
Abstract
This review introduces the concept of thermal energy storage (TES) and phase change materials (PCMs), with a special focus on organic solid-liquid PCMs, their confinement methods and their thermal management (TM) applications al low-medium temperatures (0 °C–100 °C). It then investigates the approach of embedding TES and TM functionalities in structural materials, through the development of multifunctional polymer composites that could find applications where weight saving and temperature management are equally important. The concept of structural TES composite is presented through the description of three case studies about thermoplastic structural or semi-structural composites containing a paraffinic PCM: (i) a polyamide/glass laminate containing a microencapsulated or shape-stabilized paraffin; (ii) a polyamide-based composite reinforced with discontinuous carbon fibers and containing paraffin microcapsules, and (iii) a carbon fiber laminate with a reactive thermoplastic acrylic matrix and a microencapsulated paraffin.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Thermal energy storage (TES)
The growing concerns about climate change, depletion of fossil fuels, and greenhouse gases emissions have recently attracted the attention of researchers, industries, and governments on technologies aimed at increasing efficiency in energy use. Since the routes of energy production based on fossil fuels have been lately undoubtedly related to environmental pollution and global warming, the research in the last decades has focused on sustainable and renewable energy sources, such solar, wind, and geothermal energy [1]. The wide diffusion of such sources is hindered by two main obstacles, namely their high initial plant cost and their intermittent nature. Although the first issue will be attenuated by technological development in the next years, the most effective answer to the second issue—i.e. source intermittency—relies in boosting energy storage technologies. The amount of solar energy varies on a daily and seasonal time scale, and the power and consistency of geothermal and wind sources can be unpredictable; therefore, renewable energy production plants must be supplemented with energy storage systems, to provide a significant and constant energy output even in the off-peak periods [1, 2]. Energy storage systems can also increase the efficiency of conventional energy sources, as they can help to decrease equipment sizes and initial and maintenance costs, to boost plant flexibility and efficiency, and to reduce the necessity of emergency power generators that would consume primary energy sources [3, 4].
Energy storage systems can be classified according to the intermediate energy form, which can be chemical, electrical, electrochemical, mechanical, or thermal [1]. Among energy storage systems, particularly interesting for some applications is thermal energy storage (TES), defined as the temporary storage of excess heat that can be used where and when needed. TES technologies can reduce the mismatch between thermal energy demand and availability, thereby contributing to a more efficient exploitation of intermittent energy sources [5]. Compared to other energy storage systems, the storage of energy in the form of heat (or cold) exhibits longer storage times and higher efficiency [1]. The cycle of a TES system, reported in figure 1, comprises charging, storage and discharging steps.

Figure 1. Typical working cycle of a TES system, showing the charging, storage and discharging steps and the variation of stored energy over time (adapted from [1]).
Download figure:
Standard image High-resolution imageTES technologies are currently employed for specific purposes in three main cases, namely (i) to store waste/excess heat that can be released when needed, e.g. to recover waste industrial heat [6], or in solar thermal power plants during peak periods [7]; (ii) to keep the temperature in a specific range, e.g. in buildings to store excess energy during the day and release it at nighttime [8], or for body temperature regulation through smart thermoregulating garments [9]; (iii) to temporarily store heat and prevent a temperature rise that would otherwise damage a component, as in the thermal management (TM) of electronic devices [10].
In some applications the desired product is the stored and released thermal energy, as in the cases (i) and (ii): these are generally referred to as examples of 'thermal energy storage properly said', and they normally need energy storage systems with high thermal capacity, to store as much energy as possible. In other applications, the excess heat is stored mainly to avoid a dangerous rise in temperature, as in case (iii): these are examples of 'TM', and their main requirement is usually a well-defined energy storage rate. It is not easy to distinguish between TES properly said and TM. Some other classifications categorize as TES properly said only the case (i), while the other cases are examples of TM as they must keep temperature in an optimal range. This review discusses about TES or TM without a strict distinction, although the case studies presented in section 4 could be more suitably employed for TM applications.
1.1. Classification of TES technologies
The most common classification of TES technologies is based on the way of varying the internal energy of the storage medium. Thermal energy can be stored and released through a temperature variation (sensible heat TES, SH-TES), an endo/exothermic phase change (latent heat TES, LH-TES), or a thermochemical reaction (thermochemical heat TES, TH-TES) [5]. The selection of a TES system over another depends on several parameters, such as the required heat storage period (hours, months, days), economic considerations, working temperature, available volume, as reported in table 1 [11]. The following paragraphs summarize the characteristics and the governing equations of each of the three TES classes.
Table 1. Main performance parameters of sensible, latent and thermochemical heat TES technologies (adapted from [11]).
| TES system | Capacity (kWh t−1) | Efficiency (%) | Storage period (h, d, m) | Cost (€ kWh−1) |
|---|---|---|---|---|
| Sensible heat | 10–50 | 50–90 | d/m | 0.1–10 |
| Latent heat | 50–150 | 75–90 | h/m | 10–50 |
| Thermochemical heat | 120–250 | 75–100 | h/d | 8–100 |
1.1.1. Sensible heat storage (SH-TES)
In SH-TES, energy is stored (and released) via an increase (or a decrease) in the temperature of the storage medium. The enthalpy variation is proportional to the temperature difference, as represented in figure 2(a). The energy stored in the system is defined by equation (1) as
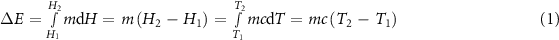

Figure 2. Variation of enthalpy of the storage medium as a function of its temperature in (a) an SH-TES unit cycle and (b) a LH-TES unit cycle (adapted from [1]).
Download figure:
Standard image High-resolution imagewhere  is the total amount of energy stored in the storage medium (J),
is the total amount of energy stored in the storage medium (J),  is the mass of the storage medium,
is the mass of the storage medium,  and
and  the initial and final enthalpy values (J/g),
the initial and final enthalpy values (J/g),  the specific heat capacity (J/(g · K)) and
the specific heat capacity (J/(g · K)) and  and
and  the initial and final temperature (°C) [12]. An analogous equation can be written for the energy release, which results in the cooling of the storage medium. The effectiveness of a storage medium and the total amount of stored/released energy depends on the available mass and volume, while a higher specific heat capacity results in increased energy storage per unit mass [13]. The medium should also be non-toxic, not expensive, and should maintain long-term stability over repeated thermal cycles. Typical materials used as sensible heat storage media are liquids such as water, oils or molten salts, or solids such as metals or rocks [14–16].
the initial and final temperature (°C) [12]. An analogous equation can be written for the energy release, which results in the cooling of the storage medium. The effectiveness of a storage medium and the total amount of stored/released energy depends on the available mass and volume, while a higher specific heat capacity results in increased energy storage per unit mass [13]. The medium should also be non-toxic, not expensive, and should maintain long-term stability over repeated thermal cycles. Typical materials used as sensible heat storage media are liquids such as water, oils or molten salts, or solids such as metals or rocks [14–16].
1.1.2. Latent heat storage (LH-TES)
LH-TES involves the storage and release of heat through the phase transition of a phase change material (PCM). The vast majority of LH-TES systems are based on the melting-solidification transition, while evaporation-condensation phase changes are generally avoided, as the associated high volume variation increases the complexity of the confinement unit [12]. The variation of enthalpy in a solid-to-liquid phase transition is illustrated in figure 2(b). During an energy storage process, the medium initially behaves like an SH-TES unit and absorbs sensible heat, which increases its temperature. This behavior continues until the medium temperature reaches the phase change temperature ( ), which is maintained up to completion of the phase transition. In this step, the absorbed energy is equal to the latent heat of phase change
), which is maintained up to completion of the phase transition. In this step, the absorbed energy is equal to the latent heat of phase change  . A further increase in absorbed energy brings a further increase in temperature: the slope of the enthalpy-temperature curve depends on the specific heat capacity of the medium in the liquid state and can be different from the slope between
. A further increase in absorbed energy brings a further increase in temperature: the slope of the enthalpy-temperature curve depends on the specific heat capacity of the medium in the liquid state and can be different from the slope between  and
and  . The total enthalpy variation
. The total enthalpy variation  (J) of such process is defined by equation (2), as
(J) of such process is defined by equation (2), as

where  is the latent heat of phase change,
is the latent heat of phase change,  is the phase change temperature and
is the phase change temperature and  and
and  are the specific heat capacity of the solid and liquid phase, respectively [12]. Generally, the absorbed latent heat is considerably higher than the sensible heat, which implies that LH-TES systems normally require less material usage, as they can store a high amount of energy in a smaller mass and volume.
are the specific heat capacity of the solid and liquid phase, respectively [12]. Generally, the absorbed latent heat is considerably higher than the sensible heat, which implies that LH-TES systems normally require less material usage, as they can store a high amount of energy in a smaller mass and volume.
1.1.3. Thermochemical heat storage (TH-TES)
Thermochemical heat storage refers to the techniques for storing and releasing heat through reversible endo/exothermic thermochemical reactions, as represented in equation (3), as

During an endothermic reaction (charging), the storage medium ( ) absorbs heat from the surrounding environment and it typically splits into two or more chemical substances (
) absorbs heat from the surrounding environment and it typically splits into two or more chemical substances ( and
and  ) which can be stored individually for a long time: The reverse reaction represents the discharging process, and the reaction between
) which can be stored individually for a long time: The reverse reaction represents the discharging process, and the reaction between  and
and  releases the same amount of heat stored during the endothermic reaction. The total exchanged heat depends on the reaction enthalpy and the quantity of material.
releases the same amount of heat stored during the endothermic reaction. The total exchanged heat depends on the reaction enthalpy and the quantity of material.
TH-TES materials can be further classified as chemical or sorption systems. In chemical systems, a considerable amount of heat is generated from an exothermic synthesis reaction, and their working principle is properly described by the model reaction reported in equation (3). In sorption systems, a gas (the sorbate) reacts with a sorbent, which can be solid (absorption reactions) or liquid (adsorption reactions) [17].
The energy storage density of TH-TES materials is up to ten times higher than that of SH-TES media, and approximately two times higher than that of the most common LH-TES systems [17], as illustrated in figure 3. The considerable heat storage density and efficiency (low heat losses) are at the basis of the great potential of TH-TES systems [18]. However, the full exploitation of this potential is only possible with an efficient heat and mass transfer to and from the storage volume. Boosting transfer efficiency and coping with slow reaction kinetics are the main issues to overcome for the scale-up and commercialization of TH-TES systems [17, 19]. Hence, the technological maturity of TH-TES is lower than that of the other two classes [20, 21].

Figure 3. Comparison of specific energy storage density values of different sensible, latent and thermochemical TES media (data from [17]).
Download figure:
Standard image High-resolution image2. Latent heat TES and phase change materials (PCMs)
LH-TES has become attractive over the other TES technologies for a wide number of applications, and this stems from three main reasons. The first is that the PCMs exhibit a high energy density, which allow them to store and release a considerable amount of heat per unit mass or volume. Therefore, LH-TES systems require considerably less material compared to the traditional sensible heat storage systems, thereby increasing the system flexibility and reducing the initial and maintenance plant costs [2, 22, 23].
The second advantage is that PCMs store and release heat at nearly constant temperature, i.e. their phase change temperature ( ). During the energy storage phase, the PCM reaches the melting temperature and maintains the temperature constant over the whole melting process, regardless of the applied heat flux or small variations of the surrounding environment. Analogously, during heat release, the temperature is kept constant until the crystallization is completed. This feature is attractive in all the TM applications, where the temperature should be stably maintained in a certain range (e.g. indoor thermal regulation of buildings) or under a critical value (e.g. cooling of electronic devices) [24, 25].
). During the energy storage phase, the PCM reaches the melting temperature and maintains the temperature constant over the whole melting process, regardless of the applied heat flux or small variations of the surrounding environment. Analogously, during heat release, the temperature is kept constant until the crystallization is completed. This feature is attractive in all the TM applications, where the temperature should be stably maintained in a certain range (e.g. indoor thermal regulation of buildings) or under a critical value (e.g. cooling of electronic devices) [24, 25].
The third advantage is represented by the technological maturity of LH-TES systems, which often makes LH-TES a preferred choice over thermochemical heat storage techniques [21]. Although TH-TES systems exceed LH-TES in energy storage density, solid-liquid PCMs are generally easier to handle and exhibit a little volume variation, thereby requiring smaller systems and less support equipment.
2.1. Selection and properties of a PCM
The number of PCMs available on the market is considerable and constantly growing. Hence, it is important to identify the key properties that make a PCM the most suitable for a specific application [26].
The primary criterion for selecting a PCM is the phase change temperature, which must be below that of the heat source but above that of the working environment. For LH-TES systems intended to avoid overheating, the most suitable PCM is the one that has the melting point just slightly below the maximum allowed temperature, as this reduces the melting rate and increases the TM window [27–29].
The phase change enthalpy is another very important property, as it represents the amount of energy that a PCM can store and release per unit mass or volume. A high phase change enthalpy leads to smaller system sizes, as less material is required to store a certain energy amount. PCMs with high melting enthalpy generally feature also a high specific heat capacity, which is desirable as it boosts the sensible heat stored before and after the melting temperature range, thereby enhancing the total exchanged heat [26, 30].
A suitable PCM should also feature a high thermal conductivity both in the solid and in the liquid phase, because this favors heat transfer from the source into the whole PCM mass [31, 32]. Other important qualities are the density and density variation upon phase transitions, the physical and chemical stability over many thermal cycles, the chemical compatibility with the container, the non-corrosiveness and non-toxicity. Finally, the ideal PCM should exhibit congruent melting, negligible supercooling effect and completely reversible melting/crystallization cycles (low hysteresis) [22, 33, 34].
The PCMs currently available on the market do not match all these criteria at once, but the recent progresses in materials research and design are opening new possibilities for selecting the most suitable PCM with enhanced performance.
2.2. Classification of PCMs
The PCMs are classified according to the type of phase transition they undergo in the TES process. As reported in figure 4, PCMs can be subjected to a gas–liquid, solid-gas, solid-liquid or solid-solid transition. The solid-liquid PCMs are by far the most widely used, because they are cheaper and more numerous than the solid-solid PCMs and do not involve gas evolution, accompanied by a large volume variation unlike the solid-gas PCMs [12, 35]. Therefore, this review will focus on solid-liquid PCMs, which can be classified as organic PCMs, inorganic PCMs, and eutectic mixtures of organic and inorganic materials, according to the generally accepted classification [12, 36].

Figure 4. Classification of solid-liquid phase change materials (adapted from [34]).
Download figure:
Standard image High-resolution image2.2.1. Organic PCMs
Organic PCMs are the most widely used in the low-medium temperature range (0 °C–100 °C) and are applied especially for the TM of buildings and electronic devices [24, 25]. They comprise oligomers or polymers with a broad range of molecular weights, thus allowing a wide choice of the working temperature. Moreover, organic PCMs are generally inexpensive and easy to handle, exhibit a relatively high energy density, do not release volatile toxic substances, and are characterized by congruent melting and negligible supercooling. On the other hand, one of the main disadvantages is represented by their low thermal conductivity; this issue is addressed in different ways, namely by increasing the heat transfer area, by using highly thermally conductive containers or by adding metallic or carbon-based micro/nano-fillers [12, 37–39]. The second disadvantage of organic PCMs is related to their flammability, which is true especially for paraffin-based PCMs and derives from their hydrocarbon nature [40, 41]. However, their flash point is approx. 150 °C–200 °C, well above their typical operating temperature range, and it increases with the molecular weight [42, 43]. Organic PCMs comprise paraffin waxes, poly(ethylene glycol)s (PEGs), and fatty acids, but also other compounds such as ketones, esters, ethers, halogen derivatives, sulphur compounds and oleochemical carbonates [44].
2.2.1.1. Paraffin waxes
Paraffin waxes are the most diffused organic PCMs and consist in saturated hydrocarbons (n-alkanes) with chemical formula CnH2n +2. Paraffins embody all the aforementioned advantages of organic PCMs: they are generally cheaper and exhibit higher heat of fusion (200–240 J g−1) and specific heat capacity (2.1–2.4 kJ (kg · K)−1) than other organic PCMs [45]. Paraffins have a remarkable thermal stability over repeated thermal cycles (also after 1000–2000 cycles), a relatively low vapor pressure and an exiguous volume change upon phase transition [46]. Paraffins are largely available on the market with a broad range of chain lengths; those between C5 (pentane) and C15 (pentadecane) are liquid at room temperature, while those containing a higher number of carbon atoms are solid with a waxy appearance. Commercial paraffinic PCMs are mixtures of different hydrocarbons that do not exhibit phase segregation even after many thermal cycles, and the formulation is designed to match a specific transition temperature while maximizing the phase change enthalpy. Paraffins are resistant to chemical and environmental degradation, but they can manifest slow oxidation when exposed to oxygen [2], which encourages the use of sealed containers.
2.2.1.2. Fatty acids
The second most popular organic solid-liquid PCM class is that of fatty acids, represented by the chemical formula CH3(CH2)2nCOOH and classified as caprylic (C8), capric (C10), lauric (C12), myristyl (C14), palmitic (C16) and stearic (C18) acid, according to the number of carbon atoms. These fatty acids show high phase change enthalpies (45–210 J g−1) and a wide range of melting temperatures (17 °C–70 °C). Fatty acids show high phase change enthalpies (45–210 J g−1) and a wide range of melting temperatures (−5 °C–70 °C) but are up to three times more expensive than paraffins and mildly corrosive [47, 48]. Their most interesting feature is that they can be produced from bio-sources and are biodegradable, and therefore they have been the subject of extensive studies to replace paraffins for low/medium temperature applications, such as the solar energy storage and TM of indoor environment [45, 49].
2.2.1.3. Poly(ethylene glycol)s (PEGs)
Known also as poly(ethylene oxide)s (PEOs), PEGs are composed of dimethylene ether chains HO–(CH2–O–CH2–)n–CH2–CH2–OH and are soluble in water and in some organic solvents, thanks to the amphiphilic nature of their chain combining hydrocarbon sequences and polar groups [44]. Also for this class of organic PCMs, the melting temperature and enthalpy increase with the molecular weight; for instance, PEG600 melts at 18.5 °C absorbing 121.1 J g−1, while PEG2000 melts at 61.2 °C absorbing 176.2 J g−1. PEGs are biodegradable and biocompatible and are also used in drug delivery systems [50]. This feature has expanded the use of PEG as a PCM in applications inside the human body, e.g. to subtract heat during the in-situ polymerization of acrylic bone cements and avoid overheating and damage to the surrounding biological tissues [51].
2.2.2. Inorganic PCMs
Inorganic PCMs exhibit an enthalpy per unit mass similar to their organic counterpart, but since they have a higher density, they can have a remarkably higher enthalpy per volume, thereby allowing the production of more compact TES systems [8]. Moreover, the thermal conductivity of inorganic PCMs can be considerably higher than that of their organic counterpart [24]. For these reasons, they are the preferred choice in the medium-high temperature range (100 °C–1000 °C) and when there are no strict requirements on non-corrosiveness. Inorganic PCMs comprise several classes of materials, such as salts, salt hydrates and metal alloys [26].
2.2.2.1. Salts and salt hydrates
Salts and salt hydrates have similar molecular structure, but the crystalline lattice of salt hydrates is not so closely packed and can easily host water molecules. Common salts and salt hydrates used as PCMs are NaNO3, KNO3, KOH, MgCl2, NaCl, MgCl2 · 6H2O, CaCl2 · 6H2O, and Na2SO4 · 10H2O, also called Glauber's Salt [26, 52]. While salts undergo a proper melting/crystallization transition, for salt hydrates the solid-liquid phase change is a dehydration-hydration process, in which the compound releases the coordinated water molecules becoming an anhydrous salt (or a lower hydrate). The vapor pressure of hydrated salts is considerably higher than that of the other solid-liquid PCMs and increases with the hydration degree; this can cause water loss and a deterioration of the TES properties. Moreover, salt hydrates often suffer from supercooling problems. Above the dehydration temperature, the anhydrous salt may segregate and settle at the bottom of the container due to its higher density, which hinders and slows down the rehydration process. This issue is usually tackled by stirring, by adding excess water to favor solubilization of the whole mass of anhydrous salt, or by adding a thickening agent (e.g. borax, graphite) that reduces the extent of phase separation and often acts as a nucleating agent [22, 27, 53]. The phase change temperature of salts and salt hydrates ranges from 10 °C to 900 °C. However, for applications requiring a melting point up to 70 °C–80 °C, organic PCMs are often preferred due to the lower cost, easier handling, lower vapor pressure, superior long term stability, lower supercooling and lower tendency to incongruent melting [22].
2.2.2.2. Metal alloys
Metals have not been extensively investigated as PCMs so far, but they are starting to attract attention thanks to their considerable thermal conductivity and thermal stability. The most promising metallic PCMs are cesium, gallium, indium, tin and bismuth for low-temperature applications, and zinc, magnesium, and aluminum for applications at higher temperatures. Metallic PCMs cover a broad range of melting temperatures, from 30 °C of neat gallium to 660 °C of aluminum. However, PCMs with a lower transition temperature are not widely used due to their low phase change enthalpy. Conversely, high-melting metals such as Al and Mg alloys also exhibit a considerable phase change enthalpy (350–520 J g−1) [7], which makes them attractive for high-temperature solar heating applications, in replacement of inorganic salts that are thermally unstable and prone to phase segregation [26].
2.2.3. Eutectic PCMs
Eutectic PCMs are mixtures of organic or inorganic compounds that melt and solidify congruently. They present a sharp melting point and a high phase change enthalpy, and their properties can be tailored to meet the requirements of a specific application. They are completely miscible in the molten state and freeze forming an intimate mixture of crystals [27], which accounts for a phase transition without segregation. As they are generally designed for a target application, they are usually more expensive than the other classes of PCMs [5, 12].
2.3. Confinement techniques for organic PCMs
Since organic solid-liquid PCMs are the most widely used materials for TES at low-medium temperatures (0 °C–100 °C), this review will be focused on this type of TES materials. Among the major drawbacks of organic PCMs is the need for confinement, to avoid leakage above the melting temperature. Confinement techniques can be divided into (i) encapsulation methods and (ii) shape-stabilization methods.
2.3.1. Encapsulation
Encapsulation methods involve a container that physically separates the PCM from the surrounding environment, is stable in the whole working temperature range and accommodates the phase transition and the associated volume change [54]. The containers can be of various sizes, shapes, and materials; one can talk about macro-, micro- or nano-encapsulation.
Macro-encapsulation is the simplest method of confining a PCM, as it involves the use of a box or a tank, made of a thermally conductive material (e.g. aluminum, stainless steel) that is chemically compatible with the PCM, to prevent any undesired chemical reactions [5, 43]. The container should be properly sealed to avoid the leakage also of the most fluid PCMs, and the design should always consider the volume expansion and contraction during the phase change. When the PCM is used for TM of buildings, the macro-capsule can be the concrete structure itself [55]. When there are no strict requirements on the strength, also thin flexible plastic (e.g. polyethylene) bags can be used, as they accommodate the volume change and do not require an ullage space [3].
On the other hand, micro- and nano-encapsulation feature microbeads with a polymeric or inorganic shell and a PCM core [43]. This confinement technique, with capsules in the micron- or sub-micron-scale range, offers two main advantages. The first is that a microencapsulated PCM is easy to handle and to embed in other materials such as gypsum and concrete by simple mixing, and it can be also added to liquids to produce PCM-enhanced heat transfer fluids [56]. The second advantage is represented by the augmented specific surface area (SSA), which increases the heat transfer surface. The capsule shells must be stable over many melting/solidification cycles and must not have any chemical interaction with the PCM.
There are several physical, physical-chemical, and chemical techniques available to produce PCM microcapsules (MCs) (table 2). Among all techniques, the most diffused, researched, and developed on industrial scale is the in-situ polymerization, which includes interfacial, suspension and emulsion polymerization [25, 57]. These techniques differ from each other mainly in terms of polarity and solubility of the monomers and the initiator. In the interfacial polymerization, the polymeric shell wall is the result of the polymerization of polar and non-polar monomers dissolved in the water and oil phase of an oil-in-water emulsion, respectively. The shell growing at the interface of the two phases becomes a barrier to diffusion and limits the reaction kinetics, thereby influencing the shell thickness and morphology. Common shell materials are polyurea, urea-formaldehyde and melamine-formaldehyde. In suspension polymerization, all the reactants are liposoluble and are dispersed in the water-based medium due to continuous agitation and addition of surfactants. As the PCM must be the oil phase, with this technique it is difficult to encapsulate hydrophilic PCMs such as PEGs or salt hydrates. The shape and size of the resulting particles is strongly influenced by the stirring speed, amount of stabilizer, fraction of the monomer phases and relative viscosity of the droplets and the water medium. The emulsion polymerization is similar to the suspension polymerization, but the main difference is the hydrophilic nature of the initiator, which is dissolved in the water phase. Common shell materials for both these techniques are acrylic and styrenic polymers such as poly(methyl methacrylate) (PMMA), polystyrene, and styrene-divinylbenzene copolymers [58, 59].
Table 2. Advantages and disadvantages of the main microencapsulation methods (adapted from [57], with data from [60]).
| Technique | Advantages | Disadvantages | Particle size (μm) | Encap. ratio (%) |
|---|---|---|---|---|
| Physical methods | ||||
| Spray drying | • Low cost | • Particle agglomeration | 0.1–5000 | 38–63 |
| • Easy to scale-up | • High temperature | |||
| • Versatility | • Uncoated particles | |||
| • Difficult control of the particle size | ||||
| Solvent evaporation | • Low cost | • Difficult to scale-up | 5–1500 | |
| Physic-chemical methods | ||||
| Coacervation | • Versatility | • Difficult to scale-up | 2–1200 | 6–68 |
| • Precise control of the particle size | • Agglomeration | |||
| Sol-gel | • Inorganic shell with high thermal conductivity | • Difficult to bring to industrial level • Complex reactions involved | 0.2–20 | 30–87 |
| Chemical methods | ||||
| Interfacial polymerization | • Wide range of shell materials | • Moderate cost • Solvent handling | 0.5–1000 | 15–88 |
| Suspension polymerization | • Already at industrial level • Versatility | 2–4000 | 7–75 | |
| Emulsion polymerization | 0.05–5 | 14–67 | ||
Another interesting technique to prepare MCs is the sol-gel method, in which a solid shell forms through the gelation of a colloidal suspension (the 'sol'). This suspension, which is often more accurately a solution, is prepared starting from a molecular precursor, such as a metal alkoxide [Mn−(OR)n] [61, 62]. Figure 5 illustrates the general sol-gel encapsulation route to obtain a silica (SiO2) shell starting from tetraethyl orthosilicate (TEOS) as the molecular precursor.

Figure 5. Encapsulation of an organic PCM via a sol-gel process starting from TEOS (adapted from [63]).
Download figure:
Standard image High-resolution imageThe PCM is first dispersed in an aqueous medium with the help of surfactants to form a stable oil-in-water (O/W) emulsion. The final micelle size and thus the capsule dimension are determined by the amount of PCM, the polarity of the aqueous medium and the type and concentration of surfactant. Separately, TEOS is dissolved in a water medium, e.g. a water-ethanol solution, and the pH of the solution is generally lowered to favor the hydrolysis reaction. Once hydrolysis is complete, the precursor solution is added to the PCM emulsion. Here, a controlled condensation reaction of the precursor happens around the PCM droplets, under basic conditions. The result of the condensation reaction is the formation of an extended silica network around the PCM droplet. The main advantage of sol-gel techniques is the possibility to form a ceramic (e.g. SiO2, TiO2, CaCO3) shell, which is generally stronger and stiffer and exhibits a higher thermal conductivity than the polymeric shells [38]. On the other hand, pure metal oxides are usually brittle and subjected to cracks. To mitigate their fragile behavior, the molecular precursor can be chosen that contains a hydrocarbon side group, which will be present also in the final network, resulting in a hybrid organo-ceramic material. For example, organosilica shells produced from methyltriethoxysilane CH3Si(OCH3)3( MTES) are less brittle and more flexible than those produced from TEOS, thanks to the side methyl group that remains in the resulting network [64, 65].
2.3.2. Shape-stabilization
In the field of PCMs, the term 'shape-stabilization' is sometimes used as a synonym of 'confinement' and also includes the microencapsulation techniques [66]. However, the vast majority of the dedicated literature refers to 'shape-stabilization' to indicate all methods to prevent PCM leakage besides encapsulation. Shape-stabilization techniques involve mixing the PCM with layered or porous materials, inorganic nanofillers, or polymer matrices, to produce a compound without any manifest leakage or exudation even when the PCM is in the molten state. Such techniques are generally simpler and cheaper than microencapsulation, but the PCM is not completely isolated from the external environment and some leakage may happen after several thermal cycles [67].
One of the simplest techniques to obtain a shape-stabilized PCM is the dispersion of nanoparticles, such as carbon nanotubes (CNTs) [37], graphene oxide (GO) [68], expanded graphite (EG) [69, 70], expanded graphite nanoplatelets (xGnPs) [71], nanoclays [53], metallic and metal oxide/nitride nanofillers [72], which increase the mixture viscosity and prevent the leakage thanks to their high SSA [66]. The shape-stabilization effect can be achieved by melt blending, vacuum impregnation or grafting of the PCM chains onto the nanofillers. Moreover, the inclusion of carbon- or metal-based fillers can enhance the thermal conductivity, thereby improving the energy storage rate and efficiency especially of in the case of organic PCMs [28].
An analogous effect of enhanced thermal conductivity can be achieved by shape-stabilizing the PCM through a highly conductive foam, where the PCM can be accommodated within the interconnected porosity. Such foams are generally metallic, ceramic, or carbon-based and are characterized by a high porosity, interesting mechanical properties and thermochemical stability [33].
Blending organic PCMs with polymer matrices is another diffused technique to prevent leakage [12]. One of the most widely used strategies consists in combining paraffinic PCMs with polyolefins like polyethylene (PE) and polypropylene (PP), due to their physical-mechanical properties and the chemical compatibility with paraffins, but other commonly used polymers are acrylics, poly(vinyl chloride), polyurethanes and elastomers as ethylene-propylene diene monomer (EPDM) rubbers [12, 73–75]. However, in many cases the satisfactory heat storage properties are accompanied by a decreasing in the mechanical performance after PCM addition [73], which is noticed especially above the PCM melting transition. This often leads to the conclusion that PCM-polymer blends are not suitable as load bearing materials [73].
2.4. Applications of PCMs for heat management
Due to their ability of store a large amount of heat at a nearly constant temperature, PCMs are interesting not only for pure thermal storage applications, for example in solar-thermal power plants, but also for helping to keep the temperature in an optimal range. Examples of PCMs applied for TM can be found in buildings [76], smart textiles [77], electronic devices [78], anti-icing coatings [79], thermoregulating packaging [80], and also biomedical applications [51]. The following sections report some of the most representative examples.
2.4.1. TM in buildings
In the last decades, energy consumption in buildings has dramatically increased due to population growth, climate change, and raised demand for thermal comfort and indoor environmental quality [81]. Housing and tertiary buildings account for the consumption more than 40% of the total primary energy and approximatively 19% of the overall CO2 emissions [5, 81]. Considering the residential buildings, among the energy end-uses, space heating and water heating are responsible for the largest portion of total energy consumption, which is 57% in the U.S., 71% in China and 80% in the E.U [81]. Hence, it is important to address and constantly improve the energy efficiency of buildings. One of the solutions implemented in the last decades involves the use of PCMs in passive or active energy storage systems.
2.4.1.1. Passive storage systems
Passive storage systems include the heating/cooling technologies without an active mechanical device and with little or no external energy inputs. An example of passive storage system is represented by the inclusion of PCMs in wallboards, ceilings or flooring materials, which can store excess energy during the day (peak hours) to release it during the night (off-peak hours), helping to regulate the temperature also in extreme weather conditions [12]. As the indoor thermal comfort is generally considered achieved in a temperature interval between 18 °C and 25 °C, the selected PCMs also manifest phase transition in this range [82].
Even though the concept of using LH-TES in buildings has been known for decades, one of the first systematic studies was performed in 1996 by Feldman and Banu [83], who fabricate PCM-enhanced lab-scale gypsum wallboard samples. The PCM phase, represented by a mixture of fatty acids, was introduced by impregnation (without further encapsulation or shape-stabilization), and it accounted for approx. 25% of the total wallboard weight. The authors evaluated the total TES performance of a room lined with such wallboards and concluded that, after the shooting off of the heating/cooling system, the room temperature could be maintained in the thermal comfort range for several hours longer than a room with traditional wallboards, without impairing the air quality [84].
Particularly attractive are the technologies that allow the storage of excessive solar thermal energy, in order to release heat during the night or to reduce overheating due to solar radiation in the peak hours. An example of this second case is provided by Wang and Zhao [85], who proposed a PCM-enhanced curtain to reduce the solar heat gain through the windows and thus the energy required for cooling, especially useful for modern glass-wall buildings. Numerical investigation demonstrated that the selection of the PCM with the most appropriate melting temperature plays a key role in determining the curtain performance, and that the heat gain of the indoor space can be reduced of up to 16.2% with a PCM layer of 5 mm.
When talking about structural or rigid building elements, there are several ways of integrating PCMs. Besides the aforementioned direct impregnation, other techniques are mixing microencapsulated PCMs with construction materials, and adding a macroencapsulated or variously stabilized PCM as a supplementary layer [24]. One of the most recent studies implementing the first approach is that of Bao et al[ 30], who developed a high performance PCM-enhanced cement composite for passive solar buildings. The PCM phase is represented by a paraffin wax ( = 28 °C) microencapsulated in polymeric shells containing graphite flakes, added during MC synthesis to enhance the thermal conductivity. Such microencapsulated PCM was mixed with the cement matrix together with nanosilica and short carbon fibers, in order to preserve the mechanical properties and further enhance the thermal conductivity. There are some studies in which the PCM is added to construction materials not with the purpose of indoor TM, but for the thermal protection of the material itself. As described in the recent review by Šavija et al[ 86], the insertion of a PCM in concrete can limit temperature fluctuations and reduce the risk of thermally induced cracks.
= 28 °C) microencapsulated in polymeric shells containing graphite flakes, added during MC synthesis to enhance the thermal conductivity. Such microencapsulated PCM was mixed with the cement matrix together with nanosilica and short carbon fibers, in order to preserve the mechanical properties and further enhance the thermal conductivity. There are some studies in which the PCM is added to construction materials not with the purpose of indoor TM, but for the thermal protection of the material itself. As described in the recent review by Šavija et al[ 86], the insertion of a PCM in concrete can limit temperature fluctuations and reduce the risk of thermally induced cracks.
2.4.1.2. Active storage systems
Active storage systems are used to store heat produced when the primary energy source is more abundant or less expensive. In buildings, active storage systems based on PCMs store heat produced by heating systems during the night, so that the energy peak is effectively reduced and shifted to nighttime when the cost of electricity is lower. Lin et al[ 87] developed a floor heating technology integrated with shape-stabilized PCM panels. Such panels, made of paraffin with a melting temperature of 52 °C (75 wt%) and PE as supporting material, were placed under a wood floor onto electric heaters. Large-scale experiments proved that the system was effective in increasing the indoor temperature and in keeping the temperature in an acceptable range long after the heaters were switched off.
2.4.2. Smart textiles
The embedment of PCMs in textile fabrics leads to the production of smart textiles that help regulating the body temperature and are particularly useful in situations of extreme weather conditions [88]. One of the first examples of PCM-enhanced textiles was produced by NASA; nonadecane (= 32 °C) was added to garment fabrics (e.g. in the space suits) to limit the impact of the extreme temperature changes to which the astronauts are subjected during space missions [89]. Later, such smart thermoregulating textiles were employed to enhance the thermal comfort of mountain outdoor clothing and apparel, but also of blankets, mattresses and pillow cases [12].
There are five main ways to embed a PCM in a synthetic textile: (a) mixing the PCM with the melt/wet spun polymer in the form of core filament; (b) producing core-sheath fibers, in which the core is composed by the PCM and the shell is the supporting polymer; (c) introducing PCM MCs in the melt/wet spun polymer; (d) applying microencapsulated PCMs on fabrics using suitable binders or coating materials; (e) adding a PCM-enriched inner layer (e.g. polyurethane foam containing PCM MCs) [77]. Among these techniques, the first three methods are often preferred as they result in versatile multifunctional polymer fibers with the PCM phase stably confined into the surrounding polymer, which reduces the risk of removal during washing [90].
The embedment of PCMs in polymeric fibers is of interest to produce not only smart textiles, but also multifunctional polymer filaments that can be co-woven with continuous reinforcing glass or carbon fibers, to produce a multifunctional yarn containing the matrix, the reinforcement and the PCM. This is an interesting route that can be explored to fabricate thermoplastic composites with TES properties.
2.4.3. TM of electronic devices
Electronic devices are well known to be sensitive to temperature, since their performance and lifetime span depend strictly on their maintenance in a precise temperature range, with a particular attention to avoid overheating. As the electronic components are being equipped with increasingly sophisticated electronics while their dimensions have decreased, the risk of overheating has also grown, and without a proper TM system the heat generation and the associated temperature rise may deteriorate the performance, bring critical components to failure and decrease the user-device interaction comfort [78]. Overheating is among the most common causes of failure of electronic components, as approximately 55% of failures can be related to high temperature problems or poor TM [91]. It has been shown that a decrease of 1 °C can decrease the failure rate of up to 4%, and an increase of 10 °C–20 °C can double the failure probability [91].
An effective TM system must also comply with the weight and size limitations, as these design parameters are increasingly important for electronic components that must be carried around, such as portable and wearable electronic devices, but also batteries and circuitry for electric vehicles (EVs). Hence, PCMs are becoming an attractive alternative to more bulky solutions such as the natural or forced convection (active cooling), also because electronic devices do not normally need to operate continuously for long periods [10]. When the device is in a heat peak and its temperature starts rising, the PCM melts and absorbs excess heat, thereby preventing an excessive temperature burst (passive cooling). When the temperature starts decreasing again, the PCM crystallizes and releases the heat back to the environment.
Pioneering experimental work of investigation on using PCMs on mobile electronic devices was carried out in 2004 by Tan and Tso [92], who assessed the efficacy of a passive cooling unit based on n-eicosane ( = 37 °C) for the TM small hand-held personal computers (personal digital assistants). The PCM was contained in an aluminum case and placed under the heaters simulating the heat generation units of such a device, i.e. the processor and other integrated circuit packaging. The authors concluded that the PCM units were indispensable to keep the working temperature of the device under an acceptable threshold of 50 °C and that the efficacy of the heat storage unit depended not only on the amount of PCM but also on its orientation, which determined the heat flux distribution in the whole device. The same concept was developed by Tomizawa et al[ 93], who developed a passive cooling unit for mobile phones containing a commercial microencapsulated paraffin with a melting temperature of 32 °C. The PCM was mixed with PE and molded as a sheet, which contributed to slow down the temperature increase, and this effect was more evident with thick sheets.
= 37 °C) for the TM small hand-held personal computers (personal digital assistants). The PCM was contained in an aluminum case and placed under the heaters simulating the heat generation units of such a device, i.e. the processor and other integrated circuit packaging. The authors concluded that the PCM units were indispensable to keep the working temperature of the device under an acceptable threshold of 50 °C and that the efficacy of the heat storage unit depended not only on the amount of PCM but also on its orientation, which determined the heat flux distribution in the whole device. The same concept was developed by Tomizawa et al[ 93], who developed a passive cooling unit for mobile phones containing a commercial microencapsulated paraffin with a melting temperature of 32 °C. The PCM was mixed with PE and molded as a sheet, which contributed to slow down the temperature increase, and this effect was more evident with thick sheets.
PCMs can be also employed for the TM of EV batteries, to support or replace the traditional cooling systems based on liquid/air circulation [27, 94]. The first attempts to integrate PCMs in automotive field date back to the early 2000s, when Al Hallaj and Selman [95] proposed a battery pack in which each cylindrical Li-ion cell was wrapped with a PCM layer with a melting temperature in the range 30 °C–60 °C. The authors demonstrated experimentally and numerically that the total temperature fluctuation was considerably lower with the PCM.
Successive approaches tried to address the low thermal conductivity of PCMs and the need for improving the thermal uniformity inside the battery pack. The most promising solutions involved metal or graphite foams as shape-stabilizers and thermal conductivity enhancers [96]. More recently, Zou et al[ 97] studied the introduction of various carbon based nanofillers (e.g. EG, CNTs, graphene) in a paraffin wax, to produce a shape-stabilized PCM to be employed in a 38120-type LiFeO4 battery pack. They found that this composite PCM was able to reduce not only the maximum operating temperature, but also the temperature oscillations.
3. Multifunctional composites
A material can be labelled as multifunctional if it features a set of properties that make it capable of sustaining different types of stimuli at the same time and ready to perform different functions when required. Multifunctional materials combine properties chosen among the structural mechanical properties, like stiffness, strength and toughness, and the non-structural properties, like sensoring functions, optical and magnetic properties, self-healing, thermal/electrical conductivity or insulation, corrosion resistance, tribological properties, and energy harvesting and storage [98–100].
Composite materials are particularly suitable to be designed as multifunctional, as they gather in one material the properties of multiple phases. Composites consist of two or more distinct materials or phases with remarkably different mechanical and/or physical properties, and therefore the properties of the resulting material are noticeably different from those of each constituent, and the composition is properly tailored to obtain the property set that best suits the specific application [101, 102].
Many natural and synthetic materials that perform multiple functions are polymer-matrix composites. They combine outstanding polymer-related properties, like toughness and low density, with the specific properties of the discontinuous phase(s), which can provide stiffness and strength but also several non-structural functions [103]. The following Sections present a general introduction on structural polymer composites and decline the concept of multifunctionality in the combination of structural and TES properties.
3.1. Structural polymer composites
Composite materials pervade our world. Besides being the most widespread material type among biological materials, composites have been produced and used by mankind for thousands of years. However, it is only in the last century, with the advent of high-strength synthetic fibers and the enormous advances in polymer chemistry and technology, that composite materials can offer a performance comparable or even superior to those of well-established structural materials [104].
Composites are generally constituted by a continuous phase, the matrix, and one or more discontinuous phases, the fillers, which are generally stronger and stiffer than the matrix and therefore can be also called reinforcements or reinforcing agents[ 105]. Composite materials can be classified according to the matrix material as polymer matrix composites (PMCs), ceramic matrix composites (CMCs) and metal matrix composites (MMCs). Each of these classes has a particular set of properties and specific application fields, as the matrix has a strong influence on several mechanical properties of the composite, such as transverse modulus and strength and the properties in shear and in compression, as well as on the maximum service temperature [101].
Polymer matrix composites are the most widely used in structural and semi-structural applications at low-medium temperatures (0 °C–250 °C), due to their lightness and high specific stiffness and strength. They combine a polymer matrix with one or more fillers, commonly added to improve stiffness, strength, toughness, and high-temperature performance. As the mechanism of property improvement strongly depends on the filler geometry, composites are generally classified according to the reinforcement type, shape, and size. The reinforcement can be in the form of fibers or particles, as observable from the commonly accepted classification reported in figure 6. Continuous carbon, glass, or aramid fibers are the reinforcements of election when high-end mechanical performance is required, while discontinuous fibers are generally use as a reinforcement for semi-structural purposes, as the properties of discontinuous-fiber polymer composites fill the property (and cost) gap between continuous fiber composites and unreinforced plastics.

Figure 6. Classification of composite materials according to the geometry of the reinforcement (adapted from [101]).
Download figure:
Standard image High-resolution imageThe polymer matrix can be either a thermoplastic or a thermosetting polymer. Although the vast majority of high performance composites have a thermosetting (e.g. epoxy) matrix [104], thermoplastic composites have received considerable attention in the last decades due to their undeniable advantages. Thermoplastics are generally tougher than thermosets, and therefore they are more damage tolerant and have a greater low-velocity impact resistance [106]. Moreover, they are fully reacted, and thus they present a low risk of chemical hazard for the worker, who is not exposed to low-molecular-weight components or by-products. Additionally, thermoplastics do not require refrigeration and have an infinite shelf life. The shaping and consolidation time for thermoplastics is shorter than that of the thermosets, as it takes minutes instead of hours. Nevertheless, thermoplastic require high processing temperatures, in the range of 250 °C–450 °C, considerably higher than the curing temperatures for thermosets (120 °C–175 °C), which demands for presses and tooling materials that can withstand this temperature regime [106]. Due to the possibility of being remelted, thermoplastics can be joined via techniques as resistance welding or ultrasonic welding. They can also be post-thermoformed, which is very attractive as it suggests the possibility of producing continuous-fiber-reinforced flat boards to be subsequently cut and thermoformed into the desired shape. However, since traditional continuous carbon, glass or aramidic fibers have very little extensibility, this can be achieved only with very simple geometries, and also the defect healing via remelting can hardly be practiced without fiber distortion [101].
The main advantages of structural polymer-matrix composites are (i) the possibility to be tailored for optimum strength and stiffness in the different loading directions, and (ii) the combination of low density and high strength and modulus. This latter aspect results in higher specific (i.e. normalized by density) mechanical properties than those of comparable aerospace metal alloys, which in turn allows designing lightweight structures, thereby leading to improved performance and fuel savings, especially in the automotive and aerospace industries. Other advantages of structural composites compared to lightweight metal alloys are the high fatigue life and the corrosion resistance [104]. Due to these features, structural composite materials are mainly applied in construction and transportation fields, but their application is expanding also in marine goods, sports equipment, and infrastructures.
On the other hand, among the main disadvantages of composite materials are the expensiveness of raw materials, fabrication, and assembly, as well as the higher sensitivity to temperature and moisture. Moreover, composite laminates exhibit poor strength in the out-of-plane direction where the matrix carries the primary load, but this aspect can be mitigated by orienting the reinforcement properly. Composites are also more susceptible than metals to impact damage, are prone to suffer from delamination, and can be challenging to repair.
3.2. Multifunctionality of polymer composites
Multifunctional materials combining structural and non-structural functions have enormous potential to impact future structural performance by reducing weight, volume, cost, and energy consumption while enhancing efficiency, versatility, and safety. Since they expand design possibilities and increase the material added value, they are attracting increasing interest from the industrial and academic point of view. New combinations of properties are being embedded in the next-generation of multifunctional materials, which can find applications in the automotive and aerospace industries, but also in civil engineering and in medicine [98]. However, the combination of properties of a multifunctional material should act synergistically and not parasitically. If the addition of self-sensing and self-healing capabilities to a structural material impairs its stiffness and strength excessively, the combination of properties in this material will not bring benefits at the system level [99].
When designing a multifunctional material, multifunctionality should be considered from the very early stages of the design process. Unlike natural materials, whose properties are the result of a locally random evolution process, in the synthetic engineering materials the design must start from the functions and then proceed in a specific direction. This is important because multifunctionality could be expressed at different dimension scales (at the level of the phase, the material or the structure), which must be taken into account during the design of the material and the prediction of final properties of the final component [103].
Composite materials achieve multifunctionality by gathering in one material the properties of multiple phases and at different dimension scales (from continuous fibers to nanometric fillers) [98]. Most of the recent developments in multifunctional materials tend to be polymer-matrix structural composites featuring one or more additional non-structural functions [107–114]. This strategy allows large weight savings at the system level, through the elimination or reduction in the number of multiple monofunctional constituents, and it gives better results than the conventional approach of optimizing weight and geometry of each individual subsystem [99].
3.3. Structural and semi-structural TES composites
Designing polymer composites as multifunctional can further expand the weight saving possibilities pursued when composites are selected as structural materials. The scientific literature reports multiple examples of multifunctional composites combining structural properties and a wide range of non-structural functions, which respond to a broad variety of needs in the most diverse applications.
An interesting field in which multifunctionality of polymer composites can be exploited, although not yet thoroughly investigated, is that of structural composites with TES capability. As explained in section 2.4, among the applications of TES materials are (i) the heat storage for temperature control, for example in the buildings industry, or to produce smart textiles for body temperature regulation, and (ii) the temporary storage of heat to prevent overheating, as in the cooling systems for electronic devices.
When a TES material is used for TM, it is usually only an extra component added to the main structure of a device. However, the resulting increase in weight and volume may be unacceptable for some applications where weight and volume reduction are crucial design parameters. In this case, it would be useful to have a multifunctional material that combines good mechanical properties and the heat storage/management function. With such materials it would be possible to build part of the structure with the 'thermal battery' material, or, in other words, to design a structure that is part of the TM system. This approach is similar to that developed for an outstanding application of multifunctionality, i.e. the structural batteries, which are devices that can carry mechanical load and store electrochemical energy simultaneously.
According to the authors' opinion, these structural or semi-structural lightweight TES composites would be attractive in four main applications:
- (a)In the transportations field, the considerable application of polymer composites in replacement of traditional materials could complicate the TM in the indoor vehicle environment, due to their different (generally lower) thermal conductivity and heat capacity. This could result in an increasing difficulty in maintaining the indoor temperature in the human thermal comfort range. This issue could be addressed by introducing a TES system able to store and release thermal energy in the temperature range around the human comfort temperature (18 °C–25 °C), and the overall performance-to-weight ratio would be maximized if this TES material is part of the structural or semi-structural components themselves. Moreover, a PCM-based material would decrease the energy consumption related to the air conditioning system and the thermal conditioning system of batteries and powertrain, which are the main auxiliary energy consumers in EVs [115]. This concept could be extended to other temperature ranges, which may be interesting for the transportation of food, biomedical items, or other perishable goods.
- (b)In the field of portable electronics, there is an increasing tendency of enhancing the performance, power and functionalities of the devices while reducing weight and volume. This trend brings issues in the thermal regulation, especially in the peak power operations. Passive cooling systems based on PCMs are attracting attention due to their capacity of maintaining the temperature under a certain threshold value and of contrasting the momentaneous but rapid heat generation during peak power operations [10]. However, in all the solutions implemented so far, the PCM is contained in an additional module, while it would be advantageous to embed it directly inside the structural components such as the phone or laptop cases. This concept can be useful for the TM of all the electronic devices that must be carried around and need TM; this group comprises not only mobile phones and laptops, but also the batteries of EVs [116]. Another interesting application in the electronics field would be the production of PCM-enhanced circuit boards, traditionally made of glass fiber (GF) reinforced epoxy resin.
- (c)Among the main drawbacks of polymer-matrix composites, compared to metals or ceramics, are the lower thermal resistance and the loss of mechanical properties with increasing temperature. This can be detrimental for some applications where the composite material is subjected to external heating in service, as in the case of luxury car chassis entirely made in carbon fiber composite. Local overheating and loss of properties could be also determined by dynamic loading and lack of heat dissipation, which can bring to premature failure by fatigue [117]. This effect, critical especially for thermoplastic composites, could be limited by adding a PCM, which absorbs excess heat and avoids temperature rise. This concept has been proven effective by Casado et al[ 118], who inserted a hydrated salt with a transition temperature of 50 °C in a polyamide-based composite reinforced with GFs. In this case, however, the PCM was not employed as an additional filler, but it was added in the gaps of the flanged plate made of the glass-reinforced polyamide. Much more capillary would be the action of the PCM if it were dispersed in the whole composite mass, or at least in those parts more subjected to fatigue damage.
- (d)One of the main problems of outdoor infrastructures operating in cold environments is the formation and accretion of ice. Critical structures such as transmission lines, wind-driven power generators, off-shore oil rigs, and means of transportation like aircrafts and ships can be damaged by the excessive weight of the ice layer and the stresses induced by freeze-thaw cycles [119]. The solutions implemented so far rely on heating methods, mechanical methods, or the circulation of de-icing fluids underneath the surface, which are effective but extremely energy-consuming [120]. More recently, slippery or superhydrophobic coatings have been developed that prevent ice adhesion or reduce ice shear strength, but they generally have low resistance to mechanical abrasion and poor durability to the outdoor weather agents. For the structures built in polymer composites, e.g. the wind turbine blades, an effective alternative could be the introduction of a PCM with phase transition at approx. 0 °C in the whole composite thickness or only in the outermost layers [79]. The heat released during crystallization of the PCM would help in decreasing the ice accretion rate, while reducing the need for heating through an external energy supply.
In all these applications, PCMs are the ideal TES materials, as they work at a nearly constant temperature and exhibit a high enthalpy per unit mass, thus being suitable for applications where weight saving is a key factor. It is therefore fundamental to understand how the PCM addition influences the mechanical properties of the host composite and how this effect varies below and above the PCM phase change.
So far, this investigation has been carried out to some extent only on construction materials like concrete and gypsum, in which a PCM phase is added to enhance the TM of indoor environments while reducing the energy consumption for indoor heating/cooling. The majority of TES systems integrated in walls or flooring materials are represented by organic PCMs with a phase transition temperature in the range 18 °C–25 °C, microencapsulated in polymeric shells. The characterization of such PCM-enhanced construction materials generally evidences that an increase in the PCM content brings an increase in TM properties and a decrease in power consumption. On the other hand, PCM addition determines a lowering of compressive and flexural properties, due to the introduction of a less mechanically strong phase. Therefore, the authors of the revised papers generally conclude that it is important to select the PCM that determines the smallest decrease in mechanical properties while providing the highest TES capability, and that it is fundamental to identify the optimal PCM content to obtain a material with the most suitable property set for a specific application [121].
The open scientific literature provides some examples of polymers containing a PCM. Organic PCMs are usually introduced into polymer matrices in three ways: (i) without further stabilization (formation of a blend) [73, 122–124], (ii) as PCM MCs [125–128], or (iii) shape-stabilized with layered materials or nanofillers, which are generally carbonaceous or ceramic [129–131]. On the other hand, very few cases are reported of PCM-enhanced polymers employable as structural materials. Excluding the field of construction materials, there is a surprisingly limited number of publications about materials designed to combine structural and TES functions. One of the first reported examples is that presented by Wirtz et al[ 132], who developed a sandwich structure made of a graphitic foam impregnated with paraffin ( = 56 °C) as the core and carbon/epoxy laminates as the skin, intended for the TM of electronic devices. The graphitic foam had the twofold function of immobilizing the molten paraffin and enhancing the thermal conductivity, while the carbon/epoxy skins increased the flexural stiffness. The literature reports several other studies on carbon foams as shape-stabilizers for organic PCMs, but they are generally employed only to enhance the thermal conductivity, and little attention is given to the mechanical performance [133–135].
= 56 °C) as the core and carbon/epoxy laminates as the skin, intended for the TM of electronic devices. The graphitic foam had the twofold function of immobilizing the molten paraffin and enhancing the thermal conductivity, while the carbon/epoxy skins increased the flexural stiffness. The literature reports several other studies on carbon foams as shape-stabilizers for organic PCMs, but they are generally employed only to enhance the thermal conductivity, and little attention is given to the mechanical performance [133–135].
More recently, a research group at RMIT University (Melbourne, Australia) developed a woven GF/epoxy laminate containing paraffin MCs and investigated the impact of the PCM on the mechanical and thermal properties of the host laminate [136–138]. An increase in the PCM weight fraction determined a decrease in the tensile, flexural and compressive properties, due to the intervention of additional damaging mechanisms as delamination, matrix cracking, and fiber/matrix debonding, and the authors indicated the weak interfacial adhesion between the capsules and the matrix as one of the causes for the decrease in mechanical performance. On the other hand, the mild processing conditions allowed retaining most of the initial PCM enthalpy, but thermogravimetric analysis (TGA) showed that a fraction of the capsules was damaged during processing. Nevertheless, these results were promising for the development of such structural TES polymer-matrix composites, which exhibited a phase change enthalpy of up to 40 J g−1.
Our group has recently expanded the investigation of structural and semi-structural TES composites, by considering and comparing several combinations of polymer matrices, reinforcements and PCMs. The research work has seen the production and characterization of epoxy/carbon composites containing paraffin stabilized with CNTs or microencapsulated paraffin [139–145], polyamide composites enhanced with a microencapsulated paraffinic PCM and reinforced with continuous or discontinuous fibers [146–150], and a fully biodegradable laminate based on thermoplastic starch, ultra-thin wood laminae and PEG [151].
4. Thermoplastic composites for TES
Thermoplastic composites have received considerable attention in the last decades, due to some advantages over their thermosetting counterpart, as explained in section 3.1 [104, 106]. Even though the use of thermoplastic composites can be advantageous for some applications, a very exiguous number of examples have been reported in the literature that deal with thermoplastic structural or semi-structural composites containing organic PCMs. The advantages and drawbacks of producing such composites are discussed through the presentation of three case studies, which illustrate the materials selection, sample preparation, and results of the characterization.
4.1. Case study 1: polyamide/GF laminates containing paraffinic PCMs
The first case study deals with the introduction of two powder-like paraffinic PCMs in polyamide12(PA12)/ GF bidirectional laminates [146]. The work aimed at comparing the performance of two PCMs with similar phase change enthalpy (≈200 J g−1) and temperature (≈43 °C) when embedded in a traditional thermoplastic matrix reinforced with continuous GFs and at studying the properties of these laminates as a function of the PCM content. The main difference between the two employed PCMs is the confinement technique. The first PCM is the microencapsulated paraffin MPCM43D produced by Microtek Laboratories Inc. (Dayton, OH, US), with an average capsule diameter of 20 µm, from now on called MC. The second is a paraffinic PCM shape-stabilized with CNTs, prepared by adding CNTs to melted paraffin in a weight content of 15 wt%. The paraffin-CNT mixture was then cryogenically grinded under liquid nitrogen and then sieved with a 300 µm metallic sieve to obtain a micrometric powder that can be easily mixed with a polymer matrix, with an average powder size of 53 ± 30 µm, from now on called ParCNT.
PA12 was melt compounded with either MC or ParCNT in different weight percentages (15–60 wt%), and the blends were hot-pressed to produce thin films for the subsequent fabrication of the laminates via film stacking. Five laminates were prepared: the neat PA-GF laminate without PCM, two laminates in which the matrix contained 30 wt% and 60 wt% of MC (PA-MC30-GF and PA-MC60-GF), and two laminates containing the same initial fractions of ParCNT (PA-ParCNT30-GF and PA-ParCNT60-GF).
One of the most important parameters during the production process of such laminates is the viscosity of the matrix, which should be low enough to allow a complete fiber wetting. The viscosity of the PA/PCM blends increases with the PCM content, as shown by a sensible decrease in the melt flow index (MFI) (figure 7). For the samples with MCs, the MFI decreases with an increase in the MC content and reaches a minimum of 5.2 g/10 min, but a more dramatic decrease is observed for the samples with ParCNT. For the highest concentrations, the measurements could be performed only with a mass of 5 or 10 kg, and the resulting MFI values were very low (0.7 g/10 min). This is probably due to the increasing content of CNTs, which are well known to heavily increase the viscosity of molten polymers [152]. This implies that, although both PCMs types decrease the fluidity of the PA, the effect of the MC is less intense than that of the ParCNT, and that it is unlikely to obtain a defect-free composite at elevated ParCNT contents.

Figure 7. Melt flow index (MFI) of the PA/PCM blends as a function of the nominal PCM content (at 230 °C and 2.16 kg; single tests at 5 kg and 10 kg are also indicated) (adapted from [146]).
Download figure:
Standard image High-resolution imageThis is evident from the optical microscope micrographs (figure 8) and the data of volume composition of the prepared laminates (table 3). The MC-containing laminates are of good quality, as they contain few matrix-rich zones and the fibers are properly wetted, but the interlaminar region is thicker than that of the neat PA-GF laminate (not shown), due to the concentration of the MC phase in this region rather than among the fibers of the same yarn. The concentration of the PCM in the interlaminar region is observable also on the ParCNT-containing laminates, but for these laminates the quality appears worse; especially in the laminate PA-ParCNT60-GF, the fibers are not properly wetted, and the fabric weaving seems distorted. The worse quality of this laminate is observable also from the lower fiber volume fraction and the considerably higher porosity (table 3), while all the other laminates show approx. the same fiber content (50–55 vol%) and void volume fraction (≈2 vol%).

Figure 8. Optical microscope micrographs of the polished cross sections of the PA/PCM/GF laminates. (a) PA-MC30-GF; (b) PA-MC60-GF; (c) PA-ParCNT30-GF; (d) PA-ParCNT60-GF.
Download figure:
Standard image High-resolution imageTable 3. Volume compositions of the PA/PCM/GF laminates and results of the DSC tests (data from [146]).
| Sample |
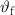 (vol%) (vol%) |
 (vol%) (vol%) |
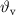 (vol%) (vol%) |
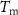 (°C) (°C) |
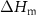 (J g−) (J g−) |
 (%) (%) |
 (°C) (°C) |
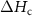 (J g−) (J g−) |
|---|---|---|---|---|---|---|---|---|
| MC | – | – | 43.5 | 200.1 | 100 | 30.0 | 200.0 | |
| ParCNT | – | – | 42.6 | 205.5 | 100 | 33.5 | 206.0 | |
| PA-GF | 53.4 | 44.5 | 2.1 | – | – | – | – | – |
| PA-MC30-GF | 55.5 | 42.2 | 2.3 | 43.9 | 6.5 | 44.2 | 25.2 | 3.1 |
| PA-MC60-GF | 53.2 | 44.3 | 2.5 | 46.3 | 17.1 | 52.7 | 23.2 | 13.8 |
| PA-ParCNT30-GF | 50.5 | 47.7 | 1.8 | 43.0 | 5.3 | 31.9 | 39.7 | 2.0 |
| PA-ParCNT60-GF | 32.4 | 57.8 | 9.8 | 43.5 | 8.4 | 15.8 | 31.3 | 5.6 |
 = volume fraction of fibers;
= volume fraction of fibers;  = volume fraction of matrix (PA/PCM system);
= volume fraction of matrix (PA/PCM system);  = volume fraction of voids;
= volume fraction of voids;  melting temperature of the PCM (°C);
melting temperature of the PCM (°C);  = PCM melting enthalpy (J/g);
= PCM melting enthalpy (J/g);  = relative PCM melting enthalpy (%);
= relative PCM melting enthalpy (%);  = crystallization temperature of the PCM (°C);
= crystallization temperature of the PCM (°C);  = PCM crystallization enthalpy (J/g).
= PCM crystallization enthalpy (J/g).
The most important test to assess the TES capability is differential scanning calorimetry (DSC), which allows the measurement of the total heat exchanged during the phase change of the PCM and the temperature interval in which it occurs. The DSC thermograms of the first heating scan, reported in figure 9, show two endothermic peaks, the first at approx. 40 °C, related to the melting of the PCM, and the second at approx. 180 °C associated to the melting of the PA phase. The melting and crystallization enthalpy of the PCMs in the prepared composites are evaluated by calculating the area of the endothermal and exothermal peak, respectively. In order to evaluate the TES efficacy of composites, the results could be compared to those of the two neat investigated PCMs (MC and ParCNT), which show a transition enthalpy of approx. 200 J g−1 either in heating or in cooling. As reported in table 3, the phase change enthalpy of the laminates is remarkably lower than expected by considering their nominal PCM mass fraction. This is evident from the values of relative melting and crystallization enthalpy ( ), calculated through equations (4
a) and (5
b) as
), calculated through equations (4
a) and (5
b) as



Figure 9. DSC thermograms (first heating scan) of the laminates PA/PCM/GF (10 °C min−1; N2) (data from [146]).
Download figure:
Standard image High-resolution imagewhere and  are the melting and crystallization enthalpy values of the PCM measured on the polymer/PCM mixtures and on the reinforced composites,
are the melting and crystallization enthalpy values of the PCM measured on the polymer/PCM mixtures and on the reinforced composites,  is the nominal weight fraction of PCM in the sample and
is the nominal weight fraction of PCM in the sample and  and
and  are the melting and crystallization enthalpy values of the neat PCM. A relative phase change enthalpy close to 100% suggests that the PCM content still present in the composite after processing is close to the nominal (initial) fraction, and that the PCM preserves its capability of melting and crystallizing also when embedded in a composite. Conversely, a low relative phase change enthalpy indicates that the PCM degrades or leaks out of the composite during processing, due to high temperatures and pressures applied or to a poor shape-stabilization. Both these phenomena occur in these composites, for which the processing conditions applied during melt compounding and hot-pressing cause a partial degradation of the PCMs. The composites with ParCNT present a much greater decrease in
are the melting and crystallization enthalpy values of the neat PCM. A relative phase change enthalpy close to 100% suggests that the PCM content still present in the composite after processing is close to the nominal (initial) fraction, and that the PCM preserves its capability of melting and crystallizing also when embedded in a composite. Conversely, a low relative phase change enthalpy indicates that the PCM degrades or leaks out of the composite during processing, due to high temperatures and pressures applied or to a poor shape-stabilization. Both these phenomena occur in these composites, for which the processing conditions applied during melt compounding and hot-pressing cause a partial degradation of the PCMs. The composites with ParCNT present a much greater decrease in  , which does not even show a trend with the ParCNT content. This suggests that the degradation of ParCNT during the process is greater than that of MC, which support the observation that a microencapsulated PCM can be more easily incorporated in a glass fiber/PA12 laminate than a shape-stabilized one, due to the generally higher thermal stability.
, which does not even show a trend with the ParCNT content. This suggests that the degradation of ParCNT during the process is greater than that of MC, which support the observation that a microencapsulated PCM can be more easily incorporated in a glass fiber/PA12 laminate than a shape-stabilized one, due to the generally higher thermal stability.
For the mechanical properties, table 4 shows the values of tensile elastic modulus, strength, and strain at break, as well as the interlaminar shear strength (ILSS). The stress at break decreases upon PCM addition, and this effect is more evident for the laminates with ParCNT, in which also the elastic modulus is lower than that of the neat PA-GF laminate. Conversely, the elastic moduli of the laminates with MC are not significantly different from that of PA-GF, which suggests that the stiffness is mainly determined by the elastic properties of the reinforcement, and the matrix fulfils its load-transfer role properly. Also the strain at break is not negatively affected by the PCM addition. Therefore, it can be concluded that the addition of PCM MCs influences the mechanical properties of the host laminate only marginally, while a sensible decrease in the mechanical performance can be appreciated with ParCNT addition. In previous works, the elastic modulus of MCs was evaluated as 28 MPa [153, 154], while the modulus, strength and strain at break of ParCNT are approx. 1.5 GPa, 5.5 MPa, and 0.7% [139].
Table 4. Main results of the mechanical investigation of the PA/PCM/GF laminates (tensile tests and short-beam shear tests) (data from [146]).
| Sample |
 (GPa) (GPa) |
 (MPa) (MPa) |
 (%) (%) | ILSS (MPa) |
|---|---|---|---|---|
| PA-GF | 13.1 ± 0.3 | 160 ± 29 | 2.7 ± 0.3 | 11.2 ± 1.7 |
| PA-MC30-GF | 12.7 ± 0.9 | 124 ± 18 | 3.0 ± 0.4 | 9.8 ± 1.7 |
| PA-MC60-GF | 13.3 ± 0.7 | 114 ± 10 | 3.0 ± 0.7 | 9.4 ± 2.1 |
| PA-ParCNT30-GF | 9.9 ± 0.4 | 92 ± 9 | 3.5 ± 0.1 | 6.7 ± 0.3 |
| PA-ParCNT60-GF | 6.5 ± 0.1 | 56 ± 15 | 3.5 ± 1.1 | 5.5 ± 1.8 |
 = elastic modulus (GPa);
= elastic modulus (GPa);  = stress at break, corresponding also to the maximum stress
= stress at break, corresponding also to the maximum stress  (MPa);
(MPa);  = strain at break, corresponding also to the strain at the maximum stress (%); ILSS = interlaminar shear strength.
= strain at break, corresponding also to the strain at the maximum stress (%); ILSS = interlaminar shear strength.
A similar observation can be made for the ILSS. The laminates with MC do not perform significantly differently from the laminate without PCM, while the ILSS of the of the laminates with ParCNT sharply decreases with an increase in the PCM content. This can be attributed to the noticeable porosity and the lower mechanical properties of the matrices with ParCNT. In conclusion, the microencapsulated PCM is more suitable to produce laminates in combination with PA and GF, since it exercises a lower influence on the matrix viscosity, has a greater thermal stability, and impacts the mechanical properties of the laminates marginally. However, due to the processing-related PCM degradation, the losses in TES capability are massive.
Two strategies can be adopted to try to produce thermoplastic composites while limiting the PCM degradation, which are presented in sections 4.2 and Par. 4.3, respectively. The first strategy consists in employing discontinuous fibers instead of a continuous fiber fabric. In this way, the composites can be produced by melt compounding followed by a single hot-pressing step, which allows avoiding one processing step necessary for the production of GF laminates. The second strategy involves a reactive thermoplastic resin, which is provided as a low viscosity precursor and can be processed at room temperature similarly to a thermosetting resin, thereby drastically reducing the risk of thermo-mechanical degradation. As this second strategy nowadays is not implementable with traditional thermoplastics, an additional approach can be considered that involves the production of commingled yarns containing PCM-enhanced thermoplastic filaments and continuous reinforcing fibers. This strategy not only allows the production of a component made of traditional thermoplastic laminate in fewer processing steps, but it also shifts the multifunctionality from the composite level to the yarn level. This approach was investigated by our group through the production of PP filaments containing paraffin MCs [155].
4.2. Case study 2: PCM MCs in semi-structural carbon/polyamide composites
The present case study deals with the preparation of discontinuous-fiber composites having the same PA12 matrix and the same microencapsulated PCM described in section 4.1, but reinforced with discontinuous carbon fibers of two different lengths, i.e. 6 mm and 100 µm and denoted as carbon fibers 'long' (CFL) and carbon fibers 'short' (CFS), respectively. PA was mixed with MC and either CFL or CFS in different weight concentrations, and the composites were prepared by melt compounding and hot-pressing. The samples are denoted as PA-MCx-CFLy or PA-MCx-CFSy, where x is the nominal MC weight fraction with respect to the total PA-MC mass and y is the weight fraction of either CLF or CFS with respect to the total mass of the composite.
Figure 10 shows the scanning electron microscopy (SEM) micrographs of the cryofracture surface of some selected compositions. The processing parameters successfully produce a homogeneous distribution of both the carbon fibers and the MCs in the matrix. The core–shell structure of this PCM is clearly observable especially in that acquired at higher magnification (figure 10(b)). The MC indicated with a dashed blue arrow shows both the thin polymer shell and the paraffinic core. The irregular morphology and the voids of the core indicate a certain aptitude of the capsules to accommodate the volume variation during the phase change and avoid the shell rupture [57]. In other cases (solid red arrow), the shell is still partly observable, but the core has been almost completely removed. The adhesion between the shell and the PA matrix is fairly good, even though some debonding is observable. The same considerations can be made for the samples containing both capsules and fibers, as appreciable from the micrograph of the sample PA-MC50-CFL20 (figures 10(c)–(d)). However, the fracture surface of this sample looks irregular, which may indicate the presence of voids and defects within the composite. This could stem from the considerably high amount of fillers (MC and CFL) and the high viscosity during compounding, which could have prevented a proper molding of the composite during the hot-pressing step.

Figure 10. SEM micrographs of the cryofracture surface of some selected compositions: (a, b) PA-MC50; (c, d) PA-MC50-CFL20.
Download figure:
Standard image High-resolution imageThe results of the DSC analysis are shown in figure 11, which reports representative thermograms of the first heating scan and the cooling scan for some selected compositions, i.e. the samples PA-MCx-CFS20. The main results of the DSC analysis of all the samples are summarized in table 5. As found for the PA-based samples described in section 4.1, the melting and crystallization peaks of MC are found at approx. 43 °C on heating and 30 °C on cooling, respectively, while the signals at higher temperature are related to the phase transition of PA. The melting/crystallization temperatures of the PCM ( ,
,  ), regarded also in this case as the peak temperatures of the endo/exothermic signals, do not vary significantly with the carbon fiber content, and the phase change enthalpy (
), regarded also in this case as the peak temperatures of the endo/exothermic signals, do not vary significantly with the carbon fiber content, and the phase change enthalpy ( ) increases with the weight fraction of MC. The value of, calculated through equation (4
a), is generally below 100%, and it decreases as the MC content increases. However, these values are mostly higher than those calculated for the samples presented in section 4.1, which highlights that applying milder processing conditions and avoiding one hot-pressing step is helpful to preserve shell integrity and minimize PCM degradation.
) increases with the weight fraction of MC. The value of, calculated through equation (4
a), is generally below 100%, and it decreases as the MC content increases. However, these values are mostly higher than those calculated for the samples presented in section 4.1, which highlights that applying milder processing conditions and avoiding one hot-pressing step is helpful to preserve shell integrity and minimize PCM degradation.

Figure 11. DSC thermograms (heating scan and cooling scan) of the samples PA-MCx-CFS20 (10 °C min−1; N2 flow = 100 ml min−1) (data from [148]).
Download figure:
Standard image High-resolution imageTable 5. Results of the DSC tests on the samples PA-MCx-CFL and PA-MCx-CFS (data from [148]).
| Sample |
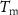 (°C) (°C) |
 (J/g) (J/g) |
 (%) (%) |
 (°C) (°C) |
 (J/g) (J/g) |
|---|---|---|---|---|---|
| MC | 44.1 | 220.1 | 100 | 30.2 | 208.2 |
| PA-MC20 | 46.2 | 44.5 | 101.1 | 26.7 | 38.1 |
| PA-MC40 | 47.3 | 91.8 | 104.3 | 26.4 | 84.1 |
| PA-MC50 | 49.9 | 100.8 | 91.6 | 25.3 | 93.8 |
| PA-MC60 | 47.8 | 116.6 | 88.3 | 24.1 | 108.9 |
| PA-MC20-CFL20 | 45.5 | 33.0 | 93.8 | 31.5 | 27.9 |
| PA-MC40-CFL20 | 45.8 | 28.9 | 41.1 | 31.4 | 23.8 |
| PA-MC50-CFL20 | 46.6 | 46.9 | 53.3 | 26.1 | 40.9 |
| PA-MC60-CFL20 | 45.8 | 44.5 | 42.1 | 30.2 | 37.6 |
| PA-MC20-CFS20 | 45 | 33.3 | 94.6 | 31.7 | 28.8 |
| PA-MC40-CFS20 | 48.1 | 58.8 | 83.5 | 25.2 | 54.3 |
| PA-MC50-CFS20 | 47.5 | 60.4 | 61.0 | 26.6 | 57.7 |
| PA-MC60-CFS20 | 44.8 | 54.6 | 51.7 | 24.5 | 51.4 |
 melting temperature of the PCM (°C);
melting temperature of the PCM (°C);  = PCM melting enthalpy (J/g);
= PCM melting enthalpy (J/g);  = relative PCM melting enthalpy (%);
= relative PCM melting enthalpy (%);  = crystallization temperature of the PCM (°C);
= crystallization temperature of the PCM (°C);  = PCM crystallization enthalpy (J/g).
= PCM crystallization enthalpy (J/g).
Nevertheless, some degradation and leakage are still present, as indicated by values of  lower than 100%. The decrease in
lower than 100%. The decrease in  is more evident with at higher MC and carbon fiber fraction. This is linked to the higher melt viscosity and the consequently higher shear stresses to which the capsules are subjected during compounding, which damages the thin capsule shell and causes the leakage and degradation of the paraffinic core. Additionally,
is more evident with at higher MC and carbon fiber fraction. This is linked to the higher melt viscosity and the consequently higher shear stresses to which the capsules are subjected during compounding, which damages the thin capsule shell and causes the leakage and degradation of the paraffinic core. Additionally,  is lower for samples containing CFL compared to those with CFS, which can be related to the higher viscosity and higher shear stresses obtained while compounding mixtures containing CFL.
is lower for samples containing CFL compared to those with CFS, which can be related to the higher viscosity and higher shear stresses obtained while compounding mixtures containing CFL.
This agrees with the results of the dynamic rheological tests, performed with a parallel plate rotational rheometer (figure 12). The viscosity increases upon filler addition, but the MC increase the viscosity significantly only at very high concentrations. On the other hand, the samples containing both MC and carbon fibers show more clearly phenomena such as the viscosity increase, the yield stress rise at low shear rates and the anticipation of the shear thinning region. Moreover, the viscosity is higher for the samples containing CFL than for those containing the same amount of CFS. This suggests that the CFL produce a higher viscosity rise also in the melt compounding process, thereby intensifying the shear stress, MC damage and paraffin degradation. This is in good agreement with DSC results, which highlight a higher PCM experimental weight fraction and enthalpy in the samples containing CFS. However, the difference in the measured viscosity seems too small to justify such discrepancies in the DSC results. It should be considered, though, that the samples analyzed by dynamic shear rheometry have been subjected to melt compounding and hot pressing, which can significantly shorten the fiber length. As fibers with a higher aspect ratio are generally more influent on the polymer melt viscosity, it is reasonable to hypothesize that the shear stresses in the melt compounder are higher at the beginning of the compounding stage, which causes an even more extensive MC damage.

Figure 12. Results of the dynamic rheological test. Values of viscosity as a function of the applied frequency for some representative compositions (parallel plate; gap 1.5 mm; diameter 8 mm; 190 °C; N2) (adapted from [148]).
Download figure:
Standard image High-resolution imageThe greater PCM degradation in CFL-containing composites can be also appreciated from TGA. As observable from figure 13, the addition of carbon fibers delays the beginning of thermal degradation of the neat PA, whilst the addition of MC shifts the thermograms to lower temperatures. The mass loss associated to MC degradation, between 200 °C and 400 °C, is less than expected by considering the nominal MC weight fraction. This is in good agreement with the DSC results, where the phase change enthalpy is less than expected due to some leakage and degradation of PCM. This effect is more evident for the CFL-containing samples than for the samples reinforced with CFS, which agrees with the results of the rheological tests and confirms that the greater increase in viscosity induced by CFL is responsible for the greater capsule breakage and paraffin degradation.

Figure 13. TGA thermograms of the samples (a) PA-MCx-CFL20; (b) PA-MCx-CFS20 (10 °C min−1; N2 flow = 15 ml min−1) (adapted from [148]).
Download figure:
Standard image High-resolution imageAnother interesting parameter of composites containing a PCM is thermal conductivity. A greater thermal conductivity helps homogenizing the temperature of the whole composite mass during heating or cooling, thereby improving the heat exchange efficiency. Figure 14 shows the values of thermal conductivity of some selected compositions determined through laser flash analysis (LFA). As expected, the introduction of carbon fibers increases the thermal conductivity. For the samples containing carbon fibers and MC, the thermal conductivity shows a maximum at 40 °C, while approaching the melting temperature of the PCM. This phenomenon, observed elsewhere in the literature [72], is more evident for the samples containing CFS; this suggests that the effective PCM content is higher for these samples compared to the CFL-reinforced ones, which is consistent with the DSC and TGA results.

Figure 14. Results of the LFA test on the samples PA, PA-CFL20, PA-MCx-CFL20 and PA-MCx-CFS20. Thermal conductivity as a function of temperature (data from [148]).
Download figure:
Standard image High-resolution imageFinally, figure 15 shows the effect of CFS, CFL and MC on the tensile elastic modulus and strength of the prepared composites. The introduction of MC decreases the elastic modulus, while the addition of the carbon fibers increases it. This effect is more evident for CFL than for CFS, because the greater load transfer length determines a higher stiffening and reinforcing capability, as thoroughly reported in the literature [156, 157]. Similar considerations can be made for  , which decreases with an increase in the MC content and increase with the carbon fiber concentration.
, which decreases with an increase in the MC content and increase with the carbon fiber concentration.

Figure 15. Results of the tensile tests. Elastic modulus and maximum stress as a function of the nominal (initial) MC volume fraction for the composites with a carbon fiber content of 0 wt% and 20 wt% (data from [148]).
Download figure:
Standard image High-resolution imageIn conclusion, the processing conditions applied for the production of these discontinuous-fiber composites were milder than those employed to produce the continuous-fiber laminates described in section 4.1, which reduces the degradation of the PCM and results in a higher final relative phase change enthalpy. However, also for these composites the paraffin degradation is not negligible and is more evident for samples reinforced with longer fibers, which cause a higher increase in viscosity. For the mechanical properties, also in this case quasi-static tensile tests highlight a decrease in elastic modulus and maximum stress upon PCM addition, while carbon fibers positively contribute to increase the elastic modulus.
4.3. Case study 3: reactive thermoplastic resin as a matrix for multifunctional carbon-fiber laminates
This case study presents a different approach to produce thermoplastic laminates containing a PCM. This approach aims at preserving the integrity of paraffin MCs by avoiding the high-temperature processing of highly viscous molten polymers [158, 159]. It is made possible by adopting the newly developed thermoplastic resin Elium® (Arkema, Lacq, France), provided as a low-viscosity liquid and processable as a thermosetting resin.
This technique is nowadays applicable only to some selected polymer formulations and not to the majority of the most common thermoplastic polymer matrices. Nevertheless, it is a powerful and effective method that is worth investigating to produce post-thermoformable PCM-enhanced composites containing continuous reinforcing fibers.
These composites were prepared at room temperature, by mixing the reactive resin precursor (EL) with the paraffin MCs presented in sections 4.1 and 4.2 in different weight fractions (0 wt%, 20 wt%, 30 wt% and 40 wt%), and by using the mixtures as matrices to prepare laminates through hand-layup and vacuum bagging techniques. The prepared laminates were labeled EL-CF, EL-MC20-CF, EL-MC30-CF and EL-MC40-CF, according to the nominal MC weight fraction in the matrix. The OM micrographs of the polished cross sections of the laminates are presented in figure 16. As for the laminates presented in section 4.1, the MC phase is preferentially located in the interlaminar region, due to the different dimensions of carbon fibers (average diameter 7 μm) and MCs (average diameter 20 μm). Moreover, the MC concentration in the interlaminar region does not increase considerably with the MC content, but what is evidently seen increasing is the thickness of the interlaminar region, and thus that of the whole laminate. This indicates that the increase in MC content causes a rise in viscosity of the EL/MC mixture, which flows out of the CF fabric slower and with more difficulty, thereby causing a final higher matrix weight (and volume) faction. Therefore, the fiber volume fraction ( ) decrease with increasing initial MC content. This is appreciable from quantitative data of fiber volume fraction (table 6), calculated with TGA, DSC, and density measurements (the details of the calculations are reported in [158]). Conversely, the volume fraction of voids (
) decrease with increasing initial MC content. This is appreciable from quantitative data of fiber volume fraction (table 6), calculated with TGA, DSC, and density measurements (the details of the calculations are reported in [158]). Conversely, the volume fraction of voids ( ) generally increases with the MC content.
) generally increases with the MC content.

Figure 16. Optical microscope micrographs of the polished cross section of the laminates (a) EL-CF; (b) EL-MC20-CF; (c) EL-MC30-CF; (d) EL-MC40-CF; (e) and (f) EL-MC30-CF, higher magnification.
Download figure:
Standard image High-resolution imageTable 6. Results of the DSC tests and volume composition of the samples EL-MCx-CF (data from [158]).
| Sample |
 (vol%) (vol%) |
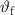 (vol%) (vol%) |
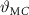 (vol%)
(vol%) |
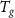 (°C) (°C) |
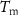 (°C) (°C) |
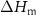 (J/g) (J/g) |
 (°C) (°C) |
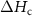 (J/g) (J/g) |
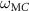 (wt%) (wt%) |
|---|---|---|---|---|---|---|---|---|---|
| MC | – | – | 1 | – | 45.0 | 208.2 | 29.8 | 208.2 | 100 |
| EL | 1.6 ± 2.1 | 56.7 ± 1.6 | 0 | 101.0 | – | – | – | – | – |
| EL-MC20-CF | 2.2 ± 1.3 | 35.6 ± 0.6 | 20.95 ± 0.19 | 102.5 | 46.6 | 30.2 | 27.5 | 31.6 | 14.5 |
| EL-MC30-CF | 1.2 ± 3.2 | 27.4 ± 1.9 | 30.41 ± 0.07 | 99.6 | 45.3 | 45.9 | 29.5 | 45.6 | 22.1 |
| EL-MC40-CF | 5.2 ± 2.4 | 25.0 ± 1.2 | 40.93 ± 0.04 | 98.6 | 46.4 | 66.8 | 27.4 | 65.6 | 32.1 |
 glass transition temperature of the PCM (°C);
glass transition temperature of the PCM (°C);  melting temperature of the PCM (°C);
melting temperature of the PCM (°C);  = PCM melting enthalpy (J/g);
= PCM melting enthalpy (J/g);  = crystallization temperature of the PCM (°C);
= crystallization temperature of the PCM (°C);  = PCM crystallization enthalpy (J/g);
= PCM crystallization enthalpy (J/g);  = experimental weight fraction of microcapsules (wt%);
= experimental weight fraction of microcapsules (wt%);  = voids volume fraction (vol%);
= voids volume fraction (vol%);  = fiber volume fraction (vol%);
= fiber volume fraction (vol%);  = MC volume fraction (vol%).
= MC volume fraction (vol%).
The decrease in  is the main reason for the decrease in mechanical performance, as observable from the data of flexural modulus, stress and strain at break reported in figure 17. The elastic modulus decreases with an increase in the MC content, but this is mainly due to the decreasing fiber volume fraction. The flexural strength follows the same trend, and this is probably due to the presence of additional failure mechanisms such as delamination and failure in the zone subjected to compression, as appreciable from a comparison between the load-displacement curves (reported in [158]). While the neat EL-CF laminate is subjected to a sudden failure after the maximum load, the laminates containing MC are subjected to a progressive failure and present a drop-plateau sequence after the maximum load. The damage was observed starting either in the mid-upper zone, subjected to compression, or in the interlaminar zone. This failure mode, commonly reported for woven fabric composites [160], is typical of materials with a tensile in-plane strength considerably higher than the ILSS, which is likely the case for these laminates.
is the main reason for the decrease in mechanical performance, as observable from the data of flexural modulus, stress and strain at break reported in figure 17. The elastic modulus decreases with an increase in the MC content, but this is mainly due to the decreasing fiber volume fraction. The flexural strength follows the same trend, and this is probably due to the presence of additional failure mechanisms such as delamination and failure in the zone subjected to compression, as appreciable from a comparison between the load-displacement curves (reported in [158]). While the neat EL-CF laminate is subjected to a sudden failure after the maximum load, the laminates containing MC are subjected to a progressive failure and present a drop-plateau sequence after the maximum load. The damage was observed starting either in the mid-upper zone, subjected to compression, or in the interlaminar zone. This failure mode, commonly reported for woven fabric composites [160], is typical of materials with a tensile in-plane strength considerably higher than the ILSS, which is likely the case for these laminates.

Figure 17. Results of the three-point bending tests on the samples EL-MCx-CF as a function of the nominal MC content in the matrix (data from [158]).
Download figure:
Standard image High-resolution imageConversely, the thermal and TES properties strongly increase with the MC content. The DSC thermograms (figure 18) show the PCM melting peak in the heating scan at approx. 45 °C and the crystallization peak in the cooling scan at approx. 30 °C, as observed for the previously reported Case Studies. The position of these peaks is not remarkably affected by the MC content, and the same is true for the glass transition temperature of the EL matrix, located at approx. 100 °C. On the other hand, the phase change enthalpy increases with the MC amount, as observable from the intensity increase of the melting and crystallization peaks. The developed enthalpy reaches 66.8 J g−1 for the sample EL-MC40-CF, which is remarkably higher than that measured on other thermoplastic composites with the same initial MC fraction. This indicates that the processing conditions of this reactive thermoplastic resin are suitable to avoid damage to the MCs and retain the heat storage ability of the paraffinic core, unlike the traditional thermoplastic matrices presented so far.

Figure 18. DSC thermograms of the samples EL-MCx-CF (10 °C min−1; N2 flow = 100 ml min−1) (data from [158]).
Download figure:
Standard image High-resolution imageThis is at the basis of the improved TM performance evidenced through a test with a thermal camera. This test was performed by heating the laminates in an oven at 70 °C and letting them cool to room temperature while measuring their surface temperature with an infrared thermal camera. Figure 19 reports the values of temperature as a function of time during the cooling stage. In the MC-enhanced laminates, the temperature trend shows plateau-like regions caused by the latent heat released during PCM crystallization, which increases the time to reach room temperature considerably.

Figure 19. Thermal camera tests on the samples EL-MCx-CF. Temperature as a function of time during cooling to room temperature (in-plane dimensions 90 × 120 mm2) (adapted from [158]).
Download figure:
Standard image High-resolution imageFinally, the prepared laminates were subjected to an in-depth characterization trough dynamic-mechanical analysis (DMA). Three testing modes were investigated, namely single-frequency scans, multifrequency scans and heating-cooling cycles. The trends of the storage modulus  and the loss modulus
and the loss modulus  acquired during single-frequency scans are illustrated in figure 20. To facilitate the comparison, the values of
acquired during single-frequency scans are illustrated in figure 20. To facilitate the comparison, the values of  of each laminate have been normalized to the value at 0 °C. The storage modulus of all laminates shows a marked decrease at the glass transition of the EL matrix, where
of each laminate have been normalized to the value at 0 °C. The storage modulus of all laminates shows a marked decrease at the glass transition of the EL matrix, where  shows a peak. The laminates containing MC shows an additional transition at the PCM melting temperature, and the drop of is more evident at high MC contents. Interestingly, the correlation between the drop amplitude and the MC weight fraction or the melting enthalpy is linear, with R2 values higher than 0.98. Another DMA parameter that correlates linearly with the melting enthalpy is the area under the
shows a peak. The laminates containing MC shows an additional transition at the PCM melting temperature, and the drop of is more evident at high MC contents. Interestingly, the correlation between the drop amplitude and the MC weight fraction or the melting enthalpy is linear, with R2 values higher than 0.98. Another DMA parameter that correlates linearly with the melting enthalpy is the area under the  peak (reported in [159]). This implies that the DSC test allows one to predict, to a certain extent, the trend of the viscoelastic properties of the composite in the temperature range around the PCM phase change.
peak (reported in [159]). This implies that the DSC test allows one to predict, to a certain extent, the trend of the viscoelastic properties of the composite in the temperature range around the PCM phase change.

Figure 20. Results of the DMA tests on the samples EL-MCx-CF. The reported values represent the peak temperatures. (a) normalized storage modulus  (solid lines) and loss modulus
(solid lines) and loss modulus  (dashed lines) (single cantilever; 1 Hz; 3 °C min−1) (adapted from [158] and [159]).
(dashed lines) (single cantilever; 1 Hz; 3 °C min−1) (adapted from [158] and [159]).
Download figure:
Standard image High-resolution imageAs structural TES composites must withstand repeated thermal cycles around the phase change temperature of the PCM, DMA tests were performed not only on heating, but also on cooling, between −40 °C and 60 °C. This is not a common approach as no other studies can be found in the literature that analyze the trend of viscoelastic parameters of PCM-enhanced polymers on cooling, to the best of the authors' knowledge. Figure 21 illustrates the trends of  during a heating-cooling-heating scan performed at 3 °C min−1 on the three prepared MC-containing laminates. The absolute values of
during a heating-cooling-heating scan performed at 3 °C min−1 on the three prepared MC-containing laminates. The absolute values of  decrease with an increase in the MC weight fraction, which was expected from the decreasing values of the elastic modulus (figure 17). The decreasing step of
decrease with an increase in the MC weight fraction, which was expected from the decreasing values of the elastic modulus (figure 17). The decreasing step of  at the PCM melting is almost completely recovered on cooling, as it reaches 90%–95% of the initial value. The recovery happens with a certain hysteresis, as the crystallization is found at lower temperatures than the melting, as also observed in DSC tests, due to reasons related both to thermal inertia and to the thermodynamics of crystallization. The trends of
at the PCM melting is almost completely recovered on cooling, as it reaches 90%–95% of the initial value. The recovery happens with a certain hysteresis, as the crystallization is found at lower temperatures than the melting, as also observed in DSC tests, due to reasons related both to thermal inertia and to the thermodynamics of crystallization. The trends of  and
and  (reported in [159]) also show that the peak on cooling (crystallization) is found approx. 30 °C lower than the melting peak. From these findings, DMA test appears as a powerful technique to evaluate material stiffness during phase transition of PCM, especially combined with DSC analysis (for a better comparison, the same rate of heating/cooling is suggested).
(reported in [159]) also show that the peak on cooling (crystallization) is found approx. 30 °C lower than the melting peak. From these findings, DMA test appears as a powerful technique to evaluate material stiffness during phase transition of PCM, especially combined with DSC analysis (for a better comparison, the same rate of heating/cooling is suggested).

Figure 21. DMA heating (1)-cooling (2)-heating (3) scans on the samples EL-MCx-CF: values of E' in the temperature range around the phase change temperature of the PCM (single cantilever; 1 Hz; 3 °C min−1) (adapted from [158] and [159]).
Download figure:
Standard image High-resolution imageFinally, multifrequency DMA analysis was performed to assess the effect of frequency on the PCM melting and the glass transition of the EL resin. Figure 22 presents the results of the sample EL-MC30-CF. The shift of the signals towards higher temperature with increasing frequency is observable not only at the glass transition of the EL resin, as commonly observed in polymer composites, but also at the PCM melting. For this transition, the frequency sensitivity is higher below the peak temperature than above it, as after the peak temperature the curves are almost overlapped. This suggests that when the core is completely molten the dependence of the signals on frequency weakens considerably.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 22. Representative DMA multifrequency thermograms of the sample EL-MC30-CF (single cantilever; 0.3-1-3-10-30 Hz; 0.3 °C min−1) (adapted from [158] and [159]).
Download figure:
Standard image High-resolution image{kind=link}
5. Conclusions and future perspectives
This review introduced the main concepts of TES and summarized the most widely used PCMs, with a special focus on organic solid-liquid PCMs and their TM applications at low and medium temperatures (0 °C–100 °C). It then explored the approach of embedding TES and TM functionalities in structural and semistructural materials, through the development of multifunctional polymer composites that could find applications where weight saving and temperature management are equally important, such as in the transportation and portable electronic fields. Moreover, PCMs embedded in polymer composites could also be useful to produce anti-icing structures for wind blades, to thermally protect the composite itself, whose mechanical properties could degrade with a temperature increase.
The concept of structural TES composites was elucidated in the second part of the review, dedicated to the presentation of three case studies. The characterization of the glass/polyamide laminates (section 4.1) evidenced that the MCs are more suitable than CNT-stabilized paraffin (ParCNT) to be compounded with a traditional thermoplastic matrix, due to their higher thermal resistance. However, the melt-compounding and the two hot-pressing operations damage the MC shells considerably, thereby causing paraffin leakage and degradation and diminishing the total final phase change enthalpy. A remarkable improvement of the final TES properties was obtained by reinforcing the PA-based composites with discontinuous carbon fibers, as described in section 4.2. This was possible because this strategy allows avoiding one of the two hot-pressing steps, but this approach is only applicable to discontinuous fiber composites. Further increases in the mechanical and TES properties were achieved by using a reactive thermoplastic matrix, as reported in section 4.3. Such resin allows mild conditions in all the processing steps, which do not cause PCM degradation.
Future work should focus on finding shape-stabilizing agents that also enhance the thermal degradation resistance of the PCM and the thermal conductivity of the composite, since the use of an appropriate shape-stabilizing filler could be a powerful and low-cost alternative to microencapsulation. Moreover, a deeper investigation should be carried out to study and optimize the mechanical and physical properties of the MCs, such as the size, mechanical strength and stiffness of the shell, and adhesion with the surrounding matrix. Furthermore, DMA proved to be a promising tool to investigate the effect of the PCM melting and crystallization on the properties of the host composite, in combination with DSC, and therefore it would be worth exploring the use of this test more in depth. Moreover, it will be fundamental to investigate the quasi-static mechanical properties of the PCM-containing composites above the melting temperature of the PCMs for a better definition of the overall performance in the working temperature range. A key factor will be the proper definition of an adequate balance between the increase in TES properties and the decrease in mechanical properties, especially in the thermal transition interval.