 glycosidic bonds, for which the degrading enzymes were rarely encountered in the PULs. The predicted PULs appeared to target complex polysaccharide polymers including xyloglucans, glucuronylxylans, and pectins, which are the main components of plant cell wall, but rarely accommodated enzymes dedicated to break down glucan homopolymers including crystalline cellulose, starch, and/or glycogen. Overall, these findings suggest a prominent diversity of PULs predicted in Bacteroidota genomes, indicating their functional specialization toward specific lignocellulosic substrates.
Metabolic classification of RUGs
We classified all RUGs according to their capacity for producing main SCFAs and utilizing monomeric carbohydrates (Supplementary Data 1). The production of butyrate, lactate, acetate, succinate, and propionate was widespread among Bacteroidota, Elusimicrobiota, and Fibrobacterota. Within Firmicutes, however, there was a significant variation in the potential to ferment SCFAs. While lactate production was widely encoded by this phylum, however, only orders of Lachnospirales, Erysipelotrichales, Peptostreptococcales, and Saccharofermentanales can potentially ferment butyrate (Supplementary Data 1). In particular, Lachnospirales are known as key rumen butyrate producers, which redirect hydrogen from reducing CO2 for methane formation to butyrate production and thus may contribute to an enhanced feed efficiency in Holstein Friesian dairy cows [19]. Acetate production was common across rumen microbiota except Thermoplasmatota (Archaea), Cyanobacteria, and Negativicutes (Firmicutes_C), which lacked genes to encode pta and acka. Acetate production is also widespread among rumen microbiota of moose [68] and dromedary camel [26]. Succinate and propionate production were widely encoded by Firmicutes, except some members of Bacilli including orders of RF39 and RFN20 lacking the required enzymes. Particularly, RF39 and RFN20 are the two major gut lineages characterized by their reduced genome size, mixotrophic lifestyle and inability to synthesize peptidoglycans as an adaptive strategy in the gut environment [74]. In ruminant animals, a greater potential for propionate, butyrate, and propionate-to-acetate fermentation are known to associate with a higher feed efficiency but also with a lower methane emission [19, 75].
In addition, the PPP, EMP, and pyruvate metabolism pathways were widely distributed across the RUGs. Certain members of the rumen microbiota including some strains of Lachnospira, Prevotella, Butyrivibrio, Bacteroidaceae, and Clostridia together with all Kiritimatiellae and Gastranaerophilaceae (Cyanobacteria) lack enolase, the enzyme that catalyze the penultimate step of the glycolysis pathway [3]. The evolutionary advantage of enolase loss is currently unknown. Energy metabolism through electron transport complex rnfABCDEG was widespread across Bacteroidota and Firmicutes (Supplementary Data 1), corroborating the finding in the moose rumen microbiota [68]. However, echABCDEF complex was the primary route for energy metabolism in Fibrobacterota, Desulfobacterota, Negativicutes (Firmicutes_C), and Thermoplasmatota, in which evidence for rnfABCDEG complex was scarce. The co-occurrence of the two complexes was rare in some Firmucutes genomes. Both Rnf and Ech are membrane-bond enzyme complexes which couple electron transfer with an H+/Na+ gradient to recycle NADH and H2 [76]. These data suggest that forage-attached microbiota show a high redundancy with respect to metabolic capabilities and their functions in the rumen. This functional redundancy is likely beneficial to the host animal as it enables the host to extract energy from diverse lignocellulosic substrates.
Conclusion
Our metagenomics data indicated that the rumen environment harbors a wealth of microbial diversity yet to be discovered, with several rare bacterial taxa playing key roles in fiber digestion. We showed that while generalist microbes and their genes are prevalent in the rumen, certain groups are specialized toward particular lignocellulosic substrates. In addition, we provided evidence for a strong temporal shift in the rumen microbial function from copiotrophy to oligotrophy along the length of rumen incubation, which may facilitate the degradation of recalcitrant lignocellulosic substrates in the rumen. The physiochemical properties of forages and the retention time of feed particulates in the rumen were the major factors determining the extent of copiotrophic–oligotrophic shifts. Taken together, the differential colonization of forages with different lignocellulosic properties demonstrates that the diversity of rumen microbiota is likely critical to the digestion of different lignocellulosic substrates, reflecting a strong niche complementarity between members of the rumen microbial community. The high taxonomic diversity, functional redundancy and metabolic partitioning are the primary characteristics of the rumen microbiome that facilitate the evolutionary adaptation to diverse lignocellulosic forages.
Data availability
The raw sequence data have been deposited in NCBI short read archive (SRA) under BioProject ID PRJNA631951. All metagenome assemblies including co-assembly and forage-specific assemblies and the sequences of RUGs have been deposited in NCBI under the same BioProject ID.
References
Morgavi DP, Kelly WJ, Janssen PH, Attwood GT. Rumen microbial (meta)genomics and its application to ruminant production. Animal. 2013;7 Suppl 1:184–201.
Gruninger RJ, Ribeiro GO, Cameron A, McAllister TA. Invited review: application of meta-omics to understand the dynamic nature of the rumen microbiome and how it responds to diet in ruminants. Animal. 2019;13:1843–54.
Seshadri R, Leahy SC, Attwood GT, Teh KH, Lambie SC, Cookson AL, et al. Cultivation and sequencing of rumen microbiome members from the Hungate1000 Collection. Nat Biotechnol. 2018;36:359–67.
Bule P, Pires VM, Fontes CM, Alves VD. Cellulosome assembly: paradigms are meant to be broken! Curr Opin Struct Biol. 2018;49:154–61.
Fontes CM, Gilbert HJ. Cellulosomes: highly efficient nanomachines designed to deconstruct plant cell wall complex carbohydrates. Annu Rev Biochem. 2010;79:655–81.
Terrapon N, Lombard V, Gilbert HJ, Henrissat B. Automatic prediction of polysaccharide utilization loci in Bacteroidetes species. Bioinformatics. 2015;31:647–55.
Grondin JM, Tamura K, Dejean G, Abbott DW, Brumer H. Polysaccharide utilization loci: fueling microbial communities. J Bacteriol. 2017;199:e00860–16.
Lapebie P, Lombard V, Drula E, Terrapon N, Henrissat B. Bacteroidetes use thousands of enzyme combinations to break down glycans. Nat Commun. 2019;10:2043.
McAllister TA, Bae HD, Jones GA, Cheng KJ. Microbial attachment and feed digestion in the rumen. J Anim Sci. 1994;72:3004–18.
Leng R. Interactions between microbial consortia in biofilms: a paradigm shift in rumen microbial ecology and enteric methane mitigation. Anim Prod Sci. 2014;54:519–43.
Wang Y, McAllister T. Rumen microbes, enzymes and feed digestion—a review. Asian Australas J Anim Sci. 2002;15:1659–76.
Huws SA, Mayorga OL, Theodorou MK, Onime LA, Kim EJ, Cookson AH, et al. Successional colonization of perennial ryegrass by rumen bacteria. Lett Appl Microbiol. 2013;56:186–96.
Liu J, Zhang M, Xue C, Zhu W, Mao S. Characterization and comparison of the temporal dynamics of ruminal bacterial microbiota colonizing rice straw and alfalfa hay within ruminants. J Dairy Sci. 2016;99:9668–81.
Huws SA, Edwards JE, Creevey CJ, Rees Stevens P, Lin W, Girdwood SE, et al. Temporal dynamics of the metabolically active rumen bacteria colonizing fresh perennial ryegrass. FEMS Microbiol Ecol. 2016;92:fiv137.
Gharechahi J, Vahidi MF, Ding XZ, Han JL, Salekdeh GH. Temporal changes in microbial communities attached to forages with different lignocellulosic compositions in cattle rumen. FEMS Microbiol Ecol. 2020;96:fiaa069.
Ellison MJ, Conant GC, Cockrum RR, Austin KJ, Truong H, Becchi M, et al. Diet alters both the structure and taxonomy of the ovine gut microbial ecosystem. DNA Res. 2014;21:115–25.
Petri RM, Schwaiger T, Penner GB, Beauchemin KA, Forster RJ, McKinnon JJ, et al. Characterization of the core rumen microbiome in cattle during transition from forage to concentrate as well as during and after an acidotic challenge. PLoS ONE. 2013;8:e83424.
Li F, Hitch TCA, Chen Y, Creevey CJ, Guan LL. Comparative metagenomic and metatranscriptomic analyses reveal the breed effect on the rumen microbiome and its associations with feed efficiency in beef cattle. Microbiome. 2019;7:6.
Shabat SK, Sasson G, Doron-Faigenboim A, Durman T, Yaacoby S, Berg Miller ME, et al. Specific microbiome-dependent mechanisms underlie the energy harvest efficiency of ruminants. ISME J. 2016;10:2958–72.
Gharechahi J, Zahiri HS, Noghabi KA, Salekdeh GH. In-depth diversity analysis of the bacterial community resident in the camel rumen. Syst Appl Microbiol. 2015;38:67–76.
Koike S, Yoshitani S, Kobayashi Y, Tanaka K. Phylogenetic analysis of fiber-associated rumen bacterial community and PCR detection of uncultured bacteria. FEMS Microbiol Lett. 2003;229:23–30.
Stewart RD, Auffret MD, Warr A, Walker AW, Roehe R, Watson M. Compendium of 4,941 rumen metagenome-assembled genomes for rumen microbiome biology and enzyme discovery. Nat Biotechnol. 2019;37:953–61.
Hess M, Sczyrba A, Egan R, Kim TW, Chokhawala H, Schroth G, et al. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science. 2011;331:463–7.
Stewart RD, Auffret MD, Warr A, Wiser AH, Press MO, Langford KW, et al. Assembly of 913 microbial genomes from metagenomic sequencing of the cow rumen. Nat Commun. 2018;9:870.
Svartstrom O, Alneberg J, Terrapon N, Lombard V, de Bruijn I, Malmsten J, et al. Ninety-nine de novo assembled genomes from the moose (Alces alces) rumen microbiome provide new insights into microbial plant biomass degradation. ISME J. 2017;11:2538–51.
Gharechahi J, Salekdeh GH. A metagenomic analysis of the camel rumen’s microbiome identifies the major microbes responsible for lignocellulose degradation and fermentation. Biotechnol Biofuels. 2018;11:216.
Khalili Ghadikolaei K, Gharechahi J, Haghbeen K, Akbari Noghabi K, Hosseini Salekdeh G, Shahbani, Zahiri H. A cold-adapted endoglucanase from camel rumen with high catalytic activity at moderate and low temperatures: an anomaly of truly cold-adapted evolution in a mesophilic environment. Extremophiles. 2018;22:315–26.
Pope PB, Denman SE, Jones M, Tringe SG, Barry K, Malfatti SA, et al. Adaptation to herbivory by the Tammar wallaby includes bacterial and glycoside hydrolase profiles different from other herbivores. PNAS. 2010;107:14793–8.
Li D, Liu CM, Luo R, Sadakane K, Lam TW. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015;31:1674–6.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–77.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Kang DD, Li F, Kirton E, Thomas A, Egan R, An H, et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ. 2019;7:e7359.
Wu YW, Simmons BA, Singer SW. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics. 2016;32:605–7.
Sieber CMK, Probst AJ, Sharrar A, Thomas BC, Hess M, Tringe SG, et al. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat Microbiol. 2018;3:836–43.
Olm MR, Brown CT, Brooks B, Banfield JF. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 2017;11:2864–8.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55.
Chaumeil PA, Mussig AJ, Hugenholtz P, Parks DH. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics. 2019;36:1925–7.
Segata N, Bornigen D, Morgan XC, Huttenhower C. PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat Commun. 2013;4:2304.
Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44:W242–5.
Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–9.
Chan PP, Lowe TM. tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. In: Kollmar M. (eds) Gene Prediction. Methods in Molecular Biology, vol 1962. Humana, New York, NY. 2019. https://doi.org/10.1007/978-1-4939-9173-0_1.
Uritskiy GV, DiRuggiero J, Taylor J. MetaWRAP-a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome. 2018;6:158.
Babicki S, Arndt D, Marcu A, Liang Y, Grant JR, Maciejewski A, et al. Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res. 2016;44:W147–53.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Oksanen J, Kindt R, Legendre P, O’Hara B, Simpson GL, Solymos P, et al. Vegan: community ecology package. R package version 2007;1:8.
Hyatt D, LoCascio PF, Hauser LJ, Uberbacher EC. Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics. 2012;28:2223–30.
Yin Y, Mao X, Yang J, Chen X, Mao F, Xu Y. dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012;40:W445–51.
Stewart RD, Auffret MD, Roehe R, Watson M. Open prediction of polysaccharide utilisation loci (PUL) in 5414 public Bacteroidetes genomes using PULpy. bioRxiv. 2018. https://doi.org/10.1101/421024.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10.
Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–6.
Bowers RM, Kyrpides NC, Stepanauskas R, Harmon-Smith M, Doud D, Reddy TBK, et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat Biotechnol. 2017;35:725–31.
Tokuda G, Mikaelyan A, Fukui C, Matsuura Y, Watanabe H, Fujishima M, et al. Fiber-associated spirochetes are major agents of hemicellulose degradation in the hindgut of wood-feeding higher termites. PNAS 2018;115:E11996–2004.
Kelly WJ, Li D, Lambie SC, Jeyanathan J, Cox F, Li Y, et al. Complete Genome Sequence of Methanogenic Archaeon ISO4-G1, a Member of the Methanomassiliicoccales, Isolated from a Sheep Rumen. Genome Announc. 2016;4:e00221–16.
Yanagita K, Kamagata Y, Kawaharasaki M, Suzuki T, Nakamura Y, Minato H. Phylogenetic analysis of methanogens in sheep rumen ecosystem and detection of Methanomicrobium mobile By fluorescence in situ hybridization. Biosci Biotechnol Biochem. 2000;64:1737–42.
Jarvis GN, Strompl C, Burgess DM, Skillman LC, Moore ER, Joblin KN. Isolation and identification of ruminal methanogens from grazing cattle. Curr Microbiol. 2000;40:327–32.
Li X, Guo J, Ji K, Zhang P. Bamboo shoot fiber prevents obesity in mice by modulating the gut microbiota. Sci Rep. 2016;6:32953.
Fierer N, Bradford MA, Jackson RB. Toward an ecological classification of soil bacteria. Ecology. 2007;88:1354–64.
Ho A, Di Lonardo DP, Bodelier PL. Revisiting life strategy concepts in environmental microbial ecology. FEMS Microbiol Ecol. 2017;93:fix006.
Nixon SL, Daly RA, Borton MA, Solden LM, Welch SA, Cole DR, et al. Genome-resolved metagenomics extends the environmental distribution of the verrucomicrobia phylum to the deep terrestrial subsurface. mSphere. 2019;4:e00613–19.
Martinez-Garcia M, Brazel DM, Swan BK, Arnosti C, Chain PS, Reitenga KG, et al. Capturing single cell genomes of active polysaccharide degraders: an unexpected contribution of Verrucomicrobia. PLoS ONE. 2012;7:e35314.
Doi RH, Kosugi A. Cellulosomes: plant-cell-wall-degrading enzyme complexes. Nat Rev Microbiol. 2004;2:541–51.
Galera-Prat A, Morais S, Vazana Y, Bayer EA, Carrion-Vazquez M. The cohesin module is a major determinant of cellulosome mechanical stability. J Biol Chem. 2018;293:7139–47.
Artzi L, Bayer EA, Morais S. Cellulosomes: bacterial nanomachines for dismantling plant polysaccharides. Nat Rev Microbiol. 2017;15:83–95.
Mechaly A, Fierobe HP, Belaich A, Belaich JP, Lamed R, Shoham Y, et al. Cohesin-dockerin interaction in cellulosome assembly: a single hydroxyl group of a dockerin domain distinguishes between nonrecognition and high affinity recognition. J Biol Chem. 2001;276:9883–8.
Mukhopadhya I, Morais S, Laverde-Gomez J, Sheridan PO, Walker AW, Kelly W, et al. Sporulation capability and amylosome conservation among diverse human colonic and rumen isolates of the keystone starch-degrader Ruminococcus bromii. Environ Microbiol. 2018;20:324–36.
Ze X, Ben David Y, Laverde-Gomez JA, Dassa B, Sheridan PO, Duncan SH, et al. Unique organization of extracellular amylases into amylosomes in the resistant starch-utilizing human colonic firmicutes bacterium ruminococcus bromii. mBio 2015;6:e01058–15.
Solden LM, Naas AE, Roux S, Daly RA, Collins WB, Nicora CD, et al. Interspecies cross-feeding orchestrates carbon degradation in the rumen ecosystem. Nat Microbiol. 2018;3:1274–84.
Liu N, Li H, Chevrette MG, Zhang L, Cao L, Zhou H, et al. Functional metagenomics reveals abundant polysaccharide-degrading gene clusters and cellobiose utilization pathways within gut microbiota of a wood-feeding higher termite. ISME J. 2019;13:104–17.
Liu J, Pu YY, Xie Q, Wang JK, Liu JX. Pectin induces an in vitro rumen microbial population shift attributed to the pectinolytic Treponema group. Curr Microbiol. 2015;70:67–74.
Etzold S, Juge N. Structural insights into bacterial recognition of intestinal mucins. Curr Opin Struct Biol. 2014;28:23–31.
Solden LM, Hoyt DW, Collins WB, Plank JE, Daly RA, Hildebrand E, et al. New roles in hemicellulosic sugar fermentation for the uncultivated Bacteroidetes family BS11. ISME J. 2017;11:691–703.
Derrien M, Collado MC, Ben-Amor K, Salminen S, de Vos WM. The mucin degrader akkermansia muciniphila is an abundant resident of the human intestinal tract. Appl Environ Microbiol. 2008;74:1646–8.
Wang Y, Huang JM, Zhou YL, Almeida A, Finn RD, Danchin A, et al. Phylogenomics of expanding uncultured environmental Tenericutes provides insights into their pathogenicity and evolutionary relationship with Bacilli. BMC Genom. 2020;21:408.
Ungerfeld EM. Shifts in metabolic hydrogen sinks in the methanogenesis-inhibited ruminal fermentation: a meta-analysis. Front Microbiol. 2015;6:37.
Buckel W, Thauer RK. Energy conservation via electron bifurcating ferredoxin reduction and proton/Na(+) translocating ferredoxin oxidation. Biochim Biophys Acta. 2013;1827:94–113.
Acknowledgements
This research was funded by the Agricultural Biotechnology Research Institute of Iran (ABRII), the international cooperation and exchange program of the National Natural Science Foundation of China (No. 31461143020), and the Chinese government contribution to CAAS-ILRI Joint Laboratory on Livestock and Forage Genetic Resources in Beijing. The paper contributes to the CGIAR Research Program on Livestock.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Gharechahi, J., Vahidi, M.F., Bahram, M. et al. Metagenomic analysis reveals a dynamic microbiome with diversified adaptive functions to utilize high lignocellulosic forages in the cattle rumen. ISME J 15, 1108–1120 (2021). https://doi.org/10.1038/s41396-020-00837-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-020-00837-2
This article is cited by
-
Gut microbiota variations in wild yellow baboons (Papio cynocephalus) are associated with sex and habitat disturbance
Scientific Reports (2024)
-
Enhanced crystalline cellulose degradation by a novel metagenome-derived cellulase enzyme
Scientific Reports (2024)
-
Trait biases in microbial reference genomes
Scientific Data (2023)
-
Exploring the rumen microbiota of Hu lambs in response to diet with paper mulberry
Applied Microbiology and Biotechnology (2023)
-
Low-abundance populations distinguish microbiome performance in plant cell wall deconstruction
Microbiome (2022)
